 Open Journal of Endocrine and Metabolic Diseases, 2014, 4, 13-23 Published Online January 2014 (http://www.scirp.org/journal/ojemd ) http://dx.doi.org/10.4236/ojemd.2014.41002 OPEN ACCESS OJEMD Androgens, Male Hypogonadism and Traumatic Brain Injury Alexandre Hohl 1,2* , Marcelo Fernando Ronsoni 1,2 , Simone van de Sande-Lee 1 , Fábio Cavalcanti de Faria Vieira 2 , Marcelo Libório Schwarzbold 2 , Alexandre Paim Diaz 2 , Roger Walz 1,2 1 Departamento de Clínica Médica, Hospital Universitário da Universidade Federal de Santa Catarina (HU-UFSC), Florianópolis, Brazil 2 Centro de Neurociências Aplicadas (CeNAp), Hospital Universitário da Universidade Federal de Santa Catarina (HU-UFSC), Florianópolis, Brazil Email: * alexandrehohl@endocrino.org.br Received December 22, 2013; revised January 10, 2014; accepted January 16, 2014 Copyright © 2014 Alexandre Hohl et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accor- dance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Alexandre Hohl et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian. ABSTRACT Traumatic brain injury (TBI) is a worldwide public health problem. Populations with a growing number of ve- hicles are experiencing many traumas and accidents. The highest-risk group is young men. Significant advances in neurosurgery and intensive therapy have resulted in increased survival rates of TBI patients. These higher survival rates, in turn, have led to an increasingly higher number of patients with neurological, cognitive, clinical, and social problems. This lack of knowledge about TBI has been called by some “the silent epidemic”. In recent years, studies of patients with moderate and severe TBI are increasing. Glasgow Coma Scale ≤ 8 and abnormal pupils at admission are used to determine the prognosis of patients with moderate or severe TBI. Several bio- markers such as interleukins, thiobarbituric acid reactive species, and some hormones have been studied in an effort to aid prognosis. Testosterone plays a key role in men. Thus, an understanding of androgens in TBI is es- sential to follow these survivors of head trauma. This review will discuss the epidemiology of TBI, its association with male hypogonadism, and possible treatments. KEYWORDS Traumatic Brain Injury; Androgens; Male; Hypogonadism; Treatment 1. Introduction The world population is growing rapidly. Similarly, the number of traffic accidents is also increasing. Unfortu- nately, public policies to reduce these accidents have not been efficient. As medicine improves, there are increa- singly more survivors of traumatic brain injury (TBI). Patients with very peculiar characteristics arise: young men who have moderate or severe TBI. Many of them undergo long periods of stay in an intensive care unit. In this context, analysis of the gonadotropic axis be- comes important. The maintenance of androgens in adult men is very important. It seems that men who have had TBI are equally reliant on androgens and these hormones can be fundamental in recovery, both physical and emo- tional. 2. Traumatic Brain Injury: An Epidemic TBI is a concern in most countries regardless of the country’s developmental state. It is a worldwide health problem because of its long-lasting disability, even in people with mild TBI. An in-depth knowledge of this disease may facilitate the development of rehabilitation services [1]. The real challenge involved in this knowledge is de- termining the number of people who suffer from TBI. Nowadays, most research articles are population-based epidemiological studies, consisting of emergency depart- Corresponding author.  A. HOHL ET AL. OPEN ACCESS OJEMD ment records, hospital admission or discharge records and death certificates. Many TBI patients do not go to hospital, because the great majority of TBI cases are mild —about 80%. The frequencies of severe and moderate cases are each about 10% [2]. This lack of knowledge about TBI has been called by some “the silent epidemic”, because the number of TBI patient is underestimated. There are several ways of do- cumenting TBI. It can be divided by gender; it is most prevalent in men. It can also be divided according to causal nexus, with transportation being the most preva- lent major cause. Also, it can be divided by age; TBI is most prevalent in young adults and older people. In 1994 in the United States, death rates due to TBI were 3 times higher for men than for women. TBI-associated death rates were also higher for African-Americans in the United States, mostly because of firearms. For all other races, transportation was the leading cause. Half of the hospitalizations in the United States in 1994 were caused by traffic accidents and 25% were due to falls. Only 10% of hospital cases resulted from firearms. Firearms do not have a high hospitalization rate because their pre-hospital fatality rate is too high. In 1996, the Centers for Disease Control and Prevention estimated that 5.3 million people were living with a disability caused by TBI (2% of the United States population), based on a model used to es- timate the number of people that have ever had a TBI that required hospitalization and resulted in a long-term disability [3]. Tagliaferri [4], in his systematic review about this theme, in 2006, showed that the incidence of TBI in Eu- rope ranges from a low of 91/100,000 (Spain) to a high of 546/100,000 (Sweden). He also showed that the second most prevalent mechanism in these events was falls (after traffic accidents). In northern Europe, falls were more prevalent, while in southern Europe vehicle accidents were the most common. Falls can be associated with alcohol consumption. The mean incidence rate for hospitalization and TBI in Europe is much larger than in the United States (235/100,000 and 103/100,000, respec- tively). The TBI mortality rate in the United States and Europe are similar, both around 15 - 20/100,000. Even though these comparisons exist, it is difficult to synthes- ize many studies, because they do not have the same methodological design. Therefore the populations ana- lyzed are not similar [4]. A recent study (2001-2007) of TBI epidemiology in Brazil showed that 12% of cases result in death. Trans- portation is the highest causes proportionally, with mo- torcycle accidents increasing rapidly. Although TBI hos- pitalizations are not the most prevalent, mortality rates of hospitalizations by TBI are the second highest, only second to cardiovascular diseases. In Brazil, the most prevalent gender for TBI was male, corresponding to 80% of the total cases. Adolescents, young adults, and older people were the most common groups of patients. The number of hospital admissions (50 - 60/100,000) is lower in comparison with other countries like the United States or European countries, which can be explained by management problems and the difficultly of immediately accessing pre-hospital care and the difficulty of moving victims from the accident site. The prevalence of TBI remained stable from 2001 to 2007, probably due to a lack of effective measures to prevent violence and acci- dents [5]. 