 Advances in Bioscience and Biotechnology, 2013, 4, 48-61 ABB http://dx.doi.org/10.4236/abb.2013.411A2007 Published Online November 2013 (http://www.scirp.org/journal/abb/) Atherogenesis, the oxidative LDL modification hypothesis revisited Dov Lichtenberg*, Ilya Pinchuk Department of Physiology and Pharmacology, Sackler School of Medicine, Tel-Aviv University, Tel-Aviv, Israel Email: *physidov@post.tau.ac.il Received 23 September 2013; revised 25 November 2013; accepted 15 December 2013 Copyright © 2013 Dov Lichtenberg, Ilya Pinchuk. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT The commonly-accepted “oxidized LDL hypothesis of atherogenesis” is based on a large number of indi- rect evidence that shows that oxidativ ely-modif ied LDL plays a role in atherogenesis. Yet, the exact role is not clear. Some researchers think that oxidatively modi- fied biomolecules initiate atherogenesis; others be- lieve that they “only” promote this multifactorial process. Regardless of the exact mechanism responsi- ble for the effect of peroxidation on atherogenesis, the “oxidative theory of AS” is apparently inconsistent with the results of meta-analysis, in which (the “ex- pected”) significant correlation between CVD and oxidative stress (OS) was found only when the OS was evaluated on the basis of the plasma concentrations of malondialdehyde (MDA), often based on the con- centration of thiobarbituric acid reactive substances (TBARS). Notably, even this association is question- able due to 1) poor reliability of the laboratory assay of MDA and 2) possible publication bias. Hence, it appears that the commonly accepted paradigm re- garding the role of oxidative damage in the patho- genesis of CVD has been overestimated. Furthermore, the hypothesis is apparently inconsistent with the disappointing results of most of the clinical trials that were designed to reduce OS by means of supplemen- tation of antioxidants, mostly vitamin E. These ap- parent inconsistencies do not contradict the oxidative modification hypothesis of AS. The source of the ap- parent contradictions is probably the oversimplified considerations on which the predictions have been based. Many reasonable arguments can be raised to explain the apparent contradictions, which mea ns tha t our current knowledge is insufficient to test the rela- tionship of oxidative stress to cardiovascular disease. Keywords: ROS; Free Radicals; Atherosclerosis; Oxidative Stress; Antioxidants 1. INTRODUCTION Reactive oxygen species (ROS) and reactive nitrogen species (RNS) play essential and diverse roles in humane physiology [1-8]. They contribute to processes such as apoptosis and mitogenesis, they help maintaining cell number homeostasis and they play a central role in im- mune responses and cell signaling, particularly at the level of redox modulation. By contrast, excessive pro- duction of these reactive species (RONS), particularly free radicals, damages DNA, proteins and lipids, thus being cytotoxic [9-11]. The concentration of RONS is therefore a critical parameter in determining their ulti- mate cellular response. ROS-induced stimulation of cells has been shown to result in changes of the cellular redox potential due to interruption of redox balance, which may adversely affect cell function. Consequently, the de- pendence of the response on the concentration of RONS is unpredictable. In fact, the prevailing reputation of free radicals is that they are the “bad guys” responsible for aging [12-16] and for many diseases [17-19]. The oxidative modification hypothesis of atherogene- sis, first proposed by Steinberg et al. in 1989, is that atherogenesis is a consequence of oxidative modification of LDL [20]. The physiological role of LDL is to carry cholesterol from the liver to cells throughout the body. Internalization of oxidized LDL into cells, including en- dothelial cells and macrophages, differs from internaliza- tion of native LDL. Whereas native LDL is recognized and internalized into cells by way of the LDL receptor, oxidized LDL is recognized by the so-called scavenger receptors, which, unlike the LDL receptor, are not down- regulated [21,22]. The liver is very rich in scavenger receptors, so that soon after intravenous injection of oxi- dized LDL it disappears from the blood, becoming ac- cumulated in endothelial cells and macrophages. As a result, these cells become “foam cells” and die, thus *Corresponding author. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 49 forming atherotic plaques [23-25]. The peroxidation-induced atherogenesis hypothesis [20] inspired the National Heart, Lung and Blood Insti- tute to fund a workshop of a group of 30 distinguished specialists in various aspects of the relevant fields to re- view all of the evidence available up to that time, in- cluding some epidemiologic data [26]. The panel found that all the available knowledge is compatible with the oxidative modification hypothesis. Hence, the panel’s final recommendation was that studies utilizing natu- rally-occurring antioxidant vitamins (e.g. vitamin E, B- carotene and vitamin C) should proceed [27,28]. In view of this conclusion, the oxidation hypothesis of athero- sclerosis became the most commonly accepted paradigm. Since the removal of oxidized LDL by the scavenger receptors is rapid, minimally oxidized LDL disappears from the circulation slower than more oxidized LDL. Consequently, the level of oxidized LDL in the blood is low and much of it is minimally oxidized LDL [29]. No- tably, the autoantibody against oxidized LDL investi- gated by Witztum et al. demonstrated that even minor modifications in the structure of LDL make it immuno- genic and that this specific antibody can be observed in arterial lesions, in agreement with the notion that most of the AS-relevant oxidation probably occurs in the artery wall itself [29]. Notably, in the presence of copper, the LDL-associated antioxidants become oxidized and only then, rapid oxidation of LDL lipids (both fatty acids and cholesterol) begins [30]. The obvious implication of this mechanism was that if peroxidation of LDL is the cause of atherogenesis, anti- oxidants should help prevent it. Unfortunately, the results of clinical trials were disappointing. Specifically, al- though several trials indicated that vitamin E supple- mentation reduces the rate of both nonfatal MI and Non- fatal Stroke, in other trials vitamin E had no significant effect on different cardiac end points. The important ar- gument of the supporters of vitamin E supplementation is that some people benefit from it, whereas “if it does not help, it does not hurt” [31]. In the words of William A. Pryor: “In view of the very low risk of reasonable sup- plementation with vitamin E, some supplementation ap- pears prudent now” [31]. Being aware of the conflicting evidence, Witztum and Steinberg recall that cardiovascular disease (CVD) is a complex and multifactorial inflammatory disease associ- ated with gradual progression of plaques. Furthermore, they raised the possibility that the “Oxidative modifica- tion Hypothesis” may not hold for humans and con- cluded that we need to design clinical trails sensitive to early lesions [29]. Moreover, in 2004, Stocker and Kea- ney, in their review on the “Role of oxidative modifica- tions in Atherogenesis”, specifically stated that it is not clear whether oxidative events are a cause or a result of AS [32]. At about the same time, two independent meta- analyses concluded that vitamin E supplementation re- sults in higher mortality [33,34], which raised the ques- tion whether antioxidant supplementation is “Good in Theory, but is the theory good?” [35]. The possibility that the theory is not good can not be ruled out, but the apparent contradiction between the predicted and observed effects of vitamin E can be ex- plained differently. Specifically, the lack of beneficial effect(s) of vitamin E supplementation may be due to at least one of many processes discussed in this critical, non-comprehensive review. For this discussion to be understood, we must first briefly describe the physio- logical roles of RONS and the evidence for their in- volvement in atherogenesis. Next, we relate to the avail- able data on the oxidative status in CVD patients in comparison to matched controls. We then take a closer look at the intervention studies, particularly vitamin E supplementation trials. In an attempt to gain understand- ing of the apparent inconsistencies between the hypothe- sis and the experimental results, we relate to a variety of possibilities with emphasis on the very complex (both protective and atherogenic) role of HDL and of lipopro- tein-associated enzymes on the peroxidation of LDL lip- ids. Based on this critical review of available data, we can only conclude that the specific roles of LDL oxida- tive modification in atherosclerosis are in a state of un- certainty. 