 Open Journal of Genetics, 2013, 3, 285-293 OJGen http://dx.doi.org/10.4236/ojgen.2013.34032 Published Online December 2013 (http://www.scirp.org/journal/ojgen/) The role of XPC protein deficiency in tobacco smoke-induced DNA hypermethylation of tumor suppressor genes Gan Wang1*, Le Wang1, Vanitha Bhoopalan1, Yue Xi1, Deepak K. Bhalla2, David Wang3, Xiao xin S. Xu1 1Institute of Environmental Health Sciences, Wayne State University, Detroit, USA 2Department of Pharmaceutical Sciences, School of Pharmacy and Health Sciences, Wayne State University, Detroit, USA 3Lafeyatte High School, Rockwood, USA Email: *g.wang@wayne.edu Received 17 July 2013; revised 16 August 2013; accepted 26 September, 2013 Copyright © 2013 Gan Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT DNA hypermethylation of tumor suppressor genes has been frequently observed in cancer patients, and therefore, may provide a valuable biomarker for can- cer prevention and treatment. DNA hypermethyla- tion may also provide an important mechanism in cancer progression. Lung cancer is strongly associ- ated with exposure to environmental carcinogens, es- pecially tobacco smoke. DNA damage generated by tobacco smoke is believed to play an important role in lung cancer development. XPC is a DNA damage re- cognition protein required for DNA repair and other DNA damage responses and attenuated XPC protein levels have been detected in many lung cancer pa- tients. We studied the role of XPC protein deficiency in tobacco smoke-caused DNA hypermethylation of important tumor suppressor genes. Using both nor- mal human fibroblasts (NF) and XPC-deficient hu man fibroblasts (XPC), our DNA methylation studies demonstrated that the XPC deficiency caused ele- vated levels of DNA hypermethylation in both Brca1 and Mlh1 tumor suppressor genes following exposure to tobacco smoke condensate (TSC). The results of our ChIP studies revealed that the XPC deficiency led to an increased binding of DNA methyltransferase 3A (DNMT3A) at the promoter region CpG island-con- taining sequences of these genes under the TSC treatment; however, this increase was partially di- minished with prior treatment with caffeine. The re- sults of our immuno-precipitation (IP) studies de- monstrated a protein-protein interaction of the ATR with DNMT3A. Our western blots revealed that the XPC deficiency caused an increase in TSC-induced ATR phosphorylation at serine 428, an indicator of ATR activation. All these results suggest that XPC deficiency causes an accelerated DNA hypermethyla- tion in important tumor suppressor genes under to- bacco smoke exposure and activation of the ATR sig- naling pathway is involved in this DNA hypermethy- lation process. Keywords: DNA Hypermethylation; Tumor Suppressors; XPC; Tobacco Smoke; DNA Damage; DNA Repair Deficiency; ATR; DNMT3A 1. INTRODUCTION DNA hypermethylations of tumor suppressors and other cancer-related genes are frequently observed in cancer patients [1-5], and therefore, may provide a valuable bio- marker for cancer diagnosis and treatment. In addition, DNA hypermethylation may also provide an important mechanism in cancer progression. Understanding the mechanism of DNA hypermethylation, therefore, would have important implications in cancer prevention and treatment. Lung cancer is one of the most common malignancies and the leading cause of cancer-related fatalities [6]. Lung cancer is strongly associated with exposure to en- vironmental factors, especially tobacco smoking [6]. The molecular mechanism through which tobacco smoking causes lung cancer to develop is not fully understood. It is believed that DNA damage generated by carcinogens in the tobacco smoke and/or their metabolites plays an important role in lung cancer development. In addition, hypermethylation of important tumor suppressor genes *Corresponding author. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 286 also is frequently observed in lung cancer patients [7,8], suggesting a possible role of DNA hypermethylation in lung cancer progression. Nucleotide excision repair (NER) is the major DNA repair pathway to repair DNA damage generated by en- vironmental carcinogens, including tobacco smoke [9,10]. The NER pathway can be further categorized into the transcription-coupled NER (TCR) and global-genome NER (GGR) sub-pathways [9,11]. In TCR the arrested transcription events initiate the NER process [12,13], whereas in GGR the DNA damage recognition of the XPC-HR23B protein complex initiates the NER process [14,15]. The DNA damage recognition signal further recruits other NER components, including XPA, RPA, TFIIH, XPG, XPF-ERCC1, to the damage site [9]. The dual-incision made by XPG and XPF-ERCC1 generates a 30 - 33 nt