3. Pituitary and Testosterone The pituitary is the “master” gland and controls the func- tions of several other glands. It has an intrinsic relation with the hypothalamus and central nervous system. It most often produces substances that can work as neuro- transmitters and/or hormones, depending on the receptor of action and releasing site of these substances. It is di- vided in an anterior pituitary (adhenohypophysis) and a posterior pituitary (neurohypophysis). It is located in the sella turcica, connected to the hypothalamus by a stalk and lined superiorly by connective tissue forming the sellar diaphragm [2]. The production of androgens depends on the coordi- nated action of the hypothalamus-pituitary-gonads axis. Basically, production involves three steps: 1) Production of the releasing hormone (GnRH) by the arcuate nucleus, at the hypothalamus and its descent to the pituitary gonadotroph cells; 2) A release of the gonadotropins; 3) Stimulation of the gonads by follicle stimulating hormone (FSH) and luteinizing hormone, producing the androgens in Leydig cells (testicles) or in the Theca In- terna (ovaries). GnRH is secreted in a pulsatile pattern every 60 to 90 minutes, thus reflecting testosterone secretion at the go- nads, also at a pulsatile pattern. Testosterone plays a fun- damental role in negative feedback to the gonadotrophs and hypothalamus. This feedback also occurs in an indi- rect way via estradiol, through aromatase conversion at the hypothalamus. GnRH descends to the gonadotrophs, at the anterior pituitary, by a small network of vessels forming the pri- mary and secondary plexuses of the hypophyseal portal system (the branches of the internal carotid). They pro- vide 70% - 90% of the pituitary blood supply. The blood reaches the anterior pituitary through the portal veins. The medium and inferior hypophyseal arteries supply the pituitary stalk and the posterior pituitary. During a TBI event, one can infer that shearing forces delivered from different angles could impair the pituitary blood flow at its portal veins, causing isolated, multiple, or partial hor-  A. HOHL ET AL. OPEN ACCESS OJEMD mone deficiencies of the anterior pituitary [6]. Another lesion that can occur is a stalk lesion. In some cases, the stalk is cut by the forces involved in the TBI and therefore interrupts the negative feedback over pro- lactin production caused by dopamine. A hyperprolacti- nemia situation can cause equal hypogonadism without necessarily any injury to the gonadotrophs. TBI can produce hormonal alterations visible at an acute phase that can persist for over 12 months after the initial trauma. Approximately 50% of patients that suffer TBI have at least one anterior pituitary hormone defi- ciency 12 months after the initial trauma. In the acute phase, gonadotropins were the most important deficiency, while after 12 months the most prevalent deficiency was somatotropin [7]. Tanriverdi also demonstrated the major prevalence of gonadotropin deficiency in patients in acute phase of TBI (40% of 95 patients) [8]. Agha related in his studies that 28% of TBI cases evolved with hypopituitarism, in which almost 12% had secondary hypogonadism [9]. According to the literature discussed earlier in this ar- ticle, TBI is three times more prevalent in men than in women. As one of the consequences of hypopituitarism is hypogonadism by lesion to the gonadotroph—mostly in the acute phase of injury—or the stalk, one should ask what the comorbidities among these patients are. Testosterone is relevant in the blood cell count, coa- gulation mechanisms, bone mineral density, muscle to- nus, sexual function, and mood and cognitive abilities [10]. Schwarzbold showed in his review that 21% of pa- tients one year after TBI received a diagnosis of psychia- tric disorder in a study in the United Kingdom [11,12]. The answer to that question could come from a paper from 2001, published by The Lancet. This study showed that the standardized mortality ratio for untreated gona- dotropin deficiencies (none from TBI causes), compared with treated deficiences or patients with an intact hypo- thalamo-pituitary gonadal axis, was 2 times greater, with significant p values (<0.0001 and <0.0005, respectively). Specific pituitary hormone deficiencies could be impor- tant in determining cardiovascular outcomes. The correct assessment and treatment benefit the rehabilitation of those patients [13]. 4. Diagnosis of Hypogonadism Although TBI-related hypogonadism may significantly increase morbidity and impair quality of life, this condi- tion remains largely under-recognized, as symptoms are often subtle and masked by the sequelae of brain damage [14]. Testosterone is a key anabolic factor and low levels of this hormone upon admission to a rehabilitation unit have been associated with longer lengths of stay and lower functional independence scores [15,16]. The diagnosis of hypogonadism following a TBI is not different from that of hypogonadism due to other causes. It is based on the presence of clinical features and bio- chemical evidence of gonadal sex-steroid deficiency. Symptoms and signs in men include decreased energy, depressed mood, loss of facial, pubic, and body hair, gy- necomastia, reduction in testicular size and muscle mass, reduced libido, infertility, and impaired sexual function. Biochemically, hypogonadotrophic or central hypogo- nadism is characterized by inappropriately low gonado- tropin levels (below the upper limit of the reference range) in the presence of gonadal hormone deficiency [9, 17], as shown in Table 1. According to a consensus statement, excluding pa- tients in a vegetative state who are unlikely to benefit from replacement therapy, prospective evaluation of the gonadotrophic axis should be routinely performed 3 and 12 months after the primary brain injury in all patients— regardless of TBI severity, as some studies indicate that this is not related to the incidence of hypogonadism. Re- trospectively, patients with any signs or symptoms of hypogonadism who have had a moderate to severe TBI more than one year ago should undergo a single hormon- al testing [18]. As such an approach may not be cost-effective due to the huge number of TBI cases all over the world, Loren- zo and collaborators [19] suggest the following criteria for selecting the ideal TBI patients to be tested, exclud- ing severely disabled patients: • Patients with initial Glasgow Coma Scale (GCS) scores of 13 or less or with GCS scores between 13 and 15 with abnormalities on brain images; • Patients who should remain under observation for at least 24 hours; • Patients with intracranial hemorrhagic lesions; • Patients who develop acute hypogonadism manifesta- tions immediately after TBI; • Patients with current hypogonadism signs or symp- toms. Similar selection criteria were suggested by a recent review [14]. Based on current evidence, the authors rec- ommend that adult patients should be assessed only one year after TBI, as there is currently no evidence that acute-phase therapy with androgens improves outcomes. However, they point out that tests in the early post-TBI period, as well as a long time after the trauma, may be important and are under evaluation [19]. Table 1. Criteria for central hypogonadism diagnosis in men. Laboratory Low morning testosterone (<300 ng/dL * ), FSH and LH inappropriately low FSH: follicle-stimulating hormone; LH: luteinizing hormone. * Normal range may vary in different laboratories.  