2. THE PHYSIOLOGICAL ROLE OF REACTIVE OXYGEN AND NITROGEN SPECIES For many years, OS has been commonly regarded a measure of a person’s probability to suffer from oxida- tive damages. Thus, OS have been commonly blamed for being involved in the pathogenesis of many diseases and antioxidants predicted to be good to us [9,18]. Now we realize that “Reactive oxygen species are ‘double-edged swords’ in cellular processes: low-dose cell signaling versus high-dose toxicity” [10,11]. Hence, “the dose re- sponse curve is unpredictable” [10]. In other words, the effects of “Free radicals and antioxidants in (both) nor- mal physiological functions and human disease” [8] de- pend upon their concentrations. At moderate concentra- tions, both the production and metabolism of RONS are tightly controlled [6] and cellular ‘redox homeostasis’ is retained. Under homeostatic conditions, ROS act as secondary messengers in intracellular signaling cascades, including the ROS-induced cell signaling and particularly the sig- naling for skeletal muscle adaptation [3]. By that, ROS contribute to the defense against infectious agents, as well as to other immune reactions, apoptosis and mito- genesis and anti-tumourigenic species. The claims made Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 50 by Ristow’s group [16] that “there is experimental basis to question Harman's Free Radical Theory of Aging” [12] and that “the existing data suggest that ROS act as essen- tial signaling molecules to promote metabolic health and longevity” [16], have yet to be evaluated but can not be ruled out. By contrast, under certain conditions free radicals may induce and maintain the oncogenic phenotype of cancer cells and this is merely one of many deleterious effects of over produced ROS, including aging and human diseases, particularly cancer, cardiovascular disease, atherosclero- sis, hypertension, ischemia/reperfusion injury, diabetes mellitus, Alzheimer’s disease and Parkinson’s disease [5,18,19]. Admittedly, it is not clear whether excessive formation of free radicals is the primary cause of any of these diseases or merely a result of inflammation or/and tissue injury. Yet, it is commonly believed that the ROS- induced damage to cell components, including mem- brane lipids, proteins and DNA contributes to all these pathologies. In short, the effects of RONS on both human function and diseases depend in a complex, non-monotonic fash- ion on their characteristics and concentrations in the relevant compartment. These, in turn depend, in a com- plex, interrelated fashion on a large number of factors. Hence, at the present time we do not have a way to pre- dict whether a given change of the redox status is likely to promote or retard our health. 3. EVIDENCE FOR THE INVOLVEMENT OF FREE RADICALS IN ATHEROGENESIS The “oxidative modification-hypothesis of AS” was origi- nally based on five findings in Steinberg’s group [20,29]: 1) That “incubation of endothelial cells in culture with high concentrations of LDL led to cell death, whereas the LDL re-isolated from the medium of a cell culture after incubation was markedly altered, such that it was taken up by monocytes/macrophages in culture much faster than native (normal) LDL [36]. 2) That the major change that occurred during the in- cubation did not take place when the medium in which the cells were grown did not contain transition metal ions [37]. 3) That addition of antioxidants to cells in plasma– containing cultures prevented the changes in the LDL. Specifically, addition of vitamin E could completely prevent oxidation of LDL induced by incubation with the cells [37]. 4) That oxidized LDL is a chemoattractant for blood monocytes, which helps recruiting them into a develop- ing lesion [24]. 5) That oxidized LDL inhibits the motility of tissue macrophages, which would tend to trap such cells in the artery wall once they got there [25]. At that time, there were several preliminary evidences that treatment of rabbits with several antioxidants re- duced the rate of atherogenesis [38]. After the publication of the oxidative modification hypothesis of AS, a large number of supporting, mainly indirect lines of evidence appeared, including evidence based on the composition of atherotic plaques [39] and data on the effect of antioxidants on the rate of athero- genesis in primates [28]. Today, we have many more lines of evidence for the involvement of LDL oxidation in atherogenesis. First, we know that there are at least ten different ways by which different oxidation products of LDL can be atherogenic via various mechanisms [4,29] and that the mode of LDL oxidation affects the biological effects of the oxidation products, as described by Levitan et al. in their compre- hensive review [40]. Although these authors found “a significant degree of specificity to different forms of oxLDL”, they note in their concluding remarks that we do not know “which of the oxLDL forms has the most pronounced effect on the development of the AS lesions” and that in spite of “the considerable progress made in recent years…there are still unresolved issues”, which are difficult to resolve because the studied oxLDL prepa- rations are “poorly defined” [40]. We also know that other processes beside oxidation can also lead to foam cell formation, irrespective of lipid peroxidation, including formation of complexes of aggre- gated [41,42] or slightly modified LDL particles with antibodies against them [29]. Introduction of the latter complexes into macrophages, by way of the immu- noglobulins (FC) receptor, results in foam cell forma- tion [43]. Foam cell formation via these and other per- oxidation-independent mechanisms is presently a topic of extensive investigations [44-46]. One of the most convincing lines of evidence for the involvement of oxidized LDL in cardio vascular disease is based on the clinically-validated association of various attributes of cardiovascular pathologies with the levels of oxidized phospholipids on apolipoprotein B (OxPL/apoB), as assayed in-vitro by the murine monoclonal antibody EO6 [47]. Using this assay, based on the specific binding of the EO6 antibody to the phosphocholine head group of oxidized phospholipids, it has been shown that the levels of oxidized phospholipids on circulating lipoproteins in the plasma: Are independent of traditional risk factors and the metabolic syndrome. Enhance the risk prediction of the Framingham Risk Score. Predict the presence and progression of coronary, femoral and carotid artery disease (particularly when amplified by lipoprotein(a) (Lp(a)) and phospholi- Copyright © 2013 SciRes. OPEN ACCESS 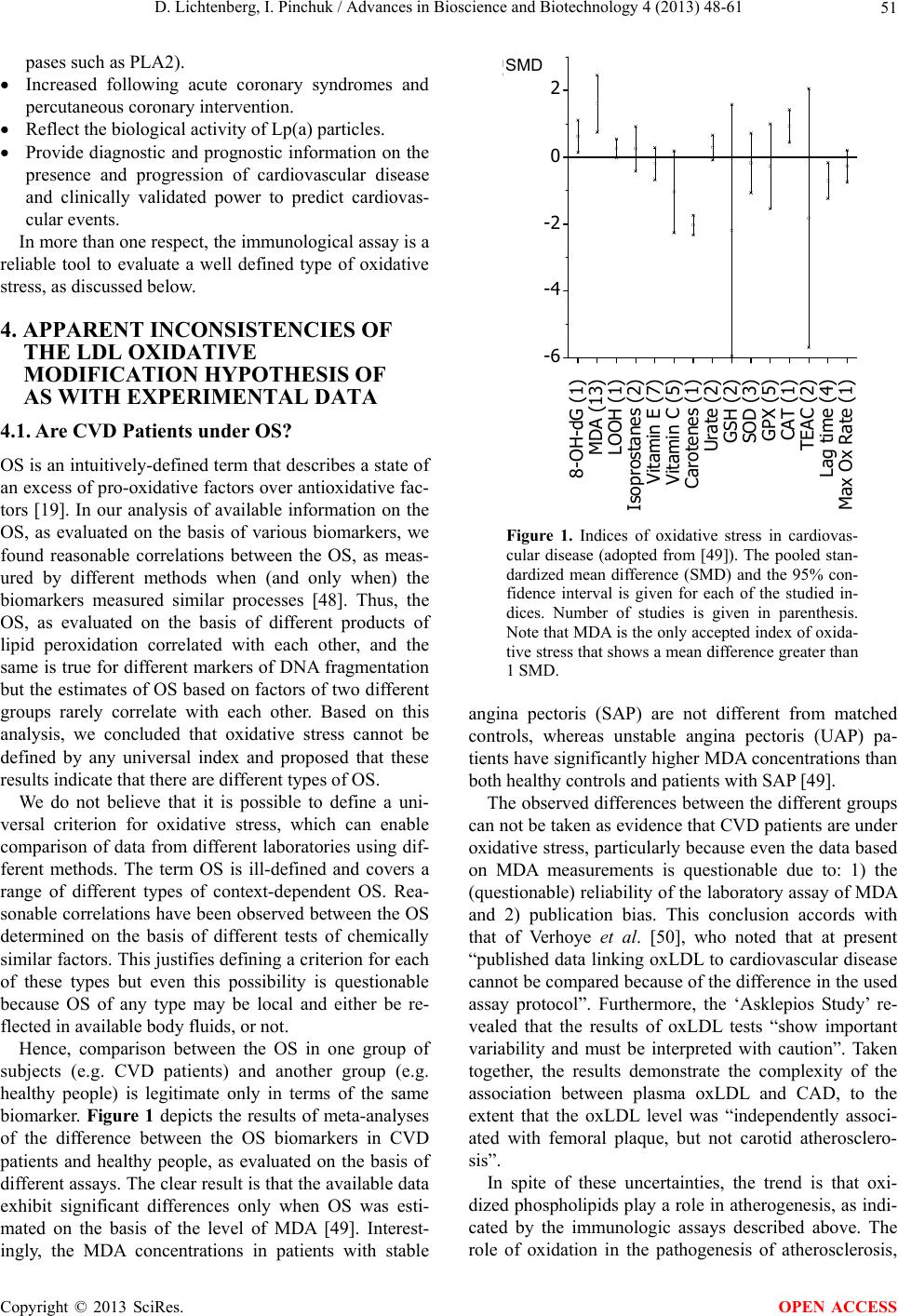 D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 51 pases such as PLA2). Increased following acute coronary syndromes and percutaneous coronary intervention. Reflect the biological activity of Lp(a) particles. Provide diagnostic and prognostic information on the presence and progression of cardiovascular disease and clinically validated power to predict cardiovas- cular events. In more than one respect, the immunological assay is a reliable tool to evaluate a well defined type of oxidative stress, as discussed below. 4. APPARENT INCONSISTENCIES OF THE LDL OXIDATIVE MODIFICATION HYPOTHESIS OF AS WITH EXPERIMENTAL DATA 4.1. Are CVD Patients under OS? OS is an intuitively-defined term that describes a state of an excess of pro-oxidative factors over antioxidative fac- tors [19]. In our analysis of available information on the OS, as evaluated on the basis of various biomarkers, we found reasonable correlations between the OS, as meas- ured by different methods when (and only when) the biomarkers measured similar processes [48]. Thus, the OS, as evaluated on the basis of different products of lipid peroxidation correlated with each other, and the same is true for different markers of DNA fragmentation but the estimates of OS based on factors of two different groups rarely correlate with each other. Based on this analysis, we concluded that oxidative stress cannot be defined by any universal index and proposed that these results indicate that there are different types of OS. We do not believe that it is possible to define a uni- versal criterion for oxidative stress, which can enable comparison of data from different laboratories using dif- ferent methods. The term OS is ill-defined and covers a range of different types of context-dependent OS. Rea- sonable correlations have been observed between the OS determined on the basis of different tests of chemically similar factors. This justifies defining a criterion for each of these types but even this possibility is questionable because OS of any type may be local and either be re- flected in available body fluids, or not. Hence, comparison between the OS in one group of subjects (e.g. CVD patients) and another group (e.g. healthy people) is legitimate only in terms of the same biomarker. Figure 1 depicts the results of meta-analyses of the difference between the OS biomarkers in CVD patients and healthy people, as evaluated on the basis of different assays. The clear result is that the available data exhibit significant differences only when OS was esti- mated on the basis of the level of MDA [49]. Interest- ingly, the MDA concentrations in patients with stable 8-OH-dG (1) MDA (13) LOOH (1) Isoprostanes (2) Vitamin E (7) Vitamin C (5) Carotenes (1) Urate (2) GSH (2) SOD (3) GPX (5) CAT (1) TEAC (2) Lag time (4) Max Ox Rate (1) -6 -4 -2 0 2 MD SMD Figure 1. Indices of oxidative stress in cardiovas- cular disease (adopted from [49]). The pooled stan- dardized mean difference (SMD) and the 95% con- fidence interval is given for each of the studied in- dices. Number of studies is given in parenthesis. Note that MDA is the only accepted index of oxida- tive stress that shows a mean difference greater