single-stranded DNA gap [9,14]. The DNA polymerases (δ or ε) fill the gap and the DNA ligase seals the gap to complete the NER process [9]. In addition to its function in DNA repair, the XPC protein also plays an important role in other DNA dam- age responses, including cell cycle arrest and apoptosis [16-18]. The presence of a functional XPC protein, therefore, is essential in determining the fate of the da- maged cells, either restoring the disrupted cellular func- tions through DNA repair or eliminating the severely da- maged cells through apoptosis. XPC protein attenuation and deficiency have been as- sociated with many types of cancer. It is well known for the high risk in developing many types of cancer, espe- cially skin cancer, in the XPC patients [10]; recent clini- cal studies reveal greatly attenuated levels of XPC pro- tein in a majority of lung and bladder cancer patients [19-21]. In addition, animal studies also reveal high pre- disposition towards skin and lung cancer when the XPC-knockout mice (XPC−/−) are exposed to chemical carcinogen acetyl-amino-fluorene [22,23]. All these re- sults suggest that XPC protein attenuation and deficiency play an important role in cancer development, especially for those cancers associated with environmental factors such as lung and bladder cancer. The mechanism through which XPC protein attenua- tion and deficiency cause cancer to develop is not known. Considering that both XPC protein attenuation and DNA hypermethylation of important tumor suppressor genes are frequently observed in cancer patients [19-21], it is possible that hypermethylation of tumor suppressor genes may be an important mechanism for XPC protein attenu- ation and deficiency in causing cancer to develop. How- ever, no studies have been done in determining the role of XPC protein attenuation and deficiency in hyperme- thylation of tumor suppressor genes and the involving mechanism. In this work, we determined the role of XPC protein deficiency in tobacco smoke-caused DNA hypermethy- lation of both Brca1 and Mlh1 tumor suppressor genes and further defined the underlying mechanism. Using fibroblasts obtained from normal individuals (NF) and XPC patients (XPC) and condensate prepared from to- bacco smoke (TSC), the results of our DNA methylation studies demonstrated that the XPC deficiency resulted in a greater increase for TSC-induced DNA hypermethyla- tion at the promote region CpG island-containing se- quences of the Brca1, Mlh1, and Xpc genes in the XPC cells than that of the NF cells. The results of our chroma- tin immunoprecipitation (ChIP) studies revealed that the XPC deficiency caused a more increase for TSC-induced DNMT3A binding at the promoter region CpG island- containing sequences of both Mlh1 and Xpc genes in the TSC-treated XPC cells than that of the NF cells; however, this increase was partially diminished when the XPC cells were treated with caffeine prior to the TSC treat- ment. The results obtained from our immune-precipita- tion (IP) studies demonstrated that the ATR protein in- teracted with the DNMT3A protein. In addition, our western blot results also indicated that the XPC defi- ciency caused a more increase for TSC-induced ATR protein phosphorylation at serine 428 in the XPC cells than in the NF cells. All these results suggest that XPC protein attenuation and deficiency results in an increase of tobacco smoke-caused DNA hypermethylation of the Brca1 and Mlh1 genes, and activation of the ATR pro- tein is involved in the TSC-caused DNA hypermethyla- tion process. 2. MATERIALS AND METHODS 2.1. Cell Lines and Oligonucleotides The GM00043, GM03021, and GM16684 human fibro- blasts were purchased from the Coriell Institute for Me- dical Research (Camden, NJ) and used in our previous studies [16-18]. The GM00043 are primary human fibro- blasts derived from a normal individual (NF) and are proficient in nucleotide excision repair (NER) pathway. The GM03021 are primary human fibroblasts derived from a group G of xeroderma pigmentosum (XPG) pa- tient and defective in XPG protein. The GM16684 are primary human fibroblasts derived from a XPC patient and deficient in XPC protein. Both NF and XPG cells were maintained in minimal essential medium (MEM) supplemented with 15% FBS, 2x essential amino acids (EAA), 2x nonessential amino acid (NEAA) and 2x vi- tamins (Vt) at 37˚C with 5% CO2. The XPC cells were maintained in MEM supplemented with 20% FBS, 2xEAA, 2xNEAA, and 2xVt at 37˚C with 5% CO2. The primers used in this study were listed in Ta b le 1 and were synthesized by Midland Certified Reagent Company, Inc. (Midland, TX). The BRCA1 primers were Copyright © 2013 SciRes. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 287 Table 1. Primers used in the DNA methylation studies. BRCA1 primer 1: 5’-CCTTGGTTTCCGTGGCAACG-3’ BRCA1 primer 2: 5’-GAGCAGAGGGTGAAGGCCTC-3’ MLH1 primer 1: 5’-CAGGAGTGAAGGAGGCCACG-3’ MLH1 primer 2: 5’-CCTGTGCCTGGTCTGTCGCCG-3’ XPC primer 1: 5’-CGTGGCCAAGCGCACCGCCTC-3’ XPC primer 2: 5’-CGAGCCATGTTGCTTGTCTGG-3’ designed to amplify a 160 bp CpG island-containing DNA sequence between the −1330 to −1170 region of the Brca1 gene. The MLH1 primers were designed to amplify a 170 bp CpG island-containing DNA sequence between the −750 to −580 of the Mlh1 gene. The XPC primers were designed to amplify a 120 bp CpG island- containing DNA sequence between the −100 to +20 of the Xpc gene. 2.2. Preparation of Tobacco Smoke Condensate (TSC) The 2R4 Reference cigarette was purchased from the University of Kentucky College of Agriculture Refer- ence Cigarette Program (Lexington, KY). The cigarettes were smoked on a standard smoking machine, taking 35-ml puffs of 2-sec duration once per min. The smoke was condensed in a trap cooled with liquid air. The col- lected material was dried onto filter paper, weighed, and dissolved in dimethylsulfoxide (DMSO) at 25 mg/ml. These values represent the equivalent of 10 cigarettes’ worth of condensate per ml. 2.3. TSC Treatment and DNA/RNA Preparation Cells were seeded in T-75 flask and grown to 40% con- fluence. The cells were treated with TSC at a concentra- tion of 25 μg/ml by adding the TSC directly into the cul- ture medium. For the Chromatin immuno-precipitation (ChIP) studies, cells were cultured in the TSC-containing medium for 48 hours. For the DNA methylation study, cells were maintained in the TSC-containing medium for up to two months with passage once every week. For the western blot study, cells were treated with TSC for 24 hours. Total RNA was isolated from cells using an RNeasy Mini kit (Qiagen, Valencia, CA). Genomic DNA was isolated from cells using a DNeasy Blood & Tissue Kit (Qiagen). 2.4. Methylated DNA Enrichment and Quantitation The methylated DNA enrichment was done using a Me- thylminer Methylated DNA Enrichment system (Invi- trogen, Carlsbad, CA). Genomic DNA was first digested with both BbvI and DpnI restriction enzymes to release the 373 bp, 390 bp, and 413 bp CpG island-containing DNA fragments from the promoter regions of the Brca1, Mlh1, and Xpc genes respectively. The methylated DNA fragments were then enriched from each DNA sample (1 μg total DNA) using a protocol recommended by the manufacturer and dissolved into 50 μl·dH 2O. The methylation-enriched DNA samples were quanti- fied using a quantitative PCR (qPCR) protocol to deter- mine the DNA levels of the Brca1, Mlh1, and Xpc pro- moter region CpG island DNA sequences in each DNA sample. The qPCR was done at the following conditions: 2 μl DNA sample was mixed with 10 μl Power Sybr green master mix (Applied Biosystems) in a total volume of 20 μl containing 2 μM of each primer for a desired target genes. The reaction was set as triplicate for each DNA sample in a 96-well plate. The plate was placed in a StepOnePlus Real Time PCR system (Applied Biosys- tems) with a setting of 95˚C for 2 minutes and then 50 cycles of 95˚C for 30 seconds, 56˚C for 30 seconds, and 72˚C for 30 seconds. The level of the β-a ctin gene DNA was also determined for each DNA sample and used as an internal control for the qPCR assay. The level of the methylation-enriched target gene DNA in the untreated cells was counted as 100% and the level of the same tar- get gene DNA in the treated cells was calculated as a fold change to that of the untreated cells for both NF and XPC cells. The qPCR data analysis was performed using a StepOne v2.1 software (Applied Biosystems). 2.5. Real Time PCR Assay A reverse transcription-based quantitative PCR (real time PCR) assay was performed using a Sybr Green-based RNA quantification method (Applied Biosystems) [16]. The real time PCR was carried out in a StepOnePlus Real Time PCR system and analyzed by a StepOne v2.1 software to determine the relative levels of mRNA for the selected genes in each RNA sample. The level of the selected target gene mRNA in the untreated NF cells was counted as 100% and the levels of the same target mRNA in the other RNA samples were calculated as fold changes in comparison to that of the untreated NF cells. 2.6. ChIP Assay The ChIP assay was performed using a previously de- scribed protocol [17]. The DNMT3A antibody (64B814) (Santa Cruz Biotechnologies, Inc., Santa Cruz, CA) was used in the ChIP study. Half of the beads obtained from the ChIP assay were analyzed by western blots to deter- mine the level of DNMT3A protein pulling down by the antibody and the remaindering beads was processed to recover the DNA that was co-