 International Journal of Organic Chemistry, 2011, 1, 1-5 Published Online March 2011 (http://www.SciRP.org/journal/ijoc) Copyright © 2011 SciRes. IJOC An Efficient α-Phosphoryloxylation of Ketones Ye Pu, Jie Yan* College of Chemical Engineering and Materials Sciences, Zhejiang University of Technology, Hangzh ou, P. R. China E-mail: jieyan87@zjut.edu.cn Received February 10, 2011; revised February 28, 2011; accepted March 4, 2011 Abstract An efficient α-phosphoryloxylation of ketones has been developed. When ketones were treated with (diace- toxyiodo)benzene and phosphates in CH2Cl2 at room temperature, the α-phosphoryloxylaction of ketones was easily be carried out, providing the ketol phosphates in good yields. Keywords: α-Phosphoryloxylation, Ketol Phosphate, (Diacetoxyiodo)benzene, Synthesis 1. Introduction Hypervalent iodine reagents have found broad applica- tion in organic chemistry and nowadays frequently used in synthesis due to they are nonmetallic oxidation re- agents and avoid the issues of toxicity of many transition metals commonly involved in such processes [1-10]. They offer high potential for the improvement of known reactions not only from the environmental point of view,they are also potentially interesting reagents for the development of completely new synthetic transfor- mations. Among hypervalent iodine reagents, [hydroxy (tosyloxy)iodo]benzene (Koser’s reagent) is the most popular and useful reagent for the direct α-tosyloxylation of ketones, and with which the α-tosyloxylation of ke- tones has been extensively studied especially in recent years [11-17]. However, to our knowledge, the analogue reaction of ketones for α-phosphoryloxylation has been rather limited and only two methods for preparation of the important ketol phosphates have been known. Using the hypervalent iodine reagent, [hydroxyl ((bis(phenyloxy) phphoryl)-oxy)iodo]benzene to react with ketones was the main method which was reported by Koser’s group in 1988, in this process hypervalent iodine reagent must be pre-prepared from (diacetoxyiodo)benzene and diphenyl phosphate [18-19]. The reaction of the prepared hyper- valent iodine reagent with terminal alkynes in aqueous MeCN was another protocol for preparation of ketol- phosphates [20]. In order to extend the scope of α- phosphoryloxylation of ketones and to prepare more ke- tol phosphates, the development of a simple, mild and efficient α-phosphoryloxylation of ketones is a much- sought process. Herein we would like to report an effi- cient one-pot α-phosphoryloxylation of ketones and a series of ketol phosphates have been prepared in good yields. 