than 1 SMD. angina pectoris (SAP) are not different from matched controls, whereas unstable angina pectoris (UAP) pa- tients have significantly higher MDA concentrations than both healthy controls and patients with SAP [49]. The observed differences between the different groups can not be taken as evidence that CVD patients are under oxidative stress, particularly because even the data based on MDA measurements is questionable due to: 1) the (questionable) reliability of the laboratory assay of MDA and 2) publication bias. This conclusion accords with that of Verhoye et al. [50], who noted that at present “published data linking oxLDL to cardiovascular disease cannot be compared because of the difference in the used assay protocol”. Furthermore, the ‘Asklepios Study’ re- vealed that the results of oxLDL tests “show important variability and must be interpreted with caution”. Taken together, the results demonstrate the complexity of the association between plasma oxLDL and CAD, to the extent that the oxLDL level was “independently associ- ated with femoral plaque, but not carotid atherosclero- sis”. In spite of these uncertainties, the trend is that oxi- dized phospholipids play a role in atherogenesis, as indi- cated by the immunologic assays described above. The role of oxidation in the pathogenesis of atherosclerosis, Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 52 as commonly described may appears to be overestimated but this may reflect homeostatic mechanisms capable of reducing the effects of various factors on the oxidative stress, as described below. 4.2. The Effect of Low Molecular Weight Antioxidants on Atherosclerosis Based on the free radical hypothesis of both longevity and atherosclerosis, supplementation of vitamin E could have been expected to have positive effects on both the rate of atherosclerosis and mortality. In fact, in animal models of atherosclerosis, antioxidants markedly de- crease the rate of progression of lesions [28,51], in ac- cordance with the oxidative modification hypothesis. By contrast, meta-analyses of the many published clinical trials show, at least apparently, higher mortality in vita- min E-treated people than in matched controls [33,34]. These meta-analyses induced serious critique (e.g. [52- 54]) and comprehensive responses to the critique [55,56]. The criticism related mostly to the following issues: 1) The choice of clinical studies to be included in the meta-analyses. 2) The choice of mortality as the only end point. 3) The heterogeneity of the participants with respect to both population and treatment. 4) The model used to analyze the data (hierarchical lo- gistic regression model vs. the traditional meta regres- sion). In short, in their communication in 2007, Blumberg and Frei described the “Clinical trials of vitamin E and cardiovascular diseases” as possibly being “fatally flawed” [54]. Decision Analysis [57,58] was designed to minimize the critique, by addressing these specific comments of the data as follows: 1) Inclusion of all the clinical trials quoted in the cri- tique of the meta-analyses. 2) The use of quality-adjusted life years (QALY) [59] as an endpoint reflects the effects of supplementation on both mortality and morbidity. 3) Using Markov model-based Monte Carlo simula- tions enable adjustment for heterogeneities (both for population and treatment) on the basis of registries. The main finding of this analysis was that vitamin E reduces the average QALY by almost four months but it also indicated that some people benefit from vitamin E supplementation [58]. Hence, the challenge was (and still is) to find criteria to differentiate between those who are likely to benefit from vitamin E supplementation and those who are not. Differentiation can be based either on identification of criteria that can be assayed in the lab or/and on data regarding the response of patients of spe- cific diseases to supplementation (see below). 4.3. The Effects of Vitamin E on HDL Peroxidation Oxidative modification of LDL is affected by HDL via different mechanisms, including acceleration of LDL peroxidation by HDL oxidation-products and inhibition of LDL peroxidation by removal of LDL peroxida- tion-products either by extraction from LDL to HDL and/or by their hydrolysis, catalyzed by HDL-associated enzymes. Hence, antioxidants can affect LDL peroxida- tion by altering the peroxidation of HDL and/or by alter- ing the effect of HDL on LDL peroxidation. In the fol- lowing discussion we first describe the results of a recent study that demonstrates the effect of vitamin E on HDL oxidation before relating to the mutual effects of peroxi- dation of HDL and LDL lipids. The very complex effect of tocopherol on the suscep- tibility of lipoproteins to oxidation is demonstrated in the recent study of its effects on both in-vitro and ex-vivo peroxidation of HDL2 & HDL3. Specifically, in their recent investigation, Wade et al. [60] found that the ef- fect of added tocopherol depends on whether its peroxi- dation was tested before or after fractionation of the se- rum. 1) When added after fractionation, tocopherol pro- tected the fraction of HDL2&3 against peroxidation. 2) By contrast, when pre-incubated with non-fraction- ated serum and tested after fractionation, tocopherol pro- moted the peroxidation of HDL2&3. 3) In another experiment, the authors tested the sus- ceptibility of the HDL2&3 fraction to peroxidation fol- lowing 6-weeks of tocopherol supplementation. The main result of this experiment was that the HDL2&3 fraction of the tocopherol-treated people were more susceptible to peroxidation than the same fraction of placebo-supple- mented controls [60]. Interestingly, the increased sensi- tivity to peroxidation was accompanied by a decrease of the activity of both HDL2&3-PON-1 and HDL2-LCAT (lecithin-cholesterol acyltransferase), probably because in the presence of higher concentrations of Toc, lower concentrations of the latter HDL-associated protective proteins were required, as described below. To conclude, tocopherol can either protect or promote HDL oxidation, depending upon the timing of exposure of serum to tocopherol. In turn, HDL (and its oxidation products) affects LDL oxidation, thus altering the com- plex effects of tocopherol on atherogenesis, as described below. 4.4. The Effect of HDL on LDL Peroxidation HDL, the beneficial form of blood cholesterol, (com- monly denoted “the good cholesterol”) is an established predictor of cardiovascular health. Its protective effect of the artery wall against atherosclerosis is well established. Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 53 Furthermore, even in individuals whose LDL level is low, HDL remains a strong predictor of the risk of coronary artery disease (CAD), indicating that HDL is an inde- pendent risk factor [51,61]. The positive effects of HDL are commonly attributed to its ability to evacuate choles- terol from the periphery to the liver. In addition to this “reverse” transport, HDL plays a modulatory role in in- flammation [62], which may also contribute to its cardio- protective effect. The importance of the antioxidative effect of HDL is emphasized by the findings of associa- tions between impaired antioxidant activity of HDL and various diseases [63,64]. In view of these facts, it is apparently surprising that under certain conditions, HDL oxidation accelerates LDL oxidation. Under such conditions, tocopherol-induced acceleration of HDL oxidation can be expected to pro- mote LDL oxidation, thus promote atherogenesis. In view of the latter possibility, much effort has been de- voted to investigate the mutual relationship between the oxidation of HDL and LDL, as described below. 4.4.1. Lipid Peroxidation in Mixtures of LDL and HDL Under many conditions, HDL is more susceptible to oxi- dation than LDL [65,66]. This is not surprising in view of the similar or higher content of Vitamin E and other antioxidants, including beta-carotene, ubiquinol (coen- zyme Q-10) and lycopene, in LDL. These antioxidants can act as the first line of defense. Furthermore, the higher surface /volume ratio in the smaller HDL particle can also accelerate the peroxidation induced by transition metal ions. It is also consistent with the finding that the smaller LDL particles are more readily oxidized than the larger ones [67]. However, the higher oxidizability of HDL is apparently inconsistent with the cardio-protective effect of HDL. Systematic kinetic studies on the oxidation in mixtures of HDL and LDL demonstrated the complexity of the effect of HDL on LDL peroxidation (and vice versa). Specifically, these mutual effects [68] depend in a com- plex fashion on the composition and physical properties of both these lipoproteins as well as on the inducer of peroxidation and the concentrations of transition metal ions and several serum proteins. The main results of the latter study were that: 1) Oxidation of LDL induced either by AAPH or by Myeloperoxidase (MPO) is inhibited by HDL under all the studied conditions, whereas. 