A. HOHL ET AL. OPEN ACCESS OJEMD 5. Acute and Chronic Hypogonadism after TBI There is considerable discrepancy among studies in the reported frequency rates of post-TBI hypogonadism and other endocrine dysfunctions. This wide variation may reflect differences in the severity of the injuries, the trauma mechanism, the study design, diagnostic criteria, the time of evaluation, and the patient selection (age, sex, body mass index, previous health impairments), which do not allow for a simple generalization of the results [20]. Most studies on endocrine alterations following TBI evaluated patients in the medium and long term. A few have focused on the acute phase and initially they simply tried to correlate the neuroendocrine changes to the se- verity of the cranial trauma [21-23]. More recently, much attention has been dedicated to determining the frequen- cy of hypogonadism and other hormonal deficiencies in cohorts of patients with TBI [7,24-30]. Gonadal hormone deficiency in clinical studies evaluating patients in the early post-TBI phase ranged from 13% to 100%. The methods and results of these studies, concerning gonadal and prolactin axes, are summarized in Table 2. The clinical impact of acutely suppressed testosterone in the setting of brain injury is unknown and warrants further investigation. Gonadotrophic axis alterations in the early phase were also reported in non-head injured extra cranial trauma patients, which suggested that these alterations may be related to adaptive responses to acute critical illness and are not specific to TBI [28]. Moreover, pituitary function may improve over time and the pres- ence of testosterone deficiency immediately after the event does not typically predict chronic hypogonadism [31]. Chronic hypopituitarism following TBI has been ex- tensively studied in recent years. The prevalence of hy- pogonadism has been reported to range from 0% to 32%. The results of 25 studies are summarized in Table 3. In the two largest series published [32,33], gonado- trophic was the second most affected among the pituitary axes and the prevalence of chronic post-TBI hypogonad- ism was 9%. A study by our group has shown similar results [34]. The role of trauma severity in predicting pituitary dys- function has been examined by several studies. Gonado- tropin deficiency was not specifically assessed in most of them, probably because reliable statistical comparisons could not be done. Regarding hypopituitarism in general, although some authors reported no association with se- verity of TBI [7,8,24,35-41], other studies, including a systematic review that comprised 1137 subjects, indicate that patients with lower GCS scores are at higher risk of developing pituitary hormone alterations [42-46]. Re- gardless, it is noteworthy that even patients with mild Table 2. Hypogonadism in TBI acute phase. Study of patients Time to testing GCS score Neuroendocrine Axis assessed Gonadal Prolactin [26] 21 7 days N/A 67.0% def. N/A cols. [25] 31 7 days 13 - 15 ↓ Testosterone N/A Agha and cols. [24] 50 12 days median 3 - 13 80.0% def. 52% high Tanriverdi and cols. [7] 52 24 hours 3 - 15 41.6% def. 12% high Kleindienst and cols. 67 Admission 3 days 7 days 3 - 15 13.0% def. 24.0% def. 18.0% def. 15% high 9% high 13% high cols. [28] 101 0 - 10 days 3 - 13 100% def. men N/A Krahulik and cols. 186 Admission 3 - 14 33.0% def. N/A Olivecrona and cols. 29 1 day 4 days ≤8 82.1% def. 100% def. GCS: Glasgow Coma Scale; N/A: not available; def.: deficient. TBI still have a substantial risk of hypopituitarism and the diagnosis should not be overlooked [31]. 6. Biochemical, Physical, and Behavioral Changes after TBI The primary damage from TBI and its subsequent im- balance may activate both secondary injury and neuro- protective cascades, which can interact with a complex biochemical network, leading to neuronal and glial sur- vival or death due to necrosis and apoptosis [55-58]. Oxidative stress has been implicated with the excito- toxic brain injury [59], including TBI [60-62], but the association between plasma markers of oxidative stress and the severity of TBI patients’ prognoses remains con- troversial [63]. An excessive amount of reactive oxygen species (ROS) generated by several combined mechan- isms, including neutrophils activation, endothelial cells, nerve and glial cells, iron ions (from hemoglobin degra- dation in the hemorrhagic areas), and brain reperfusion [55,56,58], has been implicated in brain lesions in TBI. The ROS reaction with proteins, deoxyribonucleic acids (DNA), and lipids, leading to the oxidative damage of cells and tissues in TBI, has been discussed for more than 20 years [58,64,65]. A recent study from our group investigated the associ- ation between plasma levels of lipid peroxidation (thi- obarbituric acid reactive species; TBARS) and protein oxidation (carbonyl) biomarkers and the hospital mortal- ity of patients with severe TBI. Plasma levels of TBARS 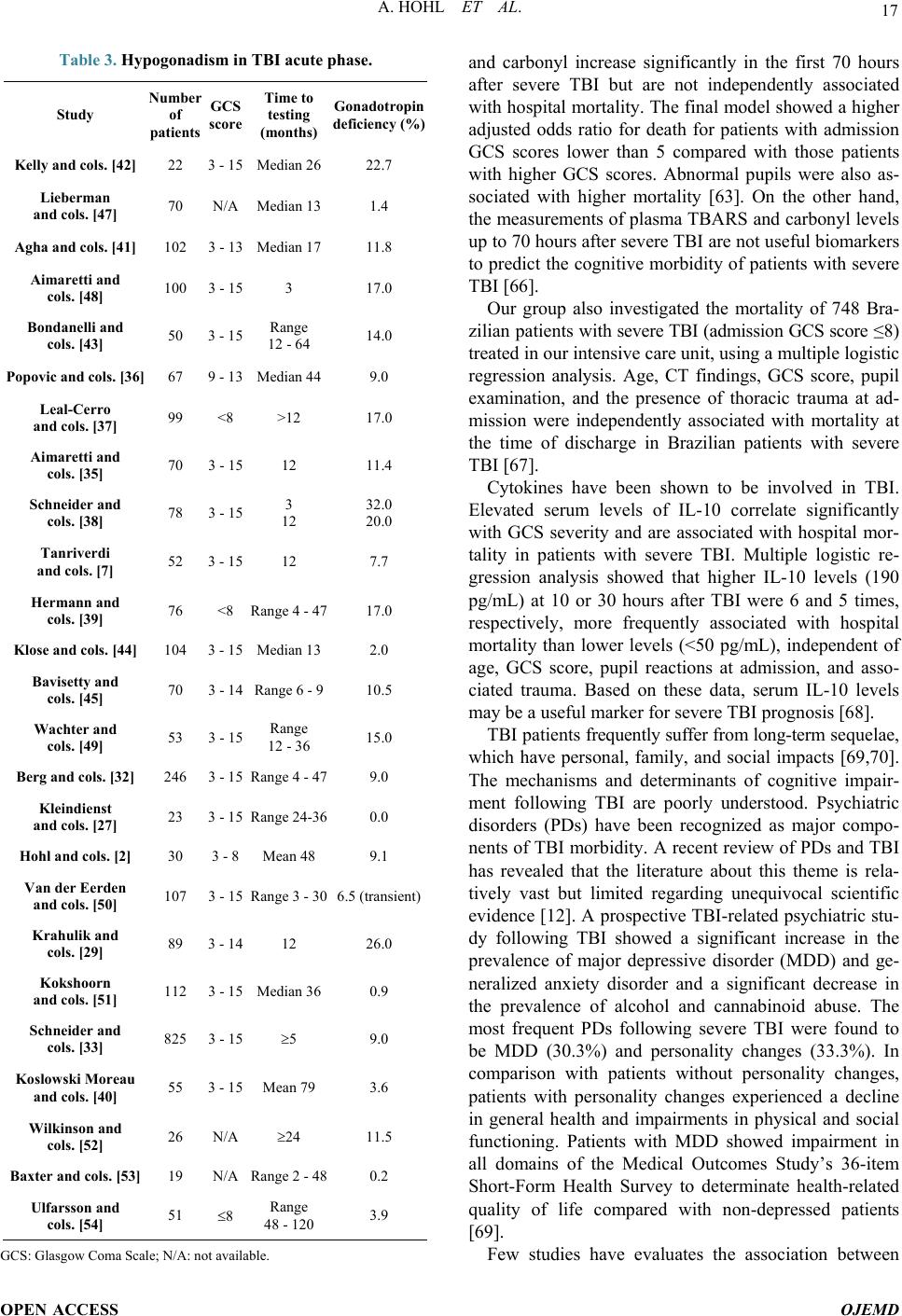 A. HOHL ET AL. OPEN ACCESS OJEMD Table 3. Hypogonadism in TBI acute phase. Study of patients GCS score Time to testing (months) deficiency (%) Kelly and cols. [42] 22 3 - 15 Median 26 22.7 Lieberman and cols. [47] 70 N/A Median 13 1.4 Agha and cols. [41] 102 3 - 13 Median 17 11.8 Aimaretti and cols. [48] 100 3 - 15 3 17.0 Bondanelli and cols. [43] 50 3 - 15 Range 12 - 64 14.0 Popovic and cols. [36] 67 9 - 13 Median 44 9.0 Leal-Cerro and cols. [37] 99 <8 >12 17.0 Aimaretti and cols. [35] 70 3 - 15 12 11.4 Schneider and cols. [38] 78 3 - 15 3 12 32.0 20.0 Tanriverdi and cols. [7] 52 3 - 15 12 7.7 Hermann and cols. [39] 76 <8 Range 4 - 47 17.0 Klose and cols. [44] 104 3 - 15 Median 13 2.0 Bavisetty and cols. [45] 70 3 - 14 Range 6 - 9 10.5 Wachter and cols. [49] 53 3 - 15 Range 12 - 36 15.0 Berg and cols. [32] 246 3 - 15 Range 4 - 47 9.0 Kleindienst and cols. [27] 23 3 - 15 Range 24-36 0.0 Hohl and cols. [2] 30 3 - 8 Mean 48 9.1 Van der Eerden and cols. [50] 107 3 - 15 Range 3 - 30 6.5 (transient) Krahulik and cols. [29] 89 