2. Results and Discussions At the beginning, we investigated the “one pot” proce- dure to improve Koser’s method due to the preparation of [hydroxyl((bis(phenyloxy)phosphoryl)oxy)iodo]bezene and its reaction with ketones were separated in two steps. When one equivalent of (diacetoxyiodo)benzene (DIB), diphenyl phosphate and acetophenone were mixed in CH2Cl2 at room temperature and stirred for 24 h, the de- sired product of α-phosphoryloxyl acetophenone was separated in 38% yield after separation. Then, a series of experiments were performed on the reaction of (diace- toxyiodo)benzene, diphenyl phosphate and acetophenone to determine the suitable reaction conditions (Scheme 1), the results are summarized in Table 1. The results indicate that the yield depended on solvent and CH2Cl2 was found to be the most preferred (entries 1-5). When (diacetoxyiodo)benzene and diphenyl phos- phate reached to two equivalents, the yield was increased to 43% (entry 6); while two equivalents of acetophenone were used to reacted with one equivalent of (diacetoxy- iodo)benzene and diphenyl phosphate, the reaction got the higher yield of 48% (entry 7). We found that the RT PhI(OAc)2HOPO(OPh)2 PhCOCH3PhCOCH2OPO(OPh)2 Scheme 1  2 Y. PU ET AL. Table 1. Optimization of the α-phosphoryloxylation of acetophenone. Entry Acetophenone (equiv.) DIB (equiv.) Diphenyl phosphate (equiv.) Solvent (2 mL) Time (h) Yield (%)a 1 1 1 1 CH3CN 24 10 2 1 1 1 THF 24 20 3 1 1 1 CH3OH 24 9 4 1 1 1 CF3CH2OH 24 9 5 1 1 1 CH2Cl2 24 38 6 1 2 2 CH2Cl2 24 43 7 2 1 1 CH2Cl2 24 48 8 2 1 1 CH2Cl2 2 8b 9 2 1 1 CH2Cl2 4 13b 10 2 1 1 CH2Cl2 8 45b 11 2 1 1 CH2Cl2 12 69b 12 2 1 1 CH2Cl2 16 72b 13 2 1 1 CH2Cl2 24 80b aIsolated yield; bMeasured by 1H NMR. product of α-phosphoryloxyl acetophenone was partly decomposed in separation and the yield was decreased much, then we used 1H NMR technique to show the true yield and after 24 h the reaction reached the highest yield of 80% (entries 8-13). Under the optimum reaction conditions, the reaction of a series of ketones (1) with (diacetoxyiodo)benzene and phosphate (2) in CH2Cl2 at room temperature was inves- tigated (Scheme 2), and several ketol phosphates (3) were obtained which all