2) Copper-induced peroxidation of LDL is inhibited by HDL at low copper/lipoprotein ratio but accelerated by HDL at high copper/lipoprotein ratio. These results indicate that the antioxidative effects of HDL are only partially due to HDL-associated enzymes, in agreement with the finding that reconstituted HDL, containing no such enzymes, inhibits peroxidation in- duced by low copper concentration [68]. Reduction of the binding of copper to LDL by competitive binding to the HDL can also contribute to the antioxidative effect of HDL. The acceleration of copper-induced oxidation of LDL by HDL, observed at high copper concentration, may be attributed to the hydroperoxides formed in the “more oxidizable” HDL, which can migrate to the “less oxidi- zable” LDL and enhance the oxidation of the LDL lipids induced by bound copper. Given the sensitivity of the rate of oxidation to many factors, this hypothesis does not necessarily contradict previous results [69], observed under different conditions, in which the migration of hydroperoxides between lipo- protein particles was too slow to accelerate the peroxida- tion in “native lipoprotein particles”. Notably, our inter- pretation of the acceleration of copper-induced LDL peroxidation by HDL is supported by the results of ex- periments in which native LDL was added to oxidizing lipoproteins at different time points. When the native LDL was added prior to decomposition of the hydroper- oxides in the oxidizing lipoprotein, the lag preceding oxidation of the LDL was much shorter than the lag ob- served when the native LDL was added at later stages (Figure 2), after the level of hydroperoxides became reduced due to their copper-catalyzed decomposition [68]. Furthermore, this interpretation accords with the ac- celeration of LDL peroxidation upon sequential exposure to copper [70]. Altogether, the observed interrelationship between the oxidation of HDL and LDL and its depend- ence on the specific conditions should be considered in future investigations regarding the oxidation of lipopro- teins. 4.4.2. Mechanisms that Contribute to the Inhibition of LDL Peroxidation by HDL The oxidative damage caused by peroxidation products can be reduced by their evacuation from the site of their production, where they may accelerate the propagation of free radicals chain reaction. Accordingly, the mecha- nisms that can contribute to the net effect of HDL can be divided to two categorize: 1) Mechanisms based on transporting peroxidation products to the liver (here denoted “the physical mode”). 2) Mechanisms based on metabolism, particularly hy- drolysis of short or/and oxidized chain phospholipids, catalyzed by LDL-associated PAF acetyl hydrolase and HDL-associated enzymes, including PON-1, PON-2 (here denoted “the chemical mode”). In relating to the chemical mode, we recall that under certain conditions the LDL-associated PAF-acetyl-hy- drolase from human plasma prevented oxidative modi- fication of LDL [71], whereas in other experiments, Copyright © 2013 SciRes. OPEN ACCESS 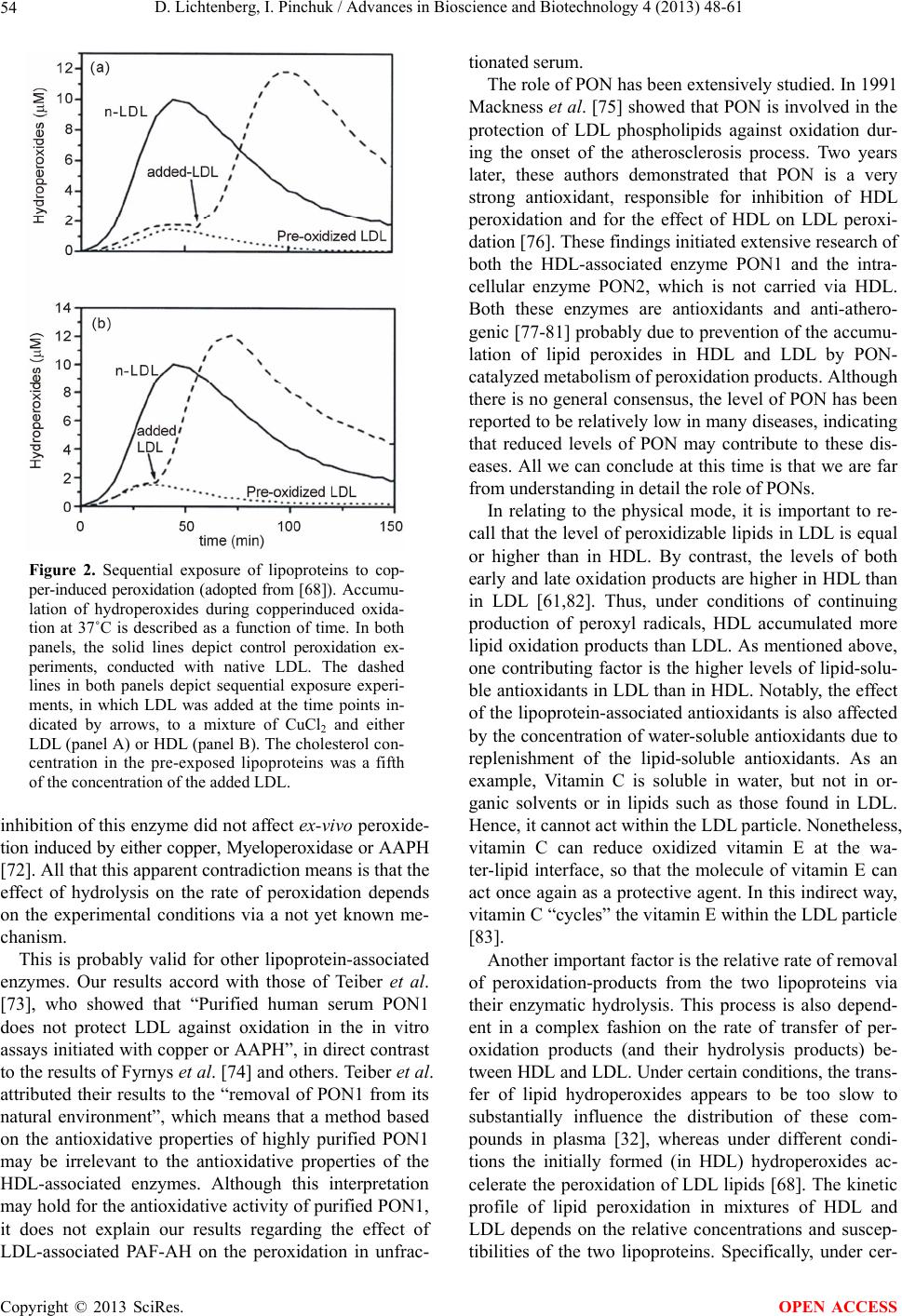 D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 54 Figure 2. Sequential exposure of lipoproteins to cop- per-induced peroxidation (adopted from [68]). Accumu- lation of hydroperoxides during copperinduced oxida- tion at 37˚C is described as a function of time. In both panels, the solid lines depict control peroxidation ex- periments, conducted with native LDL. The dashed lines in both panels depict sequential exposure experi- ments, in which LDL was added at the time points in- dicated by arrows, to a mixture of CuCl2 and either LDL (panel A) or HDL (panel B). The cholesterol con- centration in the pre-exposed lipoproteins was a fifth of the concentration of the added LDL. inhibition of this enzyme did not affect ex-vivo peroxide- tion induced by either copper, Myeloperoxidase or AAPH [72]. All that this apparent contradiction means is that the effect of hydrolysis on the rate of peroxidation depends on the experimental conditions via a not yet known me- chanism. This is probably valid for other lipoprotein-associated enzymes. Our results accord with those of Teiber et al. [73], who showed that “Purified human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH”, in direct contrast to the results of Fyrnys et al. [74] and others. Teiber et al. attributed their results to the “removal of PON1 from its natural environment”, which means that a method based on the antioxidative properties of highly purified PON1 may be irrelevant to the antioxidative properties of the HDL-associated enzymes. Although this interpretation may hold for the antioxidative activity of purified PON1, it does not explain our results regarding the effect of LDL-associated PAF-AH on the peroxidation in unfrac- tionated serum. The role of PON has been extensively studied. In 1991 Mackness et al. [75] showed that PON is involved in the protection of LDL phospholipids against oxidation dur- ing the onset of the atherosclerosis process. Two years later, these authors demonstrated that PON is a very strong antioxidant, responsible for inhibition of HDL peroxidation and for the effect of HDL on LDL peroxi- dation [76]. These findings initiated extensive research of both the HDL-associated enzyme PON1 and the intra- cellular enzyme PON2, which is not carried via HDL. Both these enzymes are antioxidants and anti-athero- genic [77-81] probably due to prevention of the accumu- lation of lipid peroxides in HDL and LDL by PON- catalyzed metabolism of peroxidation products. Although there is no general consensus, the level of PON has been reported to be relatively low in many diseases, indicating that reduced levels of PON may contribute to these dis- eases. All we can conclude at this time is that we are far from understanding in detail the role of PONs. In relating to the physical mode, it is important to re- call that the level of peroxidizable lipids in LDL is equal or higher than in HDL. By contrast, the levels of both early and late oxidation products are higher in HDL than in LDL [61,82]. Thus, under conditions of continuing production of peroxyl radicals, HDL accumulated more lipid oxidation products than LDL. As mentioned above, one contributing factor is the higher levels of lipid-solu- ble antioxidants in LDL than in HDL. Notably, the effect of the lipoprotein-associated antioxidants is also affected by the concentration of water-soluble antioxidants due to replenishment of the lipid-soluble antioxidants. As an example, Vitamin C is soluble in water, but not in or- ganic solvents or in lipids such as those found in LDL. Hence, it cannot act within the LDL particle. Nonetheless, vitamin C can reduce oxidized vitamin E at the wa- ter-lipid interface, so that the molecule of vitamin E can act once again as a protective agent. In this indirect way, vitamin C “cycles” the vitamin E within the LDL particle [83]. Another important factor is the relative rate of removal of peroxidation-products from the two lipoproteins via their enzymatic hydrolysis. This process is also depend- ent in a complex fashion on the rate of transfer of per- oxidation products (and their hydrolysis products) be- tween HDL and LDL. Under certain conditions, the trans- fer of lipid hydroperoxides appears to be too slow to substantially influence the distribution of these com- pounds in plasma [32], whereas under different condi- tions the initially formed (in HDL) hydroperoxides ac- celerate the peroxidation of LDL lipids [68]. The kinetic profile of lipid peroxidation in mixtures of HDL and LDL depends on the relative concentrations and suscep- tibilities of the two lipoproteins. Specifically, under cer- Copyright © 2013 SciRes. OPEN ACCESS 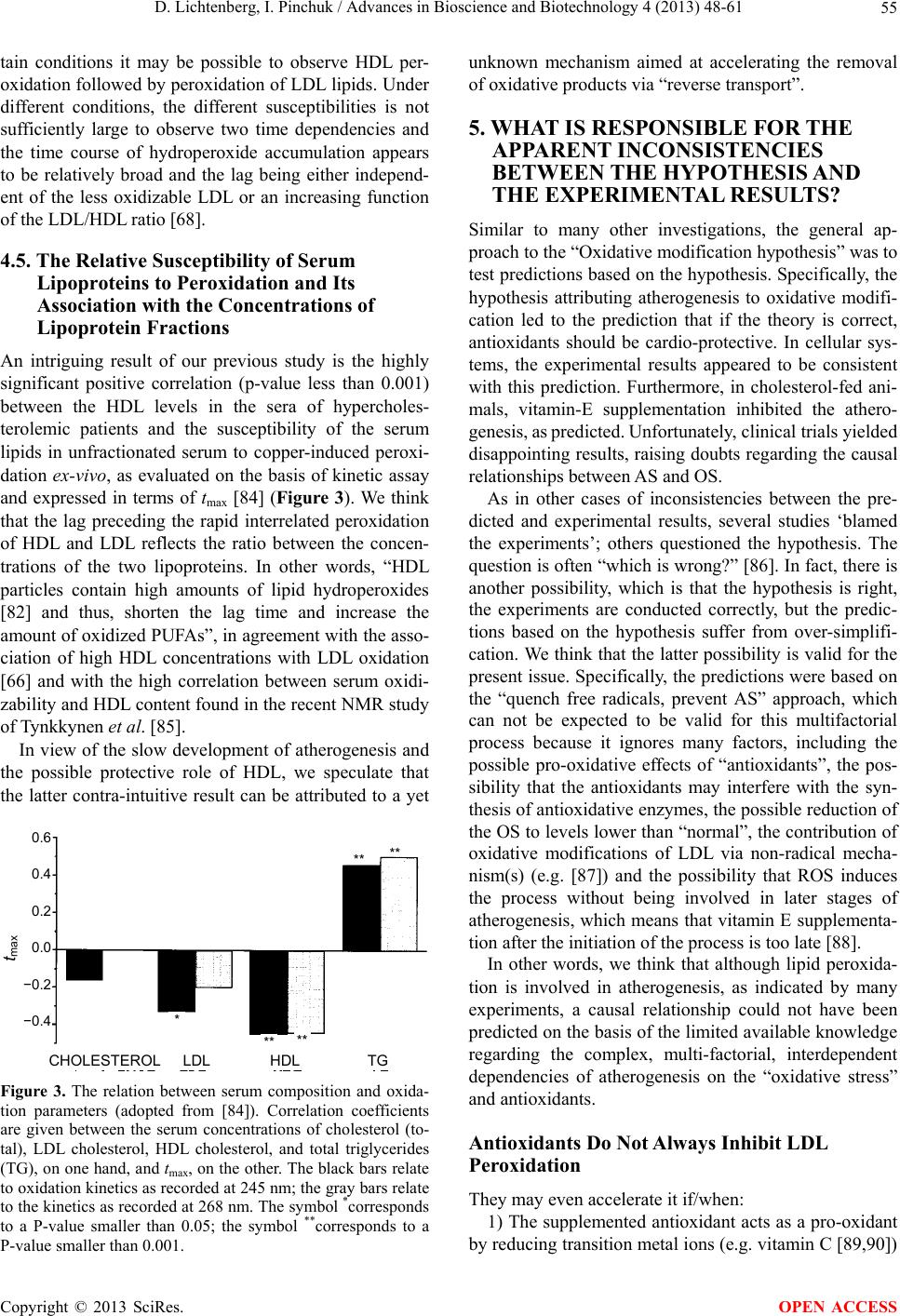 D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 55 tain conditions it may be possible to observe HDL per- oxidation followed by peroxidation of LDL lipids. Under different conditions, the different susceptibilities is not sufficiently large to observe two time dependencies and the time course of hydroperoxide accumulation appears to be relatively broad and the lag being either independ- ent of the less oxidizable LDL or an increasing function of the LDL/HDL ratio [68]. 4.5. The Relative Susceptibility of Serum Lipoproteins to Peroxidation and Its Association with the Concentrations of Lipoprotein Fractions An intriguing result of our previous study is the highly significant positive correlation (p-value less than 0.001) between the HDL levels in the sera of hypercholes- terolemic patients and the susceptibility of the serum lipids in unfractionated serum to copper-induced peroxi- dation ex-vivo, as evaluated on the basis of kinetic assay and expressed in terms of tmax [84] (Figure 3). We think that the lag preceding the rapid interrelated peroxidation of HDL and LDL reflects the ratio between the concen- trations of the two lipoproteins. In other words, “HDL particles contain high amounts of lipid hydroperoxides [82] and thus, shorten the lag time and increase the amount of oxidized PUFAs”, in agreement with the asso- ciation of high HDL concentrations with LDL oxidation [66] and with the high correlation between serum oxidi- zability and HDL content found in the recent NMR study of Tynkkynen et al. [85]. In view of the slow development of atherogenesis and the possible protective role of HDL, we speculate that the latter contra-intuitive result can be attributed to a yet CHOLESTEROL LDL HDL TG ** t max ** ** ** * 0.6 0.4 0.2 0.0 −0.2 −0.4 Figure 3. The relation between serum composition and oxida- tion parameters (adopted from [84]). Correlation coefficients are given between the serum concentrations of cholesterol (to- tal), LDL cholesterol, HDL cholesterol, and total triglycerides (TG), on one hand, and tmax, on the other. The black bars relate to oxidation kinetics as recorded at 245 nm; the gray bars relate to the kinetics as recorded at 268 nm. The symbol *corresponds to a P-value smaller than 0.05; the symbol **corresponds to a P-value smaller than 0.001. unknown mechanism aimed at accelerating the removal of oxidative products via “reverse transport”. 