3 - 14 12 26.0 Kokshoorn and cols. [51] 112 3 - 15 Median 36 0.9 Schneider and cols. [33] 825 3 - 15 ≥5 9.0 Koslowski Moreau and cols. [40] 55 3 - 15 Mean 79 3.6 Wilkinson and cols. [52] 26 N/A ≥24 11.5 Baxter and cols. [53] 19 N/A Range 2 - 48 0.2 Ulfarsson and cols. [54] 51 ≤8 Range 48 - 120 3.9 GCS: Glasgow Coma Scale; N/A: not available. and carbonyl increase significantly in the first 70 hours after severe TBI but are not independently associated with hospital mortality. The final model showed a higher adjusted odds ratio for death for patients with admission GCS scores lower than 5 compared with those patients with higher GCS scores. Abnormal pupils were also as- sociated with higher mortality [63]. On the other hand, the measurements of plasma TBARS and carbonyl levels up to 70 hours after severe TBI are not useful biomarkers to predict the cognitive morbidity of patients with severe TBI [66]. Our group also investigated the mortality of 748 Bra- zilian patients with severe TBI (admission GCS score ≤8) treated in our intensive care unit, using a multiple logistic regression analysis. Age, CT findings, GCS score, pupil examination, and the presence of thoracic trauma at ad- mission were independently associated with mortality at the time of discharge in Brazilian patients with severe TBI [67]. Cytokines have been shown to be involved in TBI. Elevated serum levels of IL-10 correlate significantly with GCS severity and are associated with hospital mor- tality in patients with severe TBI. Multiple logistic re- gression analysis showed that higher IL-10 levels (190 pg/mL) at 10 or 30 hours after TBI were 6 and 5 times, respectively, more frequently associated with hospital mortality than lower levels (<50 pg/mL), independent of age, GCS score, pupil reactions at admission, and asso- ciated trauma. Based on these data, serum IL-10 levels may be a useful marker for severe TBI prognosis [68]. TBI patients frequently suffer from long-term sequelae, which have personal, family, and social impacts [69,70]. The mechanisms and determinants of cognitive impair- ment following TBI are poorly understood. Psychiatric disorders (PDs) have been recognized as major compo- nents of TBI morbidity. A recent review of PDs and TBI has revealed that the literature about this theme is rela- tively vast but limited regarding unequivocal scientific evidence [12]. A prospective TBI-related psychiatric stu- dy following TBI showed a significant increase in the prevalence of major depressive disorder (MDD) and ge- neralized anxiety disorder and a significant decrease in the prevalence of alcohol and cannabinoid abuse. The most frequent PDs following severe TBI were found to be MDD (30.3%) and personality changes (33.3%). In comparison with patients without personality changes, patients with personality changes experienced a decline in general health and impairments in physical and social functioning. Patients with MDD showed impairment in all domains of the Medical Outcomes Study’s 36-item Short-Form Health Survey to determinate health-related quality of life compared with non-depressed patients [69]. Few studies have evaluates the association between 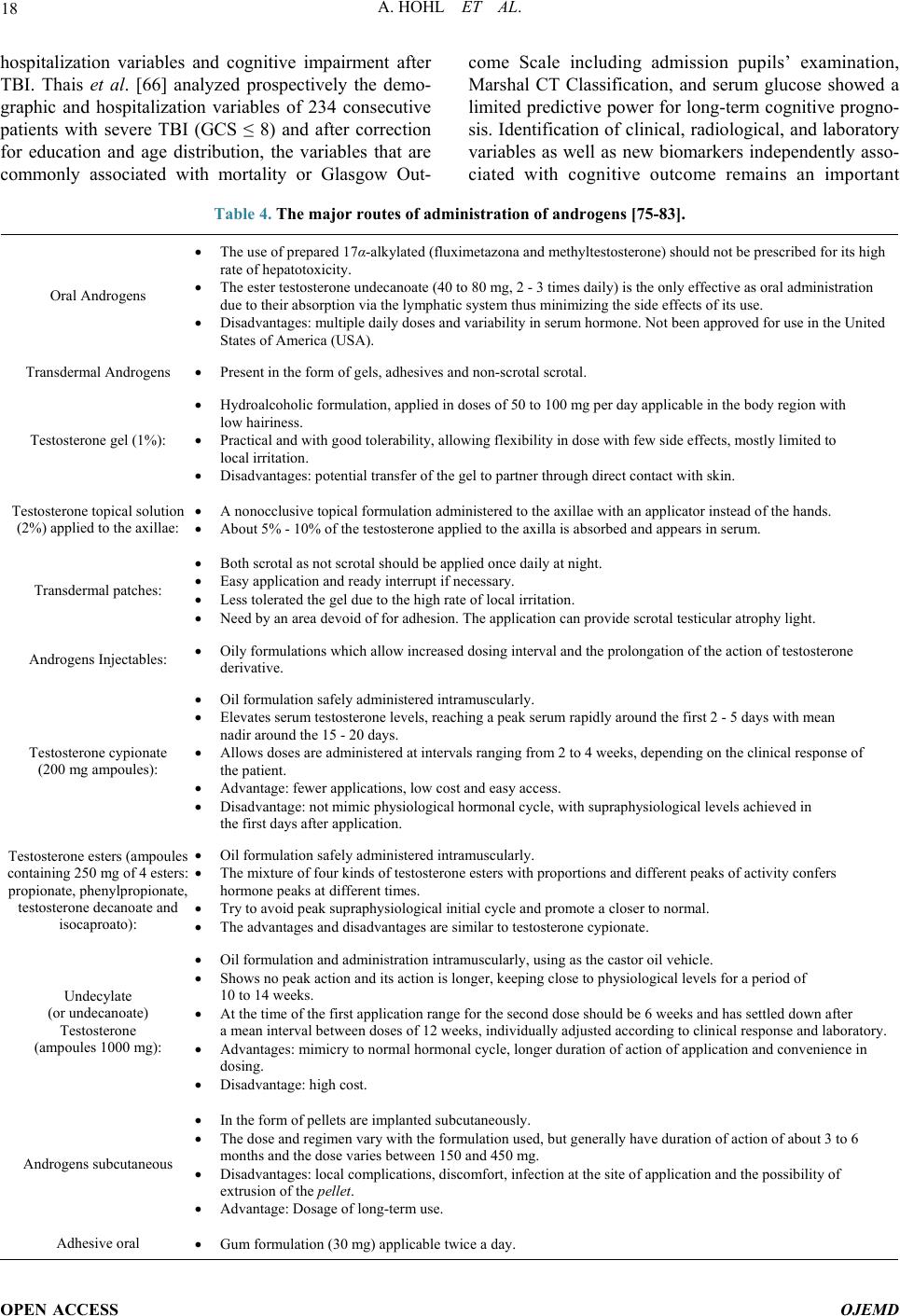 A. HOHL ET AL. OPEN ACCESS OJEMD hospitalization variables and cognitive impairment after TBI. Thais et al. [66] analyzed prospectively the demo- graphic and hospitalization variables of 234 consecutive patients with severe TBI (GCS ≤ 8) and after correction for education and age distribution, the variables that are commonly associated with mortality or Glasgow Out- come Scale including admission pupils’ examination, Marshal CT Classification, and serum glucose showed a limited predictive power for long-term cognitive progno- sis. Identification of clinical, radiological, and laboratory variables as well as new biomarkers independently asso- ciated with cognitive outcome remains an important Table 4. The major routes of administration of androgens [75-83]. Oral Androgens The use of prepared 17α-alkylated (fluximetazona and methyltestosterone) should not be prescribed for its high rate of hepatotoxicity. The ester testosterone undecanoate (40 to 80 mg, 2 - 3 times daily) is the only effective as oral administration due to their absorption via the lymphatic system thus minimizing the side effects of its use. Disadvantages: multiple daily doses and variability in serum hormone. Not been approved for use in the United States of America (USA). Transdermal Androgens Present in the form of gels, adhesives and non-scrotal scrotal. Testosterone gel (1%): Hydroalcoholic formulation, applied in doses of 50 to 100 mg per day applicable in the body region with low hairiness. Practical and with good tolerability, allowing flexibility in dose with few side effects, mostly limited to local irritation. Disadvantages: potential transfer of the gel to partner through direct contact with skin. Testosterone topical solution (2%) applied to the axillae: A nonocclusive topical formulation administered to the axillae with an applicator instead of the hands. About 5% - 10% of the testosterone applied to the axilla is absorbed and appears in serum. Transdermal patches: Both scrotal as not scrotal should be applied once daily at night. Easy application and ready interrupt if necessary. Less tolerated the gel due to the high rate of local irritation. Need by an area devoid of for adhesion. The application can provide scrotal testicular atrophy light. Androgens Injectables: Oily formulations which allow increased dosing interval and the prolongation of the action of testosterone derivative. Testosterone cypionate (200 mg ampoules): Oil formulation safely administered intramuscularly. Elevates serum testosterone levels, reaching a peak serum rapidly around the first 2 - 5 days with mean nadir around the 15 - 20 days. Allows doses are administered at intervals ranging from 2 to 4 weeks, depending on the clinical response of the patient. Advantage: fewer applications, low cost and easy access. Disadvantage: not mimic physiological hormonal cycle, with supraphysiological levels achieved in the first days after application. Testosterone esters (ampoules containing 250 mg of 4 esters: propionate, phenylpropionate, testosterone decanoate and isocaproato): Oil formulation safely administered intramuscularly. The mixture of four kinds of testosterone esters with proportions and different peaks of activity confers hormone peaks at different times. Try to avoid peak supraphysiological initial cycle and promote a closer to normal. The advantages and disadvantages are similar to testosterone cypionate. Undecylate (or undecanoate) Testosterone (ampoules 1000 mg): Oil formulation and administration intramuscularly, using as the castor oil vehicle. Shows no peak action and its action is longer, keeping close to physiological levels for a period of 10 to 14 weeks. At the time of the first application range for the second dose should be 6 weeks and has settled down after a mean interval between doses of 12 weeks, individually adjusted according to clinical response and laboratory. Advantages: mimicry to normal hormonal cycle, longer duration of action of application and convenience in dosing. Disadvantage: high cost. Androgens subcutaneous In the form of pellets are implanted subcutaneously. The dose and regimen vary with the formulation used, but generally have duration of action of about 3 to 6 months and the dose varies between 150 and 450 mg. Disadvantages: local complications, discomfort, infection at the site of application and the possibility of extrusion of the pellet. Advantage: Dosage of long-term use. Adhesive oral Gum formulation (30 mg) applicable twice a day.  