were characterized by 1H NMR, 13C NMR, IR and MS spectra, the results are summarized in Table 2. It is shown that the α-phosphoryloxylaction of ketones was easily carried out at room temperature and complete in 24 h, most provided the corresponding ketol phosphates in good to excellent yields. In com- parison with other ketones, β-diketone 1e and 1f usually needed much less time in the reaction and got the pro- ducts in excellent yields due to the high activity of α-hydrogen (entries 5-6,8). The proposed mechanism for the α-phosphoryloxy- laction of ketones is similar to the literature procedure [21], which is shown as follows (Scheme 3). 3. Experimental IR spectra were recorded on a Thermo-Nicolet 6700 in- strument, NMR spectra were measured on a Bruker ANANCE Ⅲ (500 MHz) spectrometer, and Mass spec- tra were determined on Thermo-ITQ 1100 mass spec- trometer. Ketones, (diacetoxyiodo)benzene and phos- phate are commercially available. 4. A Typical Procedure for α-Phosphoryloxylation of Ketones A mixture of acetophenone (1a) (0.5 mmol), (diacetoxy iodo)benzene (0.25 mmol) and diphenyl phosphate (2a) (0.25 mmol) in CH2Cl2 (2 mL) was stirred at room tem- perature for 24 h, then H2O (5 ml) and sat. aq Na2CO3 RCCH2R' + PhI(OAc)2 + (R''O)2PO2H RCCH(R')OP(OR'')2 OO O CH2 Cl2 RT 123 1a: R=Ph, R'=H 1b: R=Me, R'=H 1c: R=p-NO2-C6H4, R'=H 1d: R, R'=-(CH2)4- 1e: R=Ph, R'=PhCO 1f: R=Ph, R'=MeCO 3a: R=Ph, R'=H, R''=Ph 3b: R=Me, R'=H, R''=Ph 3c: R=p-NO2-C6H4, R'=H, R''=Ph 3d: R, R'=-(CH2)4-, R''=Ph 3e: R=Ph, R'=PhCO, R''=Ph 3f: R=Ph, R'=MeCO, R''=Ph 3g: R=Ph, R'=H, R''=Bz 3h: R=Ph, R'=MeCO, R''=Bz 4 2a: R''=Ph 2b: R''=Bz 2c: R''=p-NO2-C6H4 3i: R=Ph, R'=H, R''=p-NO2-C6H Scheme 2 Copyright © 2011 SciRes. IJOC 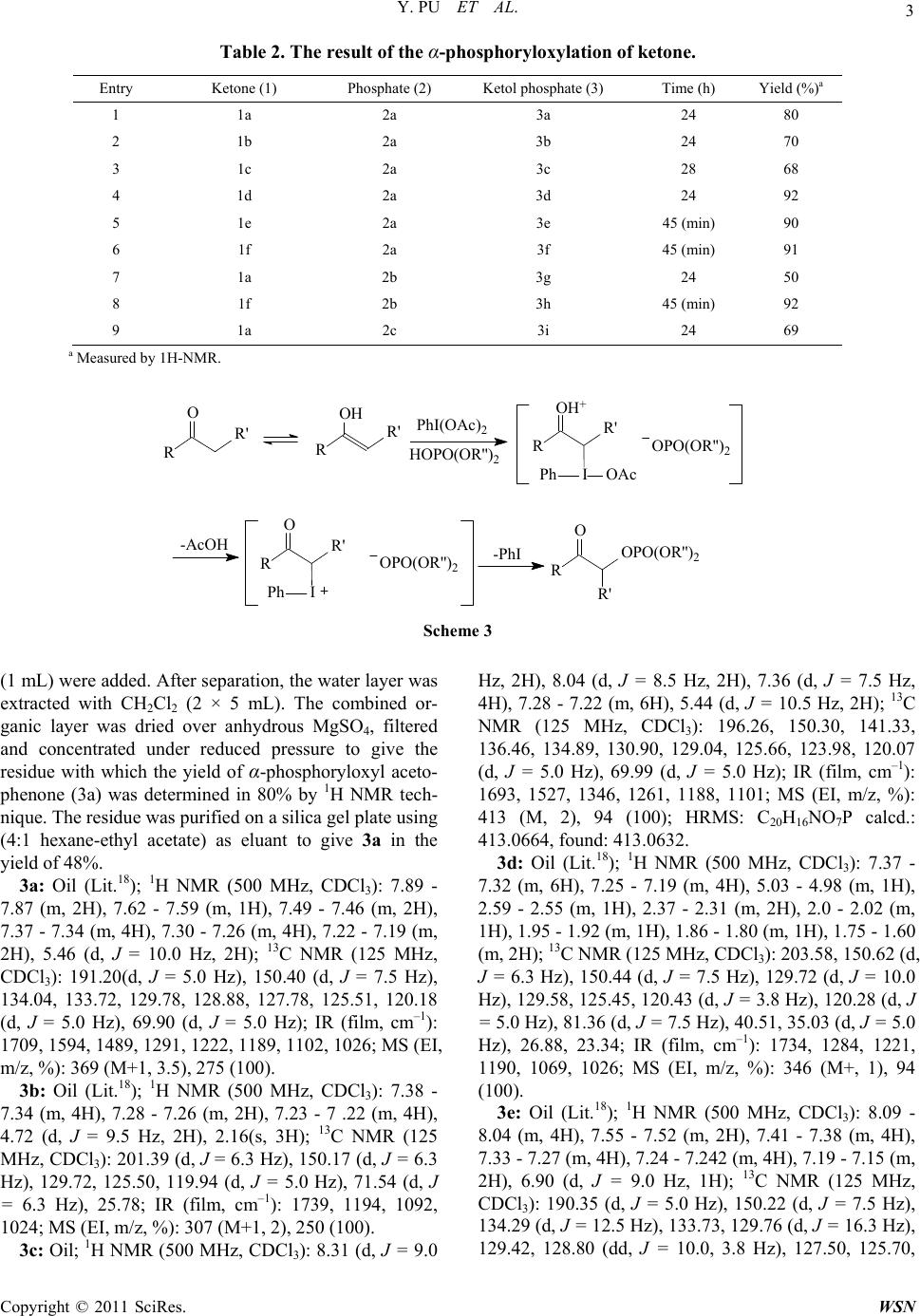 Y. PU ET AL. 3 Table 2. The result of the α-phosphoryloxylation of ketone. Entry Ketone (1) Phosphate (2) Ketol phosphate (3) Time (h) Yield (%)a 1 1a 2a 3a 24 80 2 1b 2a 3b 24 70 3 1c 2a 3c 28 68 4 1d 2a 3d 24 92 5 1e 2a 3e 45 (min) 90 6 1f 2a 3f 45 (min) 91 7 1a 2b 3g 24 50 8 1f 2b 3h 45 (min) 92 9 1a 2c 3i 24 69 a Measured by 1H-NMR. R R' OH+ IOAcPh R R' O R R' OH R OPO(OR'')2 O R' OPO(OR'')2 R R' O IPh OPO(OR'')2 -PhI -AcOH PhI(OAc)2 HOPO(OR'')2 Scheme 3 (1 mL) were added. After separation, the water layer was extracted with CH2Cl2 (2 × 5 mL). The combined or- ganic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to give the residue with which the yield of α-phosphoryloxyl aceto- phenone (3a) was determined in 80% by 1H NMR tech- nique. The residue was purified on a silica gel plate using (4:1 hexane-ethyl acetate) as eluant to give 3a in the yield of 48%. 