5. WHAT IS RESPONSIBLE FOR THE APPARENT INCONSISTENCIES BETWEEN THE HYPOTHESIS AND THE EXPERIMENTAL RESULTS? Similar to many other investigations, the general ap- proach to the “Oxidative modification hypothesis” was to test predictions based on the hypothesis. Specifically, the hypothesis attributing atherogenesis to oxidative modifi- cation led to the prediction that if the theory is correct, antioxidants should be cardio-protective. In cellular sys- tems, the experimental results appeared to be consistent with this prediction. Furthermore, in cholesterol-fed ani- mals, vitamin-E supplementation inhibited the athero- genesis, as predicted. Unfortunately, clinical trials yielded disappointing results, raising doubts regarding the causal relationships between AS and OS. As in other cases of inconsistencies between the pre- dicted and experimental results, several studies ‘blamed the experiments’; others questioned the hypothesis. The question is often “which is wrong?” [86]. In fact, there is another possibility, which is that the hypothesis is right, the experiments are conducted correctly, but the predic- tions based on the hypothesis suffer from over-simplifi- cation. We think that the latter possibility is valid for the present issue. Specifically, the predictions were based on the “quench free radicals, prevent AS” approach, which can not be expected to be valid for this multifactorial process because it ignores many factors, including the possible pro-oxidative effects of “antioxidants”, the pos- sibility that the antioxidants may interfere with the syn- thesis of antioxidative enzymes, the possible reduction of the OS to levels lower than “normal”, the contribution of oxidative modifications of LDL via non-radical mecha- nism(s) (e.g. [87]) and the possibility that ROS induces the process without being involved in later stages of atherogenesis, which means that vitamin E supplementa- tion after the initiation of the process is too late [88]. In other words, we think that although lipid peroxida- tion is involved in atherogenesis, as indicated by many experiments, a causal relationship could not have been predicted on the basis of the limited available knowledge regarding the complex, multi-factorial, interdependent dependencies of atherogenesis on the “oxidative stress” and antioxidants. Antioxidants Do Not Always Inhibit LDL Peroxidation They may even accelerate it if/when: 1) The supplemented antioxidant acts as a pro-oxidant by reducing transition metal ions (e.g. vitamin C [89,90]) Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 56 or via tocopherol-mediated peroxidation (TMP) [91]. 2) The added antioxidant reduces the OS too much, to levels below normal, thus causing toxic effects (e.g. due to promotion of the adhesion of red blood cells to endo- thelial cells [92]). 3) Modified lipoproteins are formed via free radi- cal-independent mechanisms [46,87,93]. 4) Free radicals initiate atherosclerosis but play only a minor role in later stages of the complex processes that yield AS, so that vitamin E does not alter the process when added after initiation occurred [88]. 5) The antioxidant interferes with the protective effect of HDL against LDL peroxidation [60], as discussed above. 6) The antioxidant reduces the production of other an- tioxidative factors, as indicated by the slight difference between CVD patients and healthy controls. It is also consistent with the interpretation of Ristow et al. [7] to their disappointing results concerning the (negative) ef- fects of vitamin E-supplementation on the health of sportsmen. Furthermore, this possibility is supported by the finding of high oxidizability of HDL in the serum of vitamin E-treated patients [60]. This may also be true for LDL and if it is not the case, we can still expect changes in LDL oxidation due to HDL oxidation, as described above. Given this long (yet partial) list of possible reasons for the apparent contradictions between the observed and predicted relationship of oxidative modification and CVD, we can only conclude that this relationship is cer- tainly uncertain. 6. OS IN CVD, CONCLUDING REMARKS, IMPLICATIONS AND SPECULA TIONS Atherogenesis is a “chronic” disease that develops during many years. It depends (at least partially) on alteration of the antioxidant-pro-oxidant balance, which is responsible for maintenance of the homeostatic level of the redox state. In comparison to the homeostatic level, OS means that the redox status in the relevant body compartment is “oxidative”. Antioxidants may be sufficient to shift it to normal levels but too much antioxidant may cause a shift to a “reductive state”. Alternative terminology can be based on the level of ROS in comparison to their homeo- static level. Specifically, OS may be described as being the result of “hyperOSemia”; an excess of antioxidants can be attributed to “hypoOSemia”. In fact, we have no data on the dependence of either the functions or the toxic effects of ROS on their con- centrations. In other words, the dependence of AS on OS is not well understood. This does not undermine the va- lidity of the hypothesis that peroxidation is responsible for atherogenesis [20] but it certainly justifies reconsid- eration of the hypothesis. To us, the present review indi- cates that ROS is not just the “bad guys” and not the only “bad guys” in the multi-factorial pathogenesis of coro- nary artery disease and atherogenesis. The major change in the field of free radical research, particularly in biology and medicine, is the growing un- certainties. Now that we realize the importance of the tight control of homeostatic level of OS, the paradigm of “bad free radicals versus good antioxidants” can not be retained, not only with respect to cardiovascular issues. Even Harman’s “free radical theory of aging” is ques- tionable. This state of art (in Atherosclerosis) raises at least three questions: 1) what is the sequence of proc- esses that lead to CVD and AS? 2) what is the interrela- tionship between LDL oxidation and CVD? and 3) who is likely to benefit from antioxidant supplementation? While the first two questions have been investigated and partially answered [44,45], we have no clues to an- swer the third [94]. Needless to say, high dose indis- criminate supplementation of vitamin E can not be rec- ommended to the general public (that still spends mil- lions of dollars on vitamin E supplements). Yet, it is quite clear that some specific groups may gain from vitamin E supplementation. The challenge is to define a criterion to predict who is likely to benefit from antioxidant supple- mentation. In his commentary in 1989, Witztum raised the ques- tion “To E or not to E?” [95] and proposed that antioxi- dants can be expected to be beneficial for people under oxidative stress. He then pointed out that the problem is that we “lack measures to identify high-risk groups that would theoretically benefit most from antioxidant inter- ventions” as well as a reliable measure “to determine the in vivo effectiveness of such interventions”. Unfortu- nately, not only the existing data is still insufficient to predict who is likely to benefit from vitamin E treat- ment. We even don’t know which type of oxidative stress has to be reduced. What we know is that based on the results of the population-based Cache County Study, Hayden et al. [52] advocate “further caution regarding the use of vitamin E by those with existing cardiovascu- lar disease”. This statement is inconsistent either with the assumption that people under oxidative stress can be expected to benefit from vitamin E or with the assump- tion that CVD patients are under high oxidative stress. We think that the apparent contradiction may be partially due to unrealistic estimation of the role of oxidative stress in CVD, in agreement with our conclusion regard- ing the (modest) role of OS in CVD patients [49]. Furthermore, in his 2003 communication, Frei raised the question of whether “to C or Not to C” and con- cluded that “What we know with certainty, however, is that a healthy diet and lifestyle lowers the risk of CHD, and this is what we should advocate to CHD patients and healthy people alike” [96]. Again, this kind of conclu- sions does not help in our goal of understanding the Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 57 questions defined above. In short, the ill-defined term “oxidative stress” does not differentiate between the ex- istence of “different types of oxidative stress” and the possibility that different antioxidants may have different antioxidative activity against different types of oxidative stress. Another disappointing result is that physical exercise, which is likely to cause excessive oxidation (thus, OS), does not increase the benefit from antioxidant supple- mentation. In fact, antioxidants prevent health-promoting effects of physical exercise [7]. In our future search for criteria to