A. HOHL ET AL. OPEN ACCESS OJEMD challenge for further work involving severe TBI patients. Man with moderate or severe TBI is a complex patient. Besides hormonal issue, all knowledge of clinical, emo- tional and social variables increase the chance of recov- ery and decrease the morbidity of these men. 7. Treatment of Hypogonadism after TBI The main goal of treatment of patients with hypogonad- ism is the reestablishment of sexual function and its sub- sequent maintenance, along with the secondary sexual characteristics and sexual extra effect of androgens (bone mineral density, muscle hypertrophy, and wellness, among others) [71-73]. Testosterone substitution is necessary in all hypogo- nadal patients, because androgen deficiency causes slight anemia, changes in coagulation parameters, de-creased bone density, muscle atrophy, regression of sexual func- tion, and alterations in mood and cognitive abilities. There is no universal agreement regarding target levels of replacement therapy, but most physicians aim for the mid to upper normal range [74]. Major routes of admin- istration of and rogens (Table 4) include the following: During treatment monitoring of testosterone levels, PSA and hematocrit is advised [75]. If fertility is desired, spermatogenesis can be initiated and maintained by clomiphene, gonadotropin therapy, conventionally in the form of human chorionic gonado- tropin (hCG) and human menopausal gonadotropin (hMG) or, more recently, purified or recombinant FSH [84]. Apart from this option, patients with disorders at the hy- pothalamic level can be stimulated with pulsatile gona- dotropin-releasing hormone (GnRH). Both treatment modalities have to be administered on average for 7 - 10 months until pregnancy is achieved. In individual cases, treatment may be necessary for up to 46 months [84]. 8. Conclusion Each year, the number of publications on TBI in the medical literature increases. As young men are the most affected population, it is essential to understand the func- tioning of TBI in this population. The male gonadal axis is affected in acute severe TBI and may represent a cause of late-onset hypogonadism. The treatment of patients with TBI and evolved hypogonad is misimportant for their recovery. Disclosure Statement The authors declare that they have no conflict of interest. Acknowledgements This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Apoio à Pesquisa Científica e Tec- nológica do Estado de Santa Catarina (FAPESC). Pro- grama de Apoio a Núcleos de Excelência—PRONEX (NENASC project) and Programa Pesquisas para o SUS— PPSUS. REFERENCES [1] N. Andelic “The Epidemiology of Traumatic Brain In- jury,” The Lancet Neurology, Vol. 12, No. 1, 2013, pp. 28-29. http://dx.doi.org/10.1016/S1474-4422(12)70294-6 [2] A. Hohl, T. L. Mazzuco, M. H. Coral, M. Schwarzbold and R. Walz, “Hypogonadism after Traumatic Brain In- jury,” Arquivos Brasileiros de Endocrinologia & Meta- bologia, Vol. 53, No. 8, 2009, pp. 908-914. http://dx.doi.org/10.1590/S0004-27302009000800003 [3] D. J. Thurman, C. Alverson, K. A. Dunn, J. Guerrero and J. E. Sniezek, “Traumatic Brain Injury in the United Sta- tes: A Public Health Perspective,” The Journal of Head Trauma Rehabilitation, Vol. 14, No. 6, 1999, pp. 602- 615. http://dx.doi.org/10.1097/00001199-199912000-00009 [4] F. Tagliaferri, C. Compagnone, M. Korsic, F. Servadei and J. Kraus, “A Systematic Review of Brain Injury Epi- demiology in Europe,” Acta Neurochirurgica (Wien), Vol. 148, No. 3, 2006, pp. 255-268. http://dx.doi.org/10.1007/s00701-005-0651-y [5] R. N. R. Fernandes and M. Silva, “Epidemiology of Trau- matic Brain Injury in Brazil,” Arquivos Brasileiros de Neurocirurgia, Vol. 32, No. 3, 2013, pp. 136-142. [6] R. J. Urban, “Hypopituitarism after Acute Brain Injury,” Growth Hormone & IGF Research, 2006, Vol. 16, Sup- plement A, pp. S25-S29. [7] F. Tanriverdi, H. Senyurek, K. Unluhizarci, A. Selcuklu, F. F. Casanueva and F. Kelestimur, “High Risk of Hypo- pituitarism after Traumatic Brain Injury: A Prospective Investigation of Anterior Pituitary Function in the Acute Phase and 12 Months after Trauma,” The Journal of Clinical Endocrinology and Metabolism, Vol. 91, No. 6, 2006, pp. 2105-2111. http://dx.doi.org/10.1210/jc.2005-2476 [8] F. Tanriverdi, H. Ulutabanca, K. Unluhizarci, A. Selcuklu, F. F. Casanueva and F. Kelestimur, “Pituitary Functions in the Acute Phase of Traumatic Brain Injury: Are They Related to Severity of the Injury or Mortality?” Brain In- jury, Vol. 21, No. 4, 2007, pp. 433-439. http://dx.doi.org/10.1080/02699050701311083 [9] A. Agha and C. J. Thompson, “High Risk of Hypogonad- ism after Traumatic Brain Injury: Clinical Implications,” Pituitary, Vol. 8, No. 3-4, 2005, pp. 245-249. http://dx.doi.org/10.1007/s11102-005-3463-4 [10] M. O. van Aken and S. W. Lamberts, “Diagnosis and Treatment of Hypopituitarism: An Update,” Pituitary, Vol. 8, No. 3-4, 2005, pp. 183-191. http://dx.doi.org/10.1007/s11102-006-6039-z [11] S. Deb, I. Lyons, C. Koutzoukis, I. Ali and G. McCarthy, “Rate of Psychiatric Illness 1 Year after Traumatic Brain Injury,” The American Journal of Psychiatry, Vol. 156,  A. HOHL ET AL. OPEN ACCESS OJEMD No. 3, 1999, pp. 374-378. [12] M. Schwarzbold, A. Diaz, E. T. Martins, A. Rufino, L. N. Amante, M. E. Thais, et al., “Psychiatric Disorders and Traumatic Brain Injury,” Journal of Neuropsychiatric Disease and Treatment, Vol. 4, No. 4, 2008, pp. 797-816. [13] J. W. Tomlinson, N. Holden, R. K. Hills, K. Wheatley, R. N. Clayton, A. S. Bates, et al., West Midlands Prospec- tive Hypopituitary Study Group, “Association between Premature Mortality and Hypopituitarism,” Lancet, Vol. 357, No. 9254, 2001, pp. 425-431. http://dx.doi.org/10.1016/S0140-6736(00)04006-X [14] V. Gasco, F. Prodam, L. Pagano, S. Grottoli, S. Belcastro, P. Marzullo, et al., “Hypopituitarism Following Brain In- jury: When Does It Occur and How Best to Test?” Pitui- tary, Vol. 15, No. 1, 2012, pp. 20-24. http://dx.doi.org/10.1007/s11102-010-0235-6 [15] T. P. Young, H. M. Hoaglin and D. T. Burke, “The