3a: Oil (Lit.18); 1H NMR (500 MHz, CDCl3): 7.89 - 7.87 (m, 2H), 7.62 - 7.59 (m, 1H), 7.49 - 7.46 (m, 2H), 7.37 - 7.34 (m, 4H), 7.30 - 7.26 (m, 4H), 7.22 - 7.19 (m, 2H), 5.46 (d, J = 10.0 Hz, 2H); 13C NMR (125 MHz, CDCl3): 191.20(d, J = 5.0 Hz), 150.40 (d, J = 7.5 Hz), 134.04, 133.72, 129.78, 128.88, 127.78, 125.51, 120.18 (d, J = 5.0 Hz), 69.90 (d, J = 5.0 Hz); IR (film, cm–1): 1709, 1594, 1489, 1291, 1222, 1189, 1102, 1026; MS (EI, m/z, %): 369 (M+1, 3.5), 275 (100). 3b: Oil (Lit.18); 1H NMR (500 MHz, CDCl3): 7.38 - 7.34 (m, 4H), 7.28 - 7.26 (m, 2H), 7.23 - 7 .22 (m, 4H), 4.72 (d, J = 9.5 Hz, 2H), 2.16(s, 3H); 13C NMR (125 MHz, CDCl3): 201.39 (d, J = 6.3 Hz), 150.17 (d, J = 6.3 Hz), 129.72, 125.50, 119.94 (d, J = 5.0 Hz), 71.54 (d, J = 6.3 Hz), 25.78; IR (film, cm–1): 1739, 1194, 1092, 1024; MS (EI, m/z, %): 307 (M+1, 2), 250 (100). 3c: Oil; 1H NMR (500 MHz, CDCl3): 8.31 (d, J = 9.0 Hz, 2H), 8.04 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 7.5 Hz, 4H), 7.28 - 7.22 (m, 6H), 5.44 (d, J = 10.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): 196.26, 150.30, 141.33, 136.46, 134.89, 130.90, 129.04, 125.66, 123.98, 120.07 (d, J = 5.0 Hz), 69.99 (d, J = 5.0 Hz); IR (film, cm–1): 1693, 1527, 1346, 1261, 1188, 1101; MS (EI, m/z, %): 413 (M, 2), 94 (100); HRMS: C20H16NO7P calcd.: 413.0664, found: 413.0632. 3d: Oil (Lit.18); 1H NMR (500 MHz, CDCl3): 7.37 - 7.32 (m, 6H), 7.25 - 7.19 (m, 4H), 5.03 - 4.98 (m, 1H), 2.59 - 2.55 (m, 1H), 2.37 - 2.31 (m, 2H), 2.0 - 2.02 (m, 1H), 1.95 - 1.92 (m, 1H), 1.86 - 1.80 (m, 1H), 1.75 - 1.60 (m, 2H); 13C NMR (125 MHz, CDCl3): 203.58, 150.62 (d, J = 6.3 Hz), 150.44 (d, J = 7.5 Hz), 129.72 (d, J = 10.0 Hz), 129.58, 125.45, 120.43 (d, J = 3.8 Hz), 120.28 (d, J = 5.0 Hz), 81.36 (d, J = 7.5 Hz), 40.51, 35.03 (d, J = 5.0 Hz), 26.88, 23.34; IR (film, cm–1): 1734, 1284, 1221, 1190, 1069, 1026; MS (EI, m/z, %): 346 (M+, 1), 94 (100). 3e: Oil (Lit.18); 1H NMR (500 MHz, CDCl3): 8.09 - 8.04 (m, 4H), 7.55 - 7.52 (m, 2H), 7.41 - 7.38 (m, 4H), 7.33 - 7.27 (m, 4H), 7.24 - 7.242 (m, 4H), 7.19 - 7.15 (m, 2H), 6.90 (d, J = 9.0 Hz, 1H); 13C NMR (125 MHz, CDCl3): 190.35 (d, J = 5.0 Hz), 150.22 (d, J = 7.5 Hz), 134.29 (d, J = 12.5 Hz), 133.73, 129.76 (d, J = 16.3 Hz), 129.42, 128.80 (dd, J = 10.0, 3.8 Hz), 127.50, 125.70, Copyright © 2011 SciRes. WSN  4 Y. PU ET AL. 120.21 (d, J = 5.0 Hz), 84.06 (d, J = 6.3 Hz); IR (film, cm–1): 1711, 1682, 1294, 1230, 1186, 1163, 1091, 1011; MS (EI, m/z, %): 472 (M+, 2), 94 (100). 