predict who is likely to benefit from antioxidant supplementation, we can ei- ther look for people who are under high (relevant type of) OS or/and patients suffering from a disease that have been associated with OS such as dialysis patients [97] or diabetes mellitus type 2 (DM2) patients with haptaglobin 2-2 genotype [98]. As long as we do not have such criteria, we propose basing the decision of whether or not to expect benefit from chronic supplementation of antioxidants on whether or not a relatively short period (e.g. one month) of sup- plementation to a patient with low vitamin E level results in a significant decrease of the blood level of lipid per- oxidation markers (e.g. MDA). This approach is supported by a previous study of the effect of vitamin E on AD patients, in which the clinical outcome of a group of “vitamin E respondents”, as de- termined after a short period of supplementation, was highly significant and excided by far the effect observed in the group of AD patients that were classified as “none-respondents”. In their words [99] “the decision of whether or not to expect benefit from chronic vitamin E supplementation should be based on whether or not one month of vitamin E supplementation to patient with low vitamin E level results in a significant decrease of the blood level of lipid peroxidation markers”. Regardless of the possible effects of food supplements, fruits and vegetables are beneficial and those “many pa- tients (that) view vitamins as a quick fix to compensate for poor eating habits [Tara Parker-Pope, The Wall Street Journal, March 20, 2006] should be advised of the dif- ference between food ingredients and food supplements. A glass of red wine, orange juice or a salad of vegetables or fruits along with ROS-containing food is likely to prevent absorption of unhealthy peroxides by quenching ROS in the stomach [100]. Furthermore, it is tasty. Syn- thetic supplements, commonly do not “meet” toxic food ingredients in the gastrointestinal system. Maybe this is why they are not as beneficial as expected. 7. ACKNOWLEDGEMENTS We thank Dr Edit Schnitzer and Prof Menachem Fainaru of TAU and Prof Yury Miller (UCSD) for their constructive remarks and the Lady Davis Foundation for financial support. REFERENCES [1] Terman, A. and Brunk, U.T. (2006) Oxidative stress, ac- cumulation of biological “garbage”, and aging. Antioxi- dants & Redox Signaling, 8, 197-204. http://dx.doi.org/10.1089/ars.2006.8.197 [2] Droge, W. (2002) Free radicals in the physiological con- trol of cell function. Physiological Reviews, 82, 47-95. [3] Finkel, T. (1998) Oxygen radicals and signaling. Current Opinion in Cell Biology, 10, 248-253. http://dx.doi.org/10.1016/S0955-0674(98)80147-6 [4] Halliwell, B. (2009) The wanderings of a free radical. Free Radical Biology and Medicine, 46, 531-542. http://dx.doi.org/10.1016/j.freeradbiomed.2008.11.008 [5] Halliwell, B., Gutteridge, J.M.C. and Cross, C.E. (1992) Free-radicals, antioxidants, and human-disease—Where are we now. Journal of Laboratory and Clinical Medi- cine, 119, 598-620. [6] Rattan, S.I.S. and Demirovic, D. (2010) Hormesis can and does work in humans. Dose-Response, 8, 58-63. http://dx.doi.org/10.2203/dose-response.09-041.Rattan [7] Ristow, M., Zarse, K., Oberbach, A., Kloting, N., Bir- ringer, M., Kiehntopf, M., et al. (2009) Antioxidants pre- vent health-promoting effects of physical exercise in hu- mans. Proceedings of the National Academy of Sciences of the United States of America, 106, 8665-8670. http://dx.doi.org/10.1073/pnas.0903485106 [8] Valko, M., Leibfritz, D., Moncol, J., Cronin, M.T.D., Ma- zur, M. and Telser, J. (2007) Free radicals and antioxi- dants in normal physiological functions and human dis- ease. International Journal of Biochemistry & Cell Biol- ogy, 39, 44-84. http://dx.doi.org/10.1016/j.biocel.2006.07.001 [9] Halliwell, B. and Gutteridge, G.M.C. (2007) Free radicals in biology and medicine. Oxford University Press, Ox- ford. [10] Martin, K.R. and Barrett, J.C. (2002) Reactive oxygen species as double-edged swords in cellular processes: Low- dose cell signaling versus high-dose toxicity. Human & Experimental Toxicology, 21, 71-75. http://dx.doi.org/10.1191/0960327102ht213oa [11] Yedgar, S., Krimsky, M., Cohen, Y. and Flower, R.J. (2007) Treatment of inflammatory diseases by selective eicosanoid inhibition: A double-edged sword? Trends in Pharmacological Sciences, 28, 459-464. http://dx.doi.org/10.1016/j.tips.2007.07.005 [12] Harman, D. (1956) Aging: A theory based on free radical and radiation chemistry. Journal of Gerontology, 11, 298- 300. http://dx.doi.org/10.1093/geronj/11.3.298 [13] Jang, Y.C. and Van Remmen, H. (2009) The mitochon- drial theory of aging: Insight from transgenic and knock- out mouse models. Experimental Gerontology, 44, 256- 260. http://dx.doi.org/10.1016/j.exger.2008.12.006 [14] Lyketsos, C.G., Steinberg, M., Tschanz, J.T., Norton, M.C., Steffens, D.C. and Breitner, J.C.S. (2000) Mental Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 58 and behavioral disturbances in dementia: Findings from the Cache County Study on Memory in Aging. American Journal of Psychiatry, 157, 708-714. http://dx.doi.org/10.1176/appi.ajp.157.5.708 [15] Perez, V.I., Bokov, A., Van Remmen, H., Mele, J., Ran, Q.T., Ikeno, Y., et al. (2009) Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta—General Subjects, 1790, 1005-1014. http://dx.doi.org/10.1016/j.bbagen.2009.06.003 [16] Ristow, M. and Schmeisser, S. (2011) Extending life span by increasing oxidative stress. Free Radical Biology and Medicine, 51, 327-336. http://dx.doi.org/10.1016/j.freeradbiomed.2011.05.010 [17] Cheeseman, K.H. and Slater, T.F. (1993) An introduction to free-radical biochemistry. British Medical Bulletin, 49, 481-493. [18] Gutteridge, J.M.C. (1993) Free-radicals in disease proc- esses—A compilation of cause and consequence. Free Radical Research Communications, 19, 141-158. http://dx.doi.org/10.3109/10715769309111598 [19] Sies, H. (1991) Oxidative stress—From basic research to clinical-application. American Journal of Medicine, 91, S31-S38. http://dx.doi.org/10.1016/0002-9343(91)90281-2 [20] Steinberg, D., Parthasarathy, S., Carew, T.E., Khoo, J.C. and Witztum, J.L. (1989) Beyond cholesterol—Modifi- cations of low-density lipoprotein that increase its athero- genicity. New England Journal of Medicine, 320, 915- 924. [21] Brown, M.S. and Goldstein, J.L. (1986) A receptor-me- diated pathway for cholesterol homeostasis. Science, 232, 34-47. http://dx.doi.org/10.1126/science.3513311 [22] Goldstein, J.L., Ho, Y.K., Basu, S.K. and Brown, M.S. (1979) Binding-site on macrophages that mediates uptake and degradation of acetylated low-density lipoprotein, producing massive cholesterol deposition. Proceedings of the National Academy of Sciences of the United States of America, 76, 333-337. http://dx.doi.org/10.1073/pnas.76.1.333 [23] Chisolm, G.M. and Steinberg, D. (2000) The oxidative modification hypothesis of atherogenesis: An overview. Free Radical Biology and Medicine, 28, 1815-1826. http://dx.doi.org/10.1016/S0891-5849(00)00344-0 [24] Quinn, M.T., Parthasarathy, S. and Steinberg, D. (1985) Endothelial cell-derived chemotactic activity for mouse peritoneal-macrophages and the effects of modified forms of low-density lipoprotein. Proceedings of the National Academy of Sciences of the United States of America, 82, 5949-5953. http://dx.doi.org/10.1073/pnas.82.17.5949 [25] Quinn, M.T., Parthasarathy, S., Fong, L.G. and Steinberg, D. (1987) Oxidatively modified low-density lipoproteins —A potential role in recruitment and retention of mono- cyte macrophages during atherogenesis. Proceedings of the National Academy of Sciences of the United States of America, 84, 2995-2998. http://dx.doi.org/10.1073/pnas.84.9.2995 [26] Steinberg, D., Berliner, J.A., Burton, G.W., Carew, T.E., Chait, A., Chisolm, G.M. et al. (1992) Antioxidants in the prevention of human atherosclerosis. Summary of the Proceedings of a National Heart Lung and Blood Insti- tute Workshop, 5-6 September 1991, Bethesda, Circula- tion, 85, 2338-2344. http://dx.doi.org/10.1161/01.CIR.85.6.2337 [27] Carew, T.E., Schwenke, D.C. and Steinberg, D. (1987) Antiatherogenic effect of probucol unrelated to its hypo- cholesterolemic effect—Evidence that antioxidants in vi- vo can selectively inhibit low-density-lipoprotein deg- radation in macrophage-rich fatty streaks and slow the progression of atherosclerosis in the Watanabe heritable hyperlipidemic rabbit. Proceedings of the National Aca- demy of Sciences of the United States of America, 84, 7725-7729. http://dx.doi.org/10.1073/pnas.84.21.7725 [28] Verlangieri, A.J. and Bush, M.J. (1992) Effects of D- alpha-tocopherol supplementation on experimentally in- duced primate atherosclerosis. Journal of the American College of Nutrition, 11, 131-138. [29] Witztum, J.L. and Steinberg, D. (2001) The oxidative modification hypothesis of atherosclerosis: Does it hold for humans? Trends in Cardiovascular Medicine, 11, 93- 102. http://dx.doi.org/10.1016/S1050-1738(01)00111-6 [30] Esterbauer, H., Rotheneder, M., Striegl, G., Waeg, G., Ashy, A., Sattler, W., et al. (1989) Vitamin-e and other lipophilic antioxidants protect Ldl against oxidation. Fett Wissenschaft Technologie-Fat Science Technology, 91, 316-324. [31] Pryor, W.A. (2000) Vitamin E and heart disease: Basic science to clinical intervention trials. Free