Role of Serum Testosterone and TBI in the In-Patient Rehabil- itation Setting,” Brain Injury, Vol. 21, No. 6, 2007, pp. 645-649. http://dx.doi.org/10.1080/02699050701210426 [16] N. E. Carlson, L. A. Brenner, M. E. Wierman, C. Harri- son-Felix, C. Morey, S. Gallagher, et al., “Hypogonadism on Admission to Acute Rehabilitation Is Correlated with Lower Functional Status at Admission and Discharge,” Brain Injury, Vol. 23, No. 4, 2009, pp. 336-344. http://dx.doi.org/10.1080/02699050902788535 [17] R. Clapauch, D. J. Braga, L. P. Marinheiro, S. Buksman and Y. Schrank, “Risk of Late-Onset Hypogonadism (An- dropause) in Brazilian Men over 50 Years of Age with Osteoporosis: Usefulness of Screening Questionnaires,” Arquivos Brasileiros de Endocrinologia & Metabologia, Vol. 52, No. 9, 2008, pp. 1439-1447. http://dx.doi.org/10.1590/S0004-27302008000900006 [18] E. Ghigo, B. Masel, G. Aimaretti, J. Leon-Carrion, F. F. Casanueva, M. R. Dominguez-Morales, et al., “Consen- sus Guidelines on Screening for Hypopituitarism Follow- ing Traumatic Brain Injury,” Brain Injury, Vol. 19, No. 9, 2005, pp. 711-724. http://dx.doi.org/10.1080/02699050400025315 [19] M. Lorenzo, R. Peino, A. I. Castro, M. Lage, V. Popovic, C. Dieguez, et al., “Hypopituitarism and Growth Hor- mone Deficiency in Adult Subjects after Traumatic Brain Injury: Who and When to Test,” Pituitary, Vol. 8, No. 3- 4, 2005, pp. 233-237. http://dx.doi.org/10.1007/s11102-006-6046-0 [20] N. E. Kokshoorn, M. J. Wassenaar, N. R. Biermasz, F. Roelfsema, J. W. Smit, J. A. Romijn, et al., “Hypopitui- tarism Following Traumatic Brain Injury: Prevalence Is Affected by the Use of Different Dynamic Tests and Dif- ferent Normal Values,” European Journal of Endocri- nology, Vol. 162, No. 1, 2010, pp. 11-18. http://dx.doi.org/10.1530/EJE-09-0601 [21] J. Feibel, M. Kelly, L. Lee and P. Woolf, “Loss of Adre- nocortical Suppression after Acute Brain Injury: Role of Increased Intracranial Pressure and Brain Stem Function,” The Journal of Clinical Endocrinology and Metabolism, Vol. 57, No. 6, 1983, pp. 1245-1250. http://dx.doi.org/10.1210/jcem-57-6-1245 [22] J. M. Hackl, M. Gottardis, C. Wieser, E. Rumpl, C. Stad- ler, S. Schwarz, et al., “Endocrine Abnormalities in Se- vere Traumatic Brain Injury—A Cue to Prognosis in Se- vere Craniocerebral Trauma?” Intensive Care Medicine, Vol. 17, No. 1, 1991, pp. 25-29. http://dx.doi.org/10.1007/BF01708405 [23] F. Della Corte, A. Mancini, D. Valle, F. Gallizzi, P. Car- ducci, V. Mignani, et al., “Provocative Hypothalamopi- tuitary Axis Tests in Severe Head Injury: Correlations with Severity and Prognosis,” Critical Care Medicine, Vol. 26, No. 8, 1998, pp. 1419-1426. http://dx.doi.org/10.1097/00003246-199808000-00030 [24] A. Agha, B. Rogers, D. Mylotte, F. Taleb, W. Tormey, J. Phillips, et al., Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clinical Endocrinology (Oxford), Vol. 60, No. 5, 2004, pp. 584-591. http://dx.doi.org/10.1111/j.1365-2265.2004.02023.x [25] I. Cernak, V. J. Savic, A. Lazarov, M. Joksimovic and S. Markovic, “Neuroendocrine Responses Following Graded Traumatic Brain Injury in Male Adults,” Brain Injury, Vol. 13, No. 12, 1999, pp. 1005-1015. http://dx.doi.org/10.1080/026990599121016 [26] S. C. Lee, N. D. Zasler and J. S. Kreutzer, “Male Pitui- tary-Gonadal Dysfunction Following Severe Traumatic Brain Injury,” Brain Injury, Vol. 8, No. 6, 1994, pp. 571- 577. http://dx.doi.org/10.3109/02699059409151009 [27] A. Kleindienst, G. Brabant, C. Bock, C. Maser-Gluth and M. Buchfelder, “Neuroendocrine Function Following Traumatic Brain Injury and Subsequent Intensive Care Treatment: A Prospective Longitudinal Evaluation,” Journal of Neurotrauma, Vol. 26, No. 9, 2009, pp. 1435- 1446. http://dx.doi.org/10.1089/neu.2008.0601 [28] J. Wagner, J. R. Dusick, D. L. McArthur, P. Cohan, C. Wang, R. Swerdloff, et al., “Acute Gonadotroph and So- matotroph Hormonal Suppression after Traumatic Brain Injury,” Journal of Neurotrauma, Vol. 27, No. 6, 2010, pp. 1007-1019. http://dx.doi.org/10.1089/neu.2009.1092 [29] D. Krahulik, J. Zapletalova, Z. Frysak and M. Vaverka, “Dysfunction of Hypothalamic-Hypophysial Axis after Traumatic Brain Injury in Adults,” Journal of Neurosur- gery, Vol. 113, No. 3, 2010, pp. 581-584. http://dx.doi.org/10.3171/2009.10.JNS09930 [30] Z. Olivecrona, P. Dahlqvist and L. O. Koskinen, “Acute Neuro-Endocrine Profile and Prediction of Outcome after Severe Brain Injury,” Scandinavian Journal of Trauma, Resuscitation and Emergency Medicine, Vol. 21, 2013, p. 33. http://dx.doi.org/10.1186/1757-7241-21-33 [31] N. K. Sundaram, E. B. Geer and B. D. Greenwald, “The Impact of Traumatic Brain Injury on Pituitary Function,” Endocrinology and Metabolism Clinics of North America, Vol. 42, No. 3, 2013, pp. 565-583. http://dx.doi.org/10.1016/j.ecl.2013.05.003 [32] C. Berg, A. Oeffner, P. M. Schumm-Draeger, F. Badorrek, G. Brabant, B. Gerbert, et al., “Prevalence of Anterior Pi- tuitary Dysfunction in Patients Following Traumatic Brain Injury in a German Multi-Centre Screening Program,” Ex- perimental and Clinical Endocrinology & Diabetes, Vol. 118, No. 2, 2010, pp. 139-144. http://dx.doi.org/10.1055/s-0029-1225611 [33] H. J. Schneider, M. Schneider, I. Kreitschmann-Ander-  A. HOHL ET AL. OPEN ACCESS OJEMD mahr, U. Tuschy, H. Wallaschofski, S. Fleck, et al., “Struc- tured Assessment of Hypopituitarism after Traumatic Brain Injury and Aneurysmal Subarachnoid Hemorrhage in 1242 Patients: The German Interdisciplinary Database,” Jour- nal of Neurotrauma, Vol. 28, No. 9, 2011, pp. 1693-1698. http://dx.doi.org/10.1089/neu.2011.1887 [34] A. Hohl, J. B. Daltrozo, C. G. Pereira, T. R. Weber, H. F. Pinto, J. da Silva Gullo, et al., “Late Evaluation of the Pi- tuitary-Gonadal Axis in Survivors of Severe Traumatic Brain Injury,” Arquivos Brasileiros de Endocrinologia & Metabologia, Vol. 53, No. 8, 2009, pp. 1012-1019. http://dx.doi.org/10.1590/S0004-27302009000800016 [35] G. Aimaretti, M. R. Ambrosio, C. Di Somma, M. Gasperi, S. Cannavo, C. Scaroni, et al., “Residual Pituitary Func- tion after Brain Injury-Induced Hypopituitarism: A Pros- pective 12-Month Study,” The Journal of Clinical Endo- crinology and Metabolism, Vol. 90, No. 11, 2005, pp. 6085-6092. http://dx.doi.org/10.1210/jc.2005-0504 [36] V. Popovic, S. Pekic, D. Pavlovic, N. Maric, M. Jasovic- Gasic, B. Djurovic, et al., “Hypopituitarism as a Conse- quence of Traumatic Brain Injury (TBI) and Its Possible Relation with Cognitive Disabilities and Mental Dis- tress,” Journal of Endocrinological Investigation, Vol. 27, No. 11, 2004, pp. 1048-1054. [37] A. Leal-Cerro, J. M. Flores, M. Rincon, F. Murillo, M. Pujol, F. Garcia-Pesquera, et al., “Prevalence of Hypopi- tuitarism and Growth Hormone Deficiency in Adults Long-Term after Severe Traumatic Brain Injury,” Clini- cal Endocrinology, Vol. 62, No. 5, 2005, pp. 525-532. http://dx.doi.org/10.1111/j.1365-2265.2005.02250.x [38] H. J. Schneider, M. Schneider, B. Saller, S. Petersenn, M. Uhr, B. Husemann, et al., “Prevalence of Anterior Pitui- tary Insufficiency 3 and 12 Months after Traumatic Brain Injury,” European Journal of Endocrinology, Vol. 154, No. 2, 2006, pp. 259-265. http://dx.doi.org/10.1530/eje.1.02071 [39] B. L. Herrmann, J. Rehder, S. Kahlke, H. Wiedemayer, A. Doerfler, W. Ischebeck, et al., “Hypopituitarism Following Severe Traumatic Brain Injury,” Experimental and Clini- cal Endocrinology & Diabetes, Vol. 114, No. 6, 2006, pp. 316-321. http://dx.doi.org/10.1055/s-2006-924254 [40] O. Kozlowski Moreau, E. Yollin, E. Merlen, W. Daveluy and M. Rousseaux, “Lasting Pituitary Hormone Defi- ciency after Traumatic Brain Injury,” Journal of Neuro- trauma, Vol. 29, No. 1, 2012, pp. 81-89. http://dx.doi.org/10.1089/neu.2011.2048 [41] A. Agha, B. Rogers, M. Sherlock, P. O’Kelly, W. Tormey, J. Phillips, et al., “Anterior Pituitary Dysfunction in Sur- vivors of Traumatic Brain Injury,” Journal of Clinical Endocrinology and Metabolism, Vol. 89, No. 10, 2004, pp. 4929-4936. http://dx.doi.org/10.1210/jc.2004-0511 [42] D. F. Kelly, I. T. Gonzalo, P. Cohan, N. Berman, R. Swerdloff and C. Wang, “Hypopituitarism Following Trau- matic Brain Injury and Aneurysmal Subarachnoid He- morrhage: A Preliminary Report,” Journal of Neurosur- gery, Vol. 93, No. 5, 2000, pp. 743-752. http://dx.doi.org/10.3171/jns.2000.93.5.0743 [43] M. Bondanelli, L. De Marinis, M. R. Ambrosio, M. Mo- nesi, D. Valle, M. C. Zatelli, et al., “Occurrence of Pitui- tary Dysfunction Following Traumatic Brain Injury,” Jour- nal of Neurotrauma, Vol. 21, No. 6, 2004, pp. 685-696. http://dx.doi.org/10.1089/0897715041269713 [44] M. Klose, A. Juul, L. Poulsgaard, M. Kosteljanetz, J. Brennum and U. Feldt-Rasmussen, “Prevalence and Pre- dictive Factors of Post-Traumatic Hypopituitarism,” Cli- nical Endocrinology, Vol. 67, No. 2, 2007, pp. 193-201. http://dx.doi.org/10.1111/j.1365-2265.2007.02860.x [45] S. Bavisetty, D. L. McArthur, J. R. Dusick, C. Wang, P. Cohan, W. J. Boscardin, et al., “Chronic Hypopituitarism after Traumatic Brain Injury: Risk Assessment and Rela- tionship to Outcome,” Neurosurgery, Vol. 62, No. 5, 2008, pp. 1080-1093. [46] H. J. Schneider, I. Kreitschmann-Andermahr, E. Ghigo, G. K. Stalla and A. Agha, “Hypothalamopituitary Dysfunc- tion Following Traumatic Brain Injury and Aneurysmal Subarachnoid Hemorrhage: A Systematic Review,” JAMA, Vol. 298, No. 12, 2007, pp. 1429-1438. http://dx.doi.org/10.1001/jama.298.12.1429 [47] S. A. Lieberman, A. L. Oberoi, C. R. Gilkison, B. E. Ma- sel and R. J. Urban, “Prevalence of Neuroendocrine Dys- function in Patients Recovering from Traumatic Brain Injury,” Journal of Clinical Endocrinology and Metabo- lism, Vol. 86, No. 6, 2001, pp. 2752-2756. http://dx.doi.org/10.1210/jc.86.6.2752 [48] G. Aimaretti, M. R. Ambrosio, C. Di Somma, A. Fusco, S. Cannavo, M. Gasperi, et al., “Traumatic Brain Injury and Subarachnoid Haemorrhage Are Conditions at High Risk for Hypopituitarism: Screening Study at 3 Months after the Brain Injury,” Clinical Endocrinology, Vol. 61, No. 3, 2004, pp. 320-326. http://dx.doi.org/10.1111/j.1365-2265.2004.02094.x [49] D. Wachter, K. Gundling, M. F. Oertel, H. Stracke and D. K. Boker, “Pituitary Insufficiency after Traumatic Brain Injury,” Journal of Clinical Neuroscience, Vol. 16, No. 2, 2009, pp. 202-208. http://dx.doi.org/10.1016/j.jocn.2008.01.009 [50] A. W. van der Eerden, M. T. Twickler, F. C. Sweep, T. Beems, H. T. Hendricks, A. R. Hermus, et al., “Should Anterior Pituitary Function Be Tested during Follow-Up of All Patients Presenting at the Emergency Department because of Traumatic Brain Injury?” European Journal of Endocrinology, Vol. 162, No. 1, 2010, pp. 19-28. http://dx.doi.org/10.1530/EJE-09-0436 [51] N. E. Kokshoorn, J. W. Smit, W. A. Nieuwlaat, J. Tiemen- sma, P. H. Bisschop, R. Groote Veldman, et al., “Low Prevalence of Hypopituitarism after Traumatic Brain In- jury: A Multicenter Study,” European Journal of Endo- crinology, Vol. 165, No. 2, 2011, pp. 225-231. http://dx.doi.org/10.1530/EJE-11-0365 [52] C. W. Wilkinson, K. F. Pagulayan, E. C. Petrie, C. L. Mayer, E. A. Colasurdo, J. B. Shofer, et al., “High Pre- valence of Chronic Pituitary and Target-Organ Hormone Abnormalities after Blast-Related Mild Traumatic Brain Injury,” Frontiers in Neurology, Vol. 3, 2012, p. 11. http://dx.doi.org/10.3389/fneur.2012.00011 [53] D. Baxter, D. J. Sharp, C. Feeney, D. Papadopoulou, T. E. Ham, S Jilka, et al., “Pituitary Dysfunction after Blast  A. HOHL ET AL. OPEN ACCESS OJEMD Traumatic Brain Injury: The UK BIOSAP Study,” Annals of Neurology, Vol. 74, No. 4, 2013, pp. 527-536. http://dx.doi.org/10.1002/ana.23958 [54] T. Ulfarsson, G. Arnar Gudnason, T. Rosen, C. Blomstrand, K. S. Sunnerhagen, A. Lundgren-Nilsson, et al., “Pituitary Function and Functional Outcome in Adults After Severe Traumatic Brain Injury: The Long-Term Perspective,” Journal of Neurotrauma, Vol. 30, No. 4, 2013, pp. 271- 280. http://dx.doi.org/10.1089/neu.2012.2494 [55] R. M. Friedlander, “Apoptosis and Caspases in Neurode- generative Diseases,” New England Journal of Medicine, Vol. 348, No. 14, 2003, pp. 1365-1375. http://dx.doi.org/10.1056/NEJMra022366 [56] D. P. Graham and A. L. Cardon, “An Update on Sub- stance Use and Treatment Following Traumatic Brain In- jury,” Annals of the New York Academy of Sciences, Vol. 1141, 2008, pp. 148-162. http://dx.doi.org/10.1196/annals.1441.029 [57] M. C. LaPlaca, C. M. Simon, G. R. Prado and D. K. Cul- len, “CNS Injury Biomechanics and Experimental Mo- dels,” Progress in Brain Research, Vol. 161, 2007, pp. 13-26. http://dx.doi.org/10.1016/S0079-6123(06)61002-9 [58] J. M. Spaethling, D. M. Geddes-Klein, W. J. Miller, C. R. von Reyn, P. Singh, M. Mesfin, et al., “Linking Impact to Cellular and Molecular Sequelae of CNS Injury: Modeling in Vivo Complexity with in Vitro Simplicity,” Progress in Brain Research, Vol. 161, 2007, pp. 27-39. http://dx.doi.org/10.1016/S0079-6123(06)61003-0 [59] F. Dal-Pizzol, F. Klamt, M. M. Vianna, N. Schroder, J. Quevedo, M. S. Benfato, et al., “Lipid Peroxidation in Hippocampus Early and Late after Status Epilepticus In- duced by Pilocarpine or Kainic Acid in Wistar Rats,” Neuroscience Letters, Vol. 291, No. 3, 2000, pp. 179-182. http://dx.doi.org/10.1016/S0304-3940(00)01409-9 [60] F. Petronilho, G. Feier, B. de Souza, C. Guglielmi, L. S. Constantino, R. Walz, et al., “Oxidative Stress in Brain According to Traumatic Brain Injury Intensity,” Journal of Surgical Research, Vol. 164, No. 2, 2010, pp. 316-320. http://dx.doi.org/10.1016/j.jss.2009.04.031 [61] M. L. Schwarzbold, D. Rial, T. De Bem, D. G. Machado, M. P. Cunha, A. A. dos Santos, et al., “Effects of Trau- matic Brain Injury of Different Severities on Emotional, Cognitive, and Oxidative Stress-Related Parameters in Mice,” Journal of