H. M. DIAB ET AL.
518
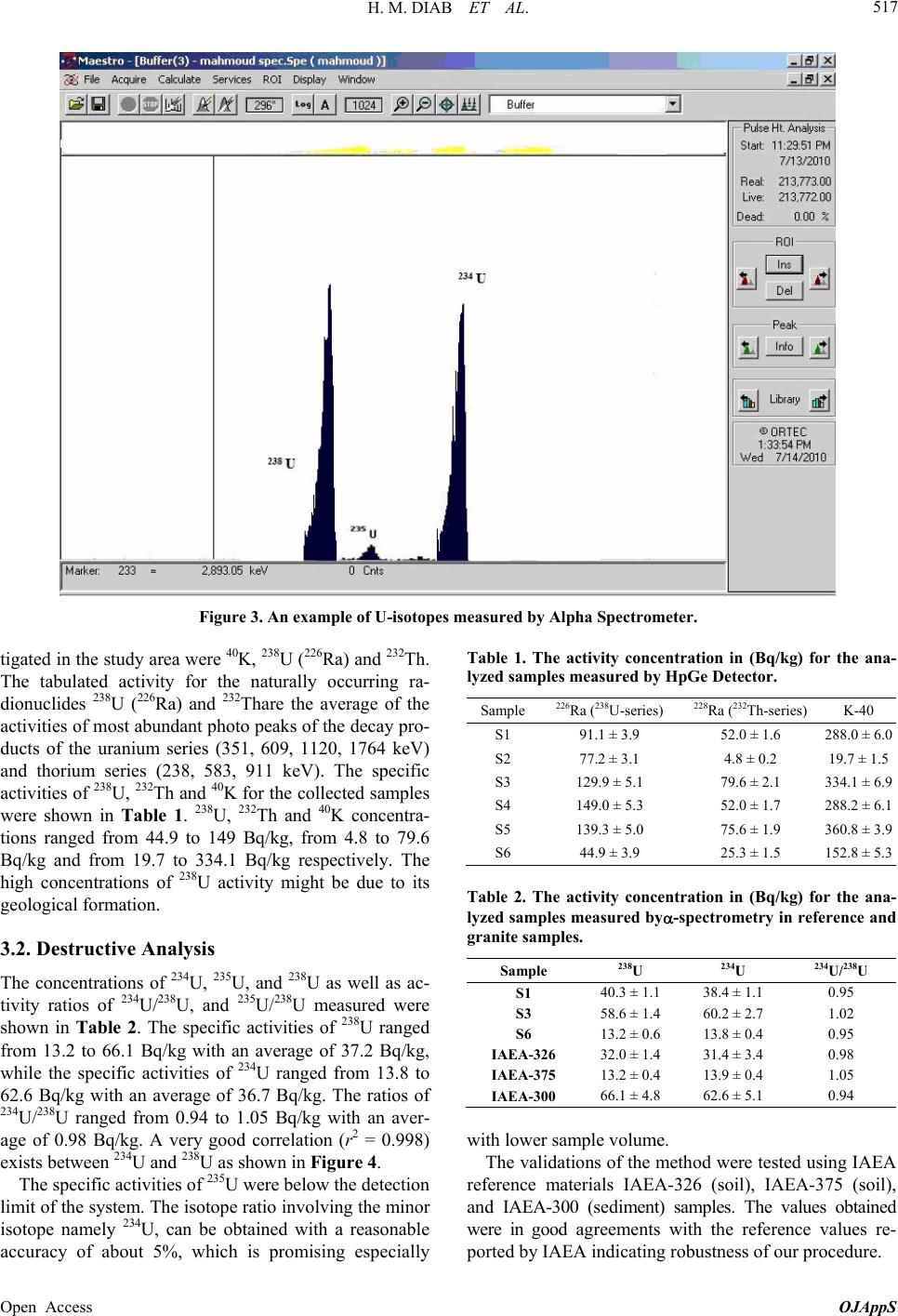
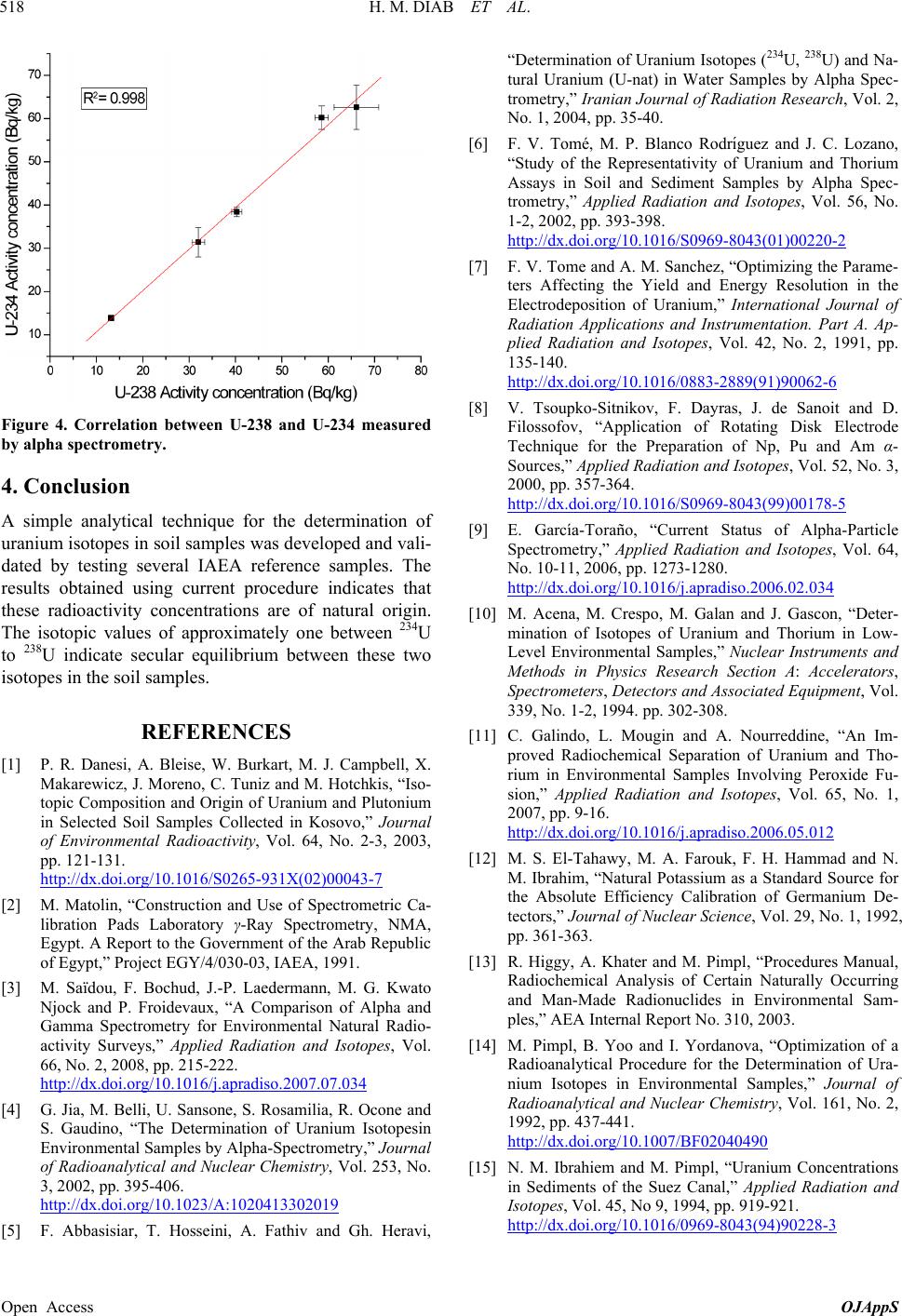
Figure 4. Correlation between U-238 and U-234 measured
by alpha spectrometry.
al technique for the determination of
soil samples was developed and
NCES
[1] P. R. Danesi, . J. Campb
Makarewicz, J.M. Hotchkis, “Iso-
4. Conclusion
A simple analytic
uranium isotopes invali-
dated by testing several IAEA reference samples. The
results obtained using current procedure indicates that
these radioactivity concentrations are of natural origin.
The isotopic values of approximately one between 234U
to 238U indicate secular equilibrium between these two
isotopes in the soil samples.
REFERE
A. Bleise, W. Burkart, M
Moreno, C. Tuniz and
ell, X.
topic Composition and Origin of Uranium and Plutonium
in Selected Soil Samples Collected in Kosovo,” Journal
of Environmental Radioactivity, Vol. 64, No. 2-3, 2003,
pp. 121-131.
http://dx.doi.org/10.1016/S0265-931X(02)00043-7
[2] M. Matolin, “
libration Pads Laboratory γ-Ray Spectrometry, N
Construction and Use of Spectrometric Ca-
MA,
lpha and
Egypt. A Report to the Government of the Arab Republic
of Egypt,” Project EGY/4/030-03, IAEA, 1991.