3f: Oil; 1H NMR (500 MHz, CDCl3): 7.94 (dd, J = 8.0, 1.0 Hz, 2H), 7.61 - 7.58 (m, 1H), 7.45 - 7.42 (m, 2H), 7.37 - 7.31 (m, 2H), 7.28 - 7.20 (m, 5H), 7.17 - 7.14 (m, 3H), 6.11 (d, J = 8.5 Hz, 1H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3): 190.35 (d, J = 5.0 Hz), 150.22 (d, J = 7.5 Hz), 134.29 (d, J = 12.5 Hz), 133.73, 129.76 (d, J = 16.3 Hz), 129.42, 128.80 (dd, J = 10.0, 3.8 Hz), 127.50, 125.70, 120.21 (d, J = 5.0 Hz), 84.06 (d, J = 6.3 Hz); IR (film, cm–1): 1730, 1688, 1218, 1024; MS (EI, m/z, %): 410 (M+, 1), 105 (100); HRMS: C22H19O6P calcd.: 410.0919, found: 410.0889. 3g: Oil; 1H NMR (500 MHz, CDCl3): 7.84 (d, J = 8.0 Hz, 2H), 7.58 - 7.52 (m, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.34 - 7.30 (m, 10H), 5.22 (d, J = 10.0 Hz, 2H), 5.18 (dd, J = 8.0, 6.0 Hz, 4H); 13C NMR (125 MHz, CDCl3): 191.99 (d, J = 5.0 Hz), 135.67 (d, J = 6.3 Hz), 133.89, 128.80, 128.51, 128.02, 127.68, 69.70 (d, J = 5.0 Hz), 68.67 (d, J = 5.0 Hz); IR (film, cm–1): 1708, 1279, 1234, 1116, 1014; MS (EI, m/z, %): 397 (M+, 1), 199 (100); HRMS: C22H21O5P calcd.: 396.1127, found: 396.1121. 3h: Oil; 1H NMR (500 MHz, CDCl3): 7.98 (d, J = 7.5 Hz, 2H), 7.59 (t, J = 7.5 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.38 - 7.29 (m, 10H), 5.97 (d, J = 8.0 Hz, 1H), 5.15 (d, J = 9.0 Hz, 2H), 5.10 - 5.01 (m, 2H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3): 191.99 (d, J = 5.0 Hz), 135.67 (d, J = 6.3 Hz), 133.89, 128.80, 128.51, 128.02, 127.68, 69.70 (d, J = 5.0 Hz), 68.67 (d, J = 5.0 Hz); IR (film, cm–1): 1708, 1279, 1234, 1116, 1014; MS (EI, m/z, %): 439 (M+1, 2), 105 (100); HRMS: C24H23O6P calcd.: 438.1232, found: 438.1201. 3i: Oil; 1H NMR (500 MHz, CDCl3): 8.28 (d, J = 9.0 Hz, 4H), 7.87 (d, J = 7.5 Hz, 2H), 7.64 - 7.62 (m,1H), 7.52 - 7.45 (m, 6H), 5.57 (d, J = 12.5Hz, 2H); 13C NMR (125 MHz, CDCl3): 190.55 (d, J = 3.8 Hz), 154.55 (d, J = 6.3 Hz), 145.19, 134.41, 132.98, 128.98, 127.62, 125.72, 120.86 (d, J = 2.5 Hz), 70.79 (d, J = 6.3 Hz); IR (film, cm–1): 1713, 1614, 1591, 1518, 1491, 1344, 1290, 1248, 1215, 1109; MS (EI, m/z, %): 458 (M+, 1), 105 (100); HRMS: C20H15N2O9P calcd.: 458.0515, found: 458.0488. 