Radical Biol- ogy and Medicine, 28, 141-164. http://dx.doi.org/10.1016/S0891-5849(99)00224-5 [32] Stocker, R. and Keaney, J.F. (2004) Role of oxidative modifications in atherosclerosis. Physiological Reviews, 84, 1381-1478. http://dx.doi.org/10.1152/physrev.00047.2003 [33] Bjelakovic, G., Nikolova, D., Simonetti, R.G. and Gluud, C. (2004) Antioxidant supplements for prevention of gas- trointestinal cancers: A systematic review and meta-ana- lysis. Lancet, 364, 1219-1228. http://dx.doi.org/10.1016/S0140-6736(04)17138-9 [34] Miller, E.R., Pastor-Barriuso, R., Dalal, D., Riemersma, R.A., Appel, L.J. and Guallar, E. (2005) Meta-analysis: High-dosage vitamin E supplementation may increase all- cause mortality. Annals of Internal Medicine, 142, 37-46. http://dx.doi.org/10.7326/0003-4819-142-1-200501040-0 0110 [35] Greenberg, E.R. (2005) Vitamin E supplements: Good in theory, but is the theory good? Annals of Internal Medi- cine, 142, 75-76. http://dx.doi.org/10.7326/0003-4819-142-1-200501040-0 0112 [36] Henriksen, T., Mahoney, E.M. and Steinberg, D. (1981) Enhanced macrophage degradation of low-density lipopro- tein previously incubated with cultured endothelial cells: Recognition by receptors for acetylated low-density lipo- proteins. Proceedings of the National Academy of Sci- ences of the United States of America, 78, 6499-6503. http://dx.doi.org/10.1073/pnas.78.10.6499 [37] Steinbrecher, U.P., Parthasarathy, S., Leake, D.S., Witztum, J.L. and Steinberg, D. (1984) Modification of low-density lipoprotein by endothelial-cells involves lipid-peroxidation Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 59 and degradation of low-density lipoprotein phospholipids. Proceedings of the National Academy of Sciences of the United States of America, 81, 3883-3887. http://dx.doi.org/10.1073/pnas.81.12.3883 [38] Kita, T., Nagano, Y., Yokode, M., Ishii, K., Kume, N., Ooshima, A., Yoshida, H. and Kawai, C. (1987) Probucol prevents the progression of atherosclerosis in watanabe heritable hyperlipidemic rabbit, an animal-model for fa- milial hypercholesterolemia. Proceedings of the National Academy of Sciences of the United States of America, 84, 5928-5931. http://dx.doi.org/10.1073/pnas.84.16.5928 [39] Vaya, J. (2013) The association between biomarkers in the blood and carotid plaque composition-focusing on oxidized lipids, oxysterols and plaque status. Biochemical Pharmacology, 86, 15-18. http://dx.doi.org/10.1016/j.bcp.2013.01.025 [40] Levitan, I., Volkov, S. and Subbaiah, P.V. (2010) Oxi- dized LDL: Diversity, patterns of recognition, and patho- physiology. Antioxidants & Redox Signaling, 13, 39-75. http://dx.doi.org/10.1089/ars.2009.2733 [41] Frank, J.S. and Fogelman, A.M. (1989) Ultrastructure of the intima in WHHL and cholesterol-fed rabbit aortas pre- pared by ultra-rapid freezing and freeze-etching. Journal of Lipid Research, 30, 967-978. [42] Khoo, J.C., Miller E., Mcloughlin P. and Steinberg D. (1988) Enhanced macrophage uptake of low-density lipo- protein after self-aggregation. Arteriosclerosis, 8, 348- 358. http://dx.doi.org/10.1161/01.ATV.8.4.348 [43] Lichtman, A.H., Binder C.J., Tsimikas S. and Witztum J.L. (2013) Adaptive immunity in atherogenesis: New in- sights and therapeutic approaches. Journal of Clinical Investigation, 123, 27-36. http://dx.doi.org/10.1172/JCI63108 [44] Halliwell, B. (2011) Free radicals and antioxidants—Quo vadis? Trends in Pharmacological Sciences, 32, 125-130. http://dx.doi.org/10.1016/j.tips.2010.12.002 [45] Curtiss, L.K. (2009) Reversing atherosclerosis? New Eng- land Journal of Medicine, 360, 1144-1146. http://dx.doi.org/10.1056/NEJMcibr0810383 [46] Lakota, K., Artenjak, A., Cucnik, S., Brguljan-Hitij, J., Cegovnik, B., Salobir, B. et al. (2012) Atherogenesis, in- flammation and autoimmunity—An overview. In: Par- thasarathy, S., Ed., Atherogenesis, InTech, 187-202. http://dx.doi.org/10.5772/26811 [47] Taleb, A., Witztum, J.L. and Tsimikas, S. (2011) Oxidi- zed phospholipids on apoB-100-containing lipoproteins: A biomarker predicting cardiovascular disease and car- diovascular events. Biomarkers in Medicine, 5, 673-694. http://dx.doi.org/10.2217/bmm.11.60 [48] Dotan, Y., Lichtenberg, D. and Pinchuk, I. (2004) Lipid peroxidation cannot be used as a universal criterion of oxi- dative stress. Progress in Lipid Research, 43, 200-227. http://dx.doi.org/10.1016/j.plipres.2003.10.001 [49] Dotan, Y., Lichtenberg, D. and Pinchuk, I. (2012) Are CVD patients under oxidative stress? In: Parthasarathy, S., Ed., Atherogenesis, InTech, pp. 413-424. http://dx.doi.org/10.5772/28181 [50] Verhoye, E. and Langlois, M.R. (2009) Circulating oxi- dized low-density lipoprotein: A biomarker of atheroscle- rosis and cardiovascular risk? Clinical Chemistry and La- boratory Medicine, 47, 128-137. http://dx.doi.org/10.1515/CCLM.2009.037 [51] Jain, S.K., Mcvie, R., Meachum, Z.D. and Smith, T. (2000) Effect of LDL plus VLDL oxidizability and hyperglycemia on blood cholesterol, phospholipid and triglyceride levels in Type-I diabetic patients. Atherosclerosis, 149, 69-73. http://dx.doi.org/10.1016/S0021-9150(99)00308-1 [52] Hayden, K.M., Welsh-Bohmer, K.A., Wengreen, H.J., Zandi, P.P., Lyketsos, C.G. and Breitner, J.C.S. (2007) Risk of mortality with vitamin E supplements: The cache county study. American Journal of Medicine, 120 , 180- 184. http://dx.doi.org/10.1016/j.amjmed.2006.03.039 [53] Jialal, I. and Devaraj, S. (2005) High-dosage vitamin E supplementation and all-cause mortality. Annals of In- ternal Medicine, 143, 155. http://dx.doi.org/10.7326/0003-4819-143-2-200507190-0 0026 [54] Blumberg, J.B. and Frei, B. (2007) Why clinical trials of vitamin E and cardiovascular diseases may be fatally flawed. Commentary on “The relationship between dose of vitamin E and suppression of oxidative stress in humans”. Free Radical Biology and Medicine, 43, 1374-1376. http://dx.doi.org/10.1016/j.freeradbiomed.2007.08.017 [55] Bjelakovic, G., Nikolova, D., Gluud, L.L., Simonetti, R.G. and Gluud, C. (2007) Mortality in randomized trials of anti- oxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA—Journal of the American Medical Association, 297, 842-857. http://dx.doi.org/10.1001/jama.297.8.842 [56] Miller, E.R., Appel, L.J., Guallar, E. and Pastor-Barriuso, R. (2005) High-dosage vitamin E supplementation and all- cause mortality—Response. Annals of Internal Medicine, 143, 156-158. http://dx.doi.org/10.7326/0003-4819-143-2-200507190-0 0029 [57] Dotan, Y., Lichtenberg, D. and Pinchuk, I. (2009) No evi- dence supports vitamin E indiscriminate supplementation. Biofactors, 35, 469-473. http://dx.doi.org/10.1002/biof.61 [58] Dotan, Y., Pinchuk, I., Lichtenberg, D. and Leshno, M. (2009) Decision analysis supports the paradigm that in- discriminate supplementation of vitamin E does more harm than good. Arteriosclerosis Thrombosis and Vascu- lar Biology, 29, 1304-1309. http://dx.doi.org/10.1161/ATVBAHA.108.178699 [59] Muennig, P.A. and Gold, M.R. (2001) Using the years- of-healthy-life measure to calculate QALYs. American Journal of Preventive Medicine, 20, 35-39. http://dx.doi.org/10.1016/S0749-3797(00)00261-0 [60] Wade, L., Nadeem, N., Young, I.S., Woodside, J.V., Mc- Ginty, A., McMaster, C. and McEneny, J. (2013) α-to- copherol induces proatherogenic changes to HDL2 & HDL3: An in vitro and ex vivo investigation. Atherosclerosis, 226, 392-397. http://dx.doi.org/10.1016/j.atherosclerosis.2012.11.032 [61] Shao, B. and Heinecke, J.W. (2009) HDL, lipid peroxide- tion, and atherosclerosis. Journal of Lipid Research, 50, 599-601. http://dx.doi.org/10.1194/jlr.E900001-JLR200 Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 60 [62] Navab, M., Reddy, S.T., Van Lenten, B.J. and Fogelman A.M. (2011) HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nature Reviews Cardi- ology, 8, 222-232. http://dx.doi.org/10.1038/nrcardio.2010.222 [63] Toker, A., Kadi, M., Yildirim, A.K., Aksoy, H. and Akcay, F. (2009) Serum lipid profile paraoxonase and aryles- terase activities in psoriasis. Cell Biochemistry and Func- tion, 27, 176-180. http://dx.doi.org/10.1002/cbf.1553 [64] Moradi, H., Pahl, M.V., Elahimehr, R. and Vaziri, N.D. (2009) Impaired antioxidant activity of high-density lipo- protein in chronic kidney disease. Translational Research, 153, 77-85. http://dx.doi.org/10.1016/j.trsl.2008.11.007 [65] Raveh, O., Pinchuk, I., Schnitzer, E., Fainaru, M., Schaffer, Z. and Lichtenberg, D. (2000) Kinetic analysis of cop- per-induced peroxidation of HDL, autoaccelerated and tocopherol-mediated peroxidation. Free Radical Biology and Medicine, 29, 131-146. http://dx.doi.org/10.1016/S0891-5849(00)00332-4 [66] Brizzi, P., Tonolo, G., Carusillo, F., Malaguarnera, M., Maioli, M. and Musumeci, S. (2003) Plasma lipid compo- sition and LDL oxidation. Clinical Chemistry and Labo- ratory Medicine, 41, 56-60. http://dx.doi.org/10.1515/CCLM.2003.010 [67] Chait, A., Brazg, R.L., Tribble, D.L. and Krauss, R.M. (1993) Susceptibility of small, dense, low-density lipo- proteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern-B. American Journal of Medicine, 94, 350-356. http://dx.doi.org/10.1016/0002-9343(93)90144-E [68] Raveh, O., Pinchuk, I., Fainaru, M. and Lichtenberg, D. (2001) Kinetics of lipid peroxidation in mixtures of HDL and LDL, mutual effects. Free Radical Biology and Medi- cine, 31, 1486-1497. http://dx.doi.org/10.1016/S0891-5849(01)00730-4 [69] Ishigaki, Y., Katagiri, H., Gao, J., Yamada, T., Imai, J., Uno, K., et al. (2008) Impact of plasma oxidized low-density lipoprotein removal on atherosclerosis. Circulation, 118, 75-83. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.7451 74 [70] Schnitzer, E., Fainaru, M. and Lichtenberg, D. (1995) Oxidation of low-density-lipoprotein upon sequential ex- posure to copper ions. Free Radical Research, 23, 137- 149. http://dx.doi.org/10.3109/10715769509064028 [71] Stafforini, D.M., Zimmerman, G.A., Mcintyre, T.M. and Prescott, S.M. (1992) The Paf acetylhydrolase from hu- man plasma prevents oxidative modification of low-den- sity-lipoprotein. Clinical Research, 40, A253. [72] Schnitzer, E., Pinchuk, I., Fainaru, M., Lichtenberg, D. and Yedgar, S. (1998) LDL-associated phospholipase A does not protect LDL against lipid peroxidation in vitro. Free Radical Biology and Medicine, 24, 1294-1303. http://dx.doi.org/10.1016/S0891-5849(97)00454-1 [73] Teiber, J.F., Draganov, D.I. and La Du, B.N. (2004) Puri- fied human serum PON1 does not protect LDL against oxidation in the in vitro assays initiated with copper or AAPH. Journal of Lipid Research, 45, 2260-2268. http://dx.doi.org/10.1194/jlr.M400213-JLR200 [74] Fyrnys, B., Blencowe, C. and Deigner, H.P. (1995) Sus- ceptibility of phospholipids of oxidizing LDL to enzyma- tic-hydrolysis modulates uptake by P388D1 macrophage- like cells. FEBS Letters, 357, 7-12. http://dx.doi.org/10.1016/0014-5793(94)01312-O [75] Mackness, M.I., Arrol, S. and Durrington, P.N. (1991) Pa- raoxonase prevents accumulation of lipoperoxides in low- density-lipoprotein. FEBS Letters, 286, 152-154. http://dx.doi.org/10.1016/0014-5793(91)80962-3 [76] Mackness, M.I., Abbott, C., Arrol, S. and Durrington, P.N. (1993) The Role of high-density-lipoprotein and lipid-so- luble antioxidant vitamins in inhibiting low-density lipo- protein oxidation. Biochemical Journal, 294, 829-834. [77] Aviram, M., Hardak, E., Vaya, J., Mahmood, S., Milo, S., Hoffman, A., et al. (2000) Human serum paraoxonases (PON1) Q and R selectively decrease lipid peroxides in human coronary and carotid atherosclerotic lesions PON1 esterase and peroxidase-like activities. Circulation, 101, 2510-2517. http://dx.doi.org/10.1161/01.CIR.101.21.2510 [78] Mackness, M.I., Mackness, B., Durrington, P.N., Fogel- man, A.M., Berliner, J., Lusis, A.J., et al. (1998) Para- oxonase and coronary heart disease. Current Opinion in Lipidology, 9, 319-324. http://dx.doi.org/10.1097/00041433-199808000-00006 [79] Mackness M.I., Durrington P.N. and Mackness B. (2000) How high-density lipoprotein protects against the effects of lipid peroxidation. Current Opinion in Lipidology, 11, 383-388. http://dx.doi.org/10.1097/00041433-200008000-00007 [80] Rosenblat, M. and Aviram, M. (2009) Paraoxonases role in the prevention of cardiovascular diseases. Biofactors, 35, 98-104. http://dx.doi.org/10.1002/biof.16 [81] Ng, C.J., Wadleigh, D.J., Gangopadhyay, A., Hama, S., Grijalva, V.R., Navab, M., Fogelman, A.M. and Reddy, S.T. (2001) Paraoxonase-2 is a ubiquitously expressed protein with antioxidant properties and is capable of pre- venting cell-mediated oxidative modification of low den- sity lipoprotein. Journal of Biological Chemistry, 276, 44444-44449. http://dx.doi.org/10.1074/jbc.M105660200 [82] Bowry, V.W., Stanley, K.K. and Stocker, R. (1992) High- density-lipoprotein is the major carrier of lipid hydroper- oxides in human blood-plasma from fasting donors. Pro- ceedings of the National Academy of Sciences of the United States of America, 89, 10316-10320. http://dx.doi.org/10.1073/pnas.89.21.10316 [83] Buettner, G.R. (1993) The pecking order of free-radicals and antioxidants: Lipid-peroxidation, α-tocopherol, and ascorbate. Archives of Biochemistry and Biophysics, 300, 535-543. http://dx.doi.org/10.1006/abbi.1993.1074 [84] Shimonov, M., Pinchuk, I., Bor, A., Beigel, I., Fainaru, M., Rubin, M., et al. (1999) Susceptibility of serum lipids to copper-induced peroxidation correlates with the level of high density lipoprotein cholesterol. Lipids, 34, 255-259. http://dx.doi.org/10.1007/s11745-999-0361-0 [85] Tynkkynen, T., Mursu, J., Nurmi, T., Tuppurainen, K., La- atikainen, R. and Soininen, P. (2012) NMR protocol for determination of oxidation susceptibility of serum lipids and application of the protocol to a chocolate study. Me- tabolomics, 8, 386-398. Copyright © 2013 SciRes. OPEN ACCESS  D. Lichtenberg, I. Pinchuk / Advances in Bioscience and Biotechnology 4 (2013) 48-61 Copyright © 2013 SciRes. 61 OPEN ACCESS http://dx.doi.org/10.1007/s11306-011-0323-2 [86] Williams, K.J. and Fisher, E.A. (2005) Oxidation, lipopro- teins, and atherosclerosis: Which is wrong, the antioxi- dants or the theory? Current Opinion in Clinical Nutri- tion and Metabolic Care, 8, 139-146. http://dx.doi.org/10.1097/00075197-200503000-00006 [87] Wu, B.J., Kathir, K., Witting, P.K., Beck, K., Choy, K., Li, C., et al. (2006) Antioxidants protect from athero- sclerosis by a heme oxygenase-1 pathway that is inde- pendent of free radical scavenging. Journal of Experi- mental Medicine, 203, 1117-1127. http://dx.doi.org/10.1084/jem.20052321 [88] Niki, E. (2011) Do free radicals play causal role in athero- sclerosis? Low density lipoprotein oxidation and vitamin E revisited. Journal of Clinical Biochemistry and Nutri- tion, 48, 3-7. http://dx.doi.org/10.3164/jcbn.11-007FR [89] Podmore, I.D., Griffiths, H.R., Herbert, K.E., Mistry, N., Mistry, P. and Lunec, J. (1998) Vitamin C exhibits pro- oxidant properties. Nature, 392, 559. http://dx.doi.org/10.1038/33308 [90] Nyyssonen, K., Porkkala Sarataho, E., Kaikkonen, J. and Salonen, J.T. (1997) Ascorbate and urate are the strongest determinants of plasma antioxidative capacity and serum lipid resistance to oxidation in Finnish men. Atheroscle- rosis, 130, 223-233. http://dx.doi.org/10.1016/S0021-9150(96)06064-9 [91] Bowry, V.W. and Stocker, R. (1993) Tocopherol-medi- ated peroxidation. The prooxidant effect of vitamin E on the radical-initiated oxidation of human low-density-lipo- protein. Journal of the American Chemical Society, 115, 6029-6044. http://dx.doi.org/10.1021/ja00067a019 [92] Koshkaryev, A., Barshtein, G. and Yedgar, S. (2010) Vi- tamin E. induces phosphatidylserine externalization and red cell adhesion to endothelial cells. Cell Biochemistry and Biophysics, 56, 109-114. http://dx.doi.org/10.1007/s12013-009-9074-3 [93] Manea, A. (2012) Vascular biology of reactive oxygen species and NADPH oxidases: Role in atherogenesis. In: Parthasarathy, S., Ed., Atherogenesis, InTech, 425-446. http://dx.doi.org/10.5772/25368 [94] Lichtenberg, D. (2011) Who is likely to gain from high dose supplementation of vitamin E? Harefuah (heb), 150, 37-40. [95] Witztum, J.L. (1998) To E or not to E—How do we tell? Circulation, 98, 2785-2787. http://dx.doi.org/10.1161/01.CIR.98.25.2785 [96] Frei, B. (2003) To C or not to C, that is the question! Jour- nal of the American College of Cardiology, 42, 253-255. http://dx.doi.org/10.1016/S0735-1097(03)00574-6 [97] Boaz, M., Smetana, S., Weinstein, T., Matas, Z., Gafter, U., Iaina, A., et al. (2000) Secondary prevention with antioxi- dants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet, 356, 1213-1218. http://dx.doi.org/10.1016/S0140-6736(00)02783-5 [98] Blum, S., Vardi, M., Brown, J.B., Russell, A., Milman, U., Shapira, C., Levy, N.S., Miller-Lotan, R., Asleh, R. and Levy, A.P. (2010) Vitamin E reduces cardiovascular disease in individuals with diabetes mellitus and the hap- toglobin 2-2 genotype. Pharmacogenomics, 11, 675-684. http://dx.doi.org/10.2217/pgs.10.17 [99] Lloret, A., Badia, M.C., Mora, N.J., Pallardo, F.V., Alonso, M.D. and Vina, J. (2009) Vitamin E paradox in Alzhei- mer’s disease: It does not prevent loss of cognition and may even be detrimental. Journal of Alzheimer’s Disease, 17, 143-149. [100] Kanner, J., Gorelik, S., Roman, S. and Kohen, R. (2012) Protection by polyphenols of postprandial human plasma and low-density lipoprotein modification: The stomach as a bioreactor. Journal of Agricultural and Food Chemistry, 60, 8790-8796. http://dx.doi.org/10.1021/jf300193g ABBREVIA TIONS AND TERMS AAPH—2,2’-Azobis(2-amidinopropane) dihydrochlo- ride; a water-soluble generator of free radicals; AS—atherosclerosis; MDA—malondialdehyde; a relatively stable peroxida- tion product; PAF-AH—platelet aggregation factor acetyl hydrolase; PON—paraoxonase; HDL associated PLA—the enzyme that catalyzes the hydrolysis of peroxidation products; QALY—quality adjusted life years; OS—oxidative stress; an ill-defined term commonly used to describe intuitively the imbalance between pro- oxidative and antioxidative species; RO(N)S—reactive oxygen (and nitrogen) species; used when we do not know which reactive oxygen (or nitro- gen) species are involved; FR—free radicals; used when we relate to established free radical mechanism(s); oxLDL—oxidatively-modified LDL; used when we mean that the relevant active species is one or more LDL oxidation product(s). HIGHLIGHTS OS is tightly controlled. In CVD patients, it is only slightly higher than normal. The weak association of OS with CVD is insufficient to prove causal relationship. Oxidative modification of LDL is one of several processes that govern atherosclerosis. Antioxidants do not necessarily inhibit the multifac- torial processes resulting in AS. Our knowledge is insufficient to predict who is likely to gain from antioxidants.
|