Neurotrauma, Vol. 27, No. 10, 2010, pp. 1883-1893. http://dx.doi.org/10.1089/neu.2010.1318 [62] A. P. Diaz, M. L. Schwarzbold, M. E. de Oliveira Thais, G. G. Cavalazzi, R. Schmoeller, J. C. Nunes, A. Hohl, R. Guarnieri, M. N. Linhares and R. Walz, “Personality Changes and Return-to-Work after Severe Traumatic Brain Injury: A Prospective Study,” Revista Brasileira de Psiquiatria, 2014, in Press. [63] A. Hohl, S. Gullo Jda, C. C. Silva, M. M. Bertotti, F. Felisberto, J. C. Nunes, et al., “Plasma Levels of Oxida- tive Stress Biomarkers and Hospital Mortality in Severe Head Injury: A Multivariate Analysis,” Journal of Criti- cal Care, Vol. 27, No. 5, 2012, pp. 523e11-523e19. [64] E. D. Hall, R. A. Vaishnav and A. G. Mustafa, “Antioxi- dant Therapies for Traumatic Brain Injury,” Neurothera- peutics, Vol. 7, No. 1, 2010, pp. 51-61. http://dx.doi.org/10.1016/j.nurt.2009.10.021 [65] E. D. Hall and J. M. Braughler, “Central Nervous System Trauma and Stroke. II. Physiological and Pharmacological Evidence for Involvement of Oxygen Radicals and Lipid Peroxidation,” Free Radical Biology and Medicine, Vol. 6, No. 3, 1989, pp. 303-313. http://dx.doi.org/10.1016/0891-5849(89)90057-9 [66] M. E. de Oliveira Thais, G. Cavallazzi, D. A. Formolo, L. D. de Castro, R. Schmoeller, R. Guarnieri, et al., “Li- mited Predictive Power of Hospitalization Variables for Long-Term Cognitive Prognosis in Adult Patients with Severe Traumatic Brain Injury,” Journal of Neuropsy- chology, Epub Ahead of Print, 2012. http://dx.doi.org/10.1111/jnp.12000 [67] E. T. Martins, M. N. Linhares, D. S. Sousa, H. K. Schroe- der, J. Meinerz, L. A. Rigo, et al., “Mortality in Severe Traumatic Brain Injury: A Multivariated Analysis of 748 Brazilian Patients from Florianopolis City,” Journal of Trauma, Vol. 67, No. 1, 2009, pp. 85-90. http://dx.doi.org/10.1097/TA.0b013e318187acee [68] F. M. Schneider Soares, N. Menezes de Souza, M. Libo- rio Schwarzbold, A. Paim Diaz, J. Costa Nunes, A. Hohl, et al., “Interleukin-10 Is an Independent Biomarker of Severe Traumatic Brain Injury Prognosis,” Neuroimmu- nomodulation, Vol. 19, No. 6, 2012, pp. 377-385. http://dx.doi.org/10.1159/000342141 [69] A. P. Diaz, M. L. Schwarzbold, M. E. Thais, A. Hohl, M. M. Bertotti, R. Schmoeller, et al., “Psychiatric Disorders and Health-Related Quality of Life after Severe Traumatic Brain Injury: A Prospective Study,” Journal of Neuro- trauma, Vol. 29, No. 6, 2012, pp. 1029-1037. http://dx.doi.org/10.1089/neu.2011.2089 [70] N. V. Marsh and D. A. Kersel, “Frequency of Behavioural Problems at One Year Following Traumatic Brain Injury: Correspondence between Patient and Caregiver Reports,” Neuropsychological Rehabilitation, Vol. 16, No. 6, 2006, pp. 684-694. http://dx.doi.org/10.1080/09602010500220290 [71] N. E. Skakkebaek, J. Toppari, O. Soder, C. M. Gordon, S. Divall and M. Draznin, “The Exposure of Fetuses and Children to Endocrine Disrupting Chemicals: A European Society for Paediatric Endocrinology (ESPE) and Pedia- tric Endocrine Society (PES) Call to Action Statement,” The Journal of Clinical Endocrinology and Metabolism, Vol. 96, No. 10, 2011, pp. 3056-3058. http://dx.doi.org/10.1210/jc.2011-1269 [72] S. H. Swan, K. M. Main, F. Liu, S. L. Stewart, R. L. Kruse, A. M. Calafat, et al., “Decrease in Anogenital Dis- tance among Male Infants with Prenatal Phthalate Expo- sure,” Environmental Health Perspectives, Vol. 113, No. 8, 2005, pp. 1056-1061. http://dx.doi.org/10.1289/ehp.8100 [73] F. H. Pierik, A. Burdorf, J. A. Deddens, R. E. Juttmann and R. F. Weber, “Maternal and Paternal Risk Factors for Cryptorchidism and Hypospadias: A Case-Control Study in Newborn Boys,” Environmental Health Perspectives, Vol. 112, No. 15, 2004, pp. 1570-1576. http://dx.doi.org/10.1289/ehp.7243  A. HOHL ET AL. OPEN ACCESS OJEMD [74] E. L. Rhoden and A. Morgentaler, “Risks of Testoste- rone-Replacement Therapy and Recommendations for Monitoring,” New England Journal of Medicine, Vol. 350, No. 5, 2004, pp. 482-492. http://dx.doi.org/10.1056/NEJMra022251 [75] S. Bhasin, G. R. Cunningham, F. J. Hayes, A. M. Mat- sumoto, P. J. Snyder, R. S. Swerdloff, et al., “Testoste- rone Therapy in Men with Androgen Deficiency Syn- dromes: An Endocrine Society Clinical Practice Guide- line,” The Journal of Clinical Endocrinology and Meta- bolism, Vol. 95, No. 6, 2010, pp. 2536-2559. http://dx.doi.org/10.1210/jc.2009-2354 [76] G. Corona, G. Rastrelli, G. Forti and M. Maggi, “Update in Testosterone Therapy for Men,” Journal of Sexual Medicine, Vol. 8, No. 3, 2011, pp. 639-654. http://dx.doi.org/10.1111/j.1743-6109.2010.02200.x [77] V. A. Giagulli, V. Triggiani, G. Corona, D. Carbone, B. Licchelli, E. Tafaro, et al., “Evidence-Based Medicine Update on Testosterone Replacement Therapy (TRT) in Male Hypogonadism: Focus on New Formulations,” Cur- rent Pharmaceutical Design, Vol. 17, No. 15, 2011, pp. 1500-1511. http://dx.doi.org/10.2174/138161211796197160 [78] G. Corona, G. Rastrelli, L. Vignozzi and M. Maggi, “Emerging Medication for the Treatment of Male Hypo- gonadism,” Expert Opinion on Emerging Drugs, Vol. 17, No. 2, 2012, pp. 239-259. http://dx.doi.org/10.1517/14728214.2012.683411 [79] C. Wang, R. S. Swerdloff, A. Iranmanesh, A. Dobs, P. J. Snyder, G. Cunningham, et al., “Transdermal Testosterone Gel Improves Sexual Function, Mood, Muscle Strength, and Body Composition Parameters in Hypogonadal Men,” The Journal of Clinical Endocrinology and Metabolism, Vol. 85, No. 8, 2000, pp. 2839-2853. http://dx.doi.org/10.1210/jc.85.8.2839 [80] C. Wang, N. Ilani, S. Arver, R. I. McLachlan, T. Soulis and A. Watkinson, “Efficacy and Safety of the 2% For- mulation of Testosterone Topical Solution Applied to the Axillae in Androgen-Deficient Men,” Clinical Endocri- nology, Vol. 75, No. 6, 2011, pp. 836-843. http://dx.doi.org/10.1111/j.1365-2265.2011.04152.x [81] C. Fennell, G. Sartorius, L. P. Ly, L. Turner, P. Y. Liu, A. J. Conway, et al., “Randomized Cross-Over Clinical Trial of Injectable vs. Implantable Depot Testosterone for Main- tenance of Testosterone Replacement Therapy in Androgen Deficient Men,” Clinical Endocrinology, Vol. 73, No. 1, 2010, pp. 102-109. [82] A. Hohl, M. O. Marques, M. H. Coral and R. Walz, “Evaluation of Late-Onset Hypogonadism (Andropause) Treatment Using Three Different Formulations of Inject- able Testosterone,” Arquivos Brasileiros de Endocrinolo- gia & Metabologia, Vol. 53, No. 8, 2009, pp. 989-995. http://dx.doi.org/10.1590/S0004-27302009000800013 [83] R. M. Meirelles and A. Hohl, “Men’s Health: So Neg- lected, Mainly by Men,” Arquivos Brasileiros de Endo- crinologia & Metabologia, Vol. 53, No. 8, 2009, pp. 899- 900. http://dx.doi.org/10.1590/S0004-27302009000800001 [84] M. Zitzmann and E. Nieschlag, “Hormone Substitution in Male Hypogonadism,” Molecular and Cellular Endocri- nology, Vol. 161, No. 1-2, 2000, p. 73-88. http://dx.doi.org/10.1016/S0303-7207(99)00227-0
|