[3] M. Saïdou, F. Bochud, J.-P. Laedermann, M. G. Kwato
Njock and P. Froidevaux, “A Comparison of A
Gamma Spectrometry for Environmental Natural Radio-
activity Surveys,” Applied Radiation and Isotopes, Vol.
66, No. 2, 2008, pp. 215-222.
http://dx.doi.org/10.1016/j.apradiso.2007.07.034
[4] G. Jia, M. Belli, U. Sansone, S
S. Gaudino, “The Determination of Uranium Is
. Rosamilia, R. Ocone and
otopesin
Environmental Samples by Alpha-Spectrometry,” Journal
of Radioanalytical and Nuclear Chemistry, Vol. 253, No.
3, 2002, pp. 395-406.
http://dx.doi.org/10.1023/A:1020413302019
[5] F. Abbasisiar, T. Hosseini, A. Fathiv and Gh. Heravi,
co Rodrı́guez and J. C. Lozano,
/S0969-8043(01)00220-2
“Determination of Uranium Isotopes (234U, 238U) and Na-
tural Uranium (U-nat) in Water Samples by Alpha Spec-
trometry,” Iranian Journal of Radiation Research, Vol. 2,
No. 1, 2004, pp. 35-40.
[6] F. V. Tomé, M. P. Blan
“Study of the Representativity of Uranium and Thorium
Assays in Soil and Sediment Samples by Alpha Spec-
trometry,” Applied Radiation and Isotopes, Vol. 56, No.
1-2, 2002, pp. 393-398.
http://dx.doi.org/10.1016
rame-
.org/10.1016/0883-2889(91)90062-6
[7] F. V. Tome and A. M. Sanchez, “Optimizing the Pa
ters Affecting the Yield and Energy Resolution in the
Electrodeposition of Uranium,” International Journal of
Radiation Applications and Instrumentation. Part A. Ap-
plied Radiation and Isotopes, Vol. 42, No. 2, 1991, pp.
135-140.
http://dx.doi
and D.
016/S0969-8043(99)00178-5
[8] V. Tsoupko-Sitnikov, F. Dayras, J. de Sanoit
Filossofov, “Application of Rotating Disk Electrode
Technique for the Preparation of Np, Pu and Am α-
Sources,” Applied Radiation and Isotopes, Vol. 52, No. 3,
2000, pp. 357-364.
http://dx.doi.org/10.1
article
o.2006.02.034
[9] E. García-Toraño, “Current Status of Alpha-P
Spectrometry,” Applied Radiation and Isotopes, Vol. 64,
No. 10-11, 2006, pp. 1273-1280.
http://dx.doi.org/10.1016/j.apradis
Deter-
An Im-
/10.1016/j.apradiso.2006.05.012
[10] M. Acena, M. Crespo, M. Galan and J. Gascon, “
mination of Isotopes of Uranium and Thorium in Low-
Level Environmental Samples,” Nuclear Instruments and
Methods in Physics Research Section A: Accelerators,
Spectrometers, Detectors and Associated Equipment, Vol.
339, No. 1-2, 1994. pp. 302-308.
[11] C. Galindo, L. Mougin and A. Nourreddine, “
proved Radiochemical Separation of Uranium and Tho-
rium in Environmental Samples Involving Peroxide Fu-
sion,” Applied Radiation and Isotopes, Vol. 65, No. 1,
2007, pp. 9-16.
http://dx.doi.org
and N.
Khater and M. Pimpl, “Procedures Manual,
mization of a
007/BF02040490
[12] M. S. El-Tahawy, M. A. Farouk, F. H. Hammad
M. Ibrahim, “Natural Potassium as a Standard Source for
the Absolute Efficiency Calibration of Germanium De-
tectors,” Journal of Nuclear Science, Vol. 29, No. 1, 1992,
pp. 361-363.
[13] R. Higgy, A.
Radiochemical Analysis of Certain Naturally Occurring
and Man-Made Radionuclides in Environmental Sam-
ples,” AEA Internal Report No. 310, 2003.
[14] M. Pimpl, B. Yoo and I. Yordanova, “Opti
Radioanalytical Procedure for the Determination of Ura-
nium Isotopes in Environmental Samples,” Journal of
Radioanalytical and Nuclear Chemistry, Vol. 161, No. 2,
1992, pp. 437-441.
http://dx.doi.org/10.1
m Concentrations
8-3
[15] N. M. Ibrahiem and M. Pimpl, “Uraniu
in Sediments of the Suez Canal,” Applied Radiation and
Isotopes, Vol. 45, No 9, 1994, pp. 919-921.
http://dx.doi.org/10.1016/0969-8043(94)9022
Open Access OJAppS