5. Acknowledgements Financial supports from the Natural Science Foundation of China (Project 21072176) and Zhejiang Province Natural Science Foundation of China (Project Y4100231) are gratefully acknowledged. 6. References [1] A. Varvoglis, “Chemical Transformations Induced by Hypervalent Iodine Reagents,” Tetrahedron, Vol. 53, No. 4, 1997, pp. 1179-1255. doi:10.1016/S0040-4020(96)00970-2 [2] P. J. Stang and V. V. Zhdankin, “Organic Polyvalent Iodine Compounds,” Chemical Reviews, Vol. 96, No. 3, 1996, pp. 1123-1178. doi:10.1021/cr940424+ [3] V. V. Zhdankin and P. J. Stang, “Recent Developments in the Chemistry of Polyvalent Iodine Compounds,” Chemical Reviews, Vol. 102, No. 7, 2002, pp. 2523-2584. doi:10.1021/cr010003+ [4] T. Wirth and U. H. Hirt, “Hypervalent Iodine Compounds: Recent Advances in Synthetic Applications,” Synthesis, No. 8, 1999, pp. 1271-1287. doi:10.1055/s-1999-3540 [5] A. Kirschning, “Hypervalent Iodine and Carbohydrates - A New Liaison,” European Journal of Organic Chemis- try, No. 11, 1998, pp. 2267-2274. doi:10.1002/(SICI)1099-0690(199811)1998:11<2267::AI D-EJOC2267>3.0.CO;2-E [6] M. Ochiai, “Nucleophilic Vinylic Substitutions of λ3-Vinyliodanes,” Journal of Organometallic Chemistry, Vol. 611, No. 1-2, 2000, pp. 494-508. doi:10.1016/S0022-328X(00)00487-3 [7] T. Okuyama, “Solvolysis of Vinyl Iodonium Salts. New Insights into Vinyl Cation Intermediates,” Accounts of Chemical Research, Vol. 35, No. 1, 2002, pp. 12-18. doi:10.1021/ar0100374 [8] V. V. Zhdankin and P. J. Stang, “Alkynyliodonium Salts in Organic Synthesis,” Tetrahedron, Vol. 54, No. 37, 1998, pp. 10927-10966. doi:10.1016/S0040-4020(98)00410-4 [9] V. V. Grushin, “Cyclic Diaryliodonium Ions: Old Mysteries Solved Andnew Applications Envisaged,” Chemical Society Reviews, Vol. 29, No. 5, 2000, pp. 315-324. doi:10.1039/a909041j [10] A. C. Boye, D. Meyer, C. K. Ingison, A. N. French and T. Wirth, “Novel Lactonization with Phenonium Ion Par- ticipation Induced by Hypervalent Iodine Reagents,” Or- ganic Letters, Vol. 5, No. 12, 2003, pp. 2157-2159. doi:10.1021/ol034616f [11] R. M. Moriarty, R. K. Vaid and G. F. Koser, “[Hy- droxy(organosulfonyloxy)iodo]arenes in Organic Synthe- sis,” Synlett, No. 7, 1990, pp. 365-383. doi:10.1055/s-1990-21097 [12] J. C. Lee and J. H. Choi, “Highly Efficient α-Organo- sulfonyloxylation of Carbonyl Compounds under Micro- wave Irradiation,” Synlett, No. 2, 2001, pp. 234-235. [13] J. Akiike, Y. Yamamoto and H. Togo, “Efficient Con- version of Ketones to α-Tosyloxyketones with m-Chloroperbenzoic Acid and p-Toluenesulfonic Acid in the Presence of Catalytic Amount of IL-Supported PhI in [emim]Ots,” Synlett, No. 14, 2007, pp. 2168-2172. [14] M. Ueno, T. Nabana and H. Togo, “Novel Oxidative α-Tosyloxylation of Alcohols with Iodosylbenzene and p-Toluenesulfonic Acid and Its Synthetic Use for Direct Preparation of Heteroaromatics,” Journal of Organic Chemistry, Vol. 68, No. 16, 2003, pp. 6424-6426. doi:10.1021/jo030045t Copyright © 2011 SciRes. IJOC  Y. PU ET AL. Copyright © 2011 SciRes. WSN 5 [15] Y. Yamamoto, Y. Kawano, P. H. Toy and H. Togo, “PhI- and Polymer-supported PhI-catalyzed Oxidative Conver- sion of Ketones and Alcohols to α-Tosyloxyketones with m-Chloroperbenzoic Acid and p-Toluenesulfonic Acid,” Tetrahedron, Vol. 63, No. 22, 2007, pp. 4680-4687. doi:10.1016/j.tet.2007.03.091 [16] T. Dohi and Y. Kita, “Hypervalent Iodine Reagents as A New Entrance to Organocatalysts,” Chemical Communi- cations, No. 16, 2009, pp. 2073-2085. [17] A. Tanaka and H. Togo, “4-MeC6H4I-Mediated Efficient α-Tosyloxylation of Ketones with Oxone® and p-Toluenesulfonic Acid in Acetonitrile,” Synlett, No. 20, 2009, pp. 3360-3364. [18] G. F. Koser, J. S. Lodaya, D. G. Ray and P. B. Kokil, “Direct α-Phosphoryloxylation of Ketones and (Phos- phoryloxy)lactonization of Pentenoic Acids with [Hy- droxy[(bis(phenyloxy)phosphoryl)oxy]iodo]benzene,” Journal of the American Chemical Society, Vol. 110, No. 9, 1988, pp. 2987-2988. doi:10.1021/ja00217a058 [19] T. Nabana and H. Togo, “Reactivities of Novel [Hy- droxy(tosyloxy)iodo]arenes and [Hdroxy(phophoryloxy)- iodo]arenes for α-Tosyloxylation and α-Phosphory- loxylation of Ketones,” Journal of Organic Chemistry, Vol. 67, No. 12, 2002, pp. 4362-4365. doi:10.1021/jo0200670 [20] P. J. Stang, M. Boeshar and J. Li, “Acetylenic Esters. Preparation and Characterization of Hitherto Unknown Alkynyl Carboxylate, RC.tplbond.COCOR1, and Alkynyl Phosphate, RC.tplbond.COPO(OR1)2, Esters,” Journal of the American Chemical Society, Vol. 108, No. 24, 1986, pp. 7832-7834. doi:10.1021/ja00284a057 [21] R. M. Moriarty, C. Condeiu, A. Tao and O. Prakash, “New Organohypervalent Iodine Reagents for α-Methylphosphonylations and α-Diphenyl- and α-Dimethylphosphinylations,” Tetrahedron Letters, Vol. 38, No. 14, 1997, pp. 2401-2404. doi:10.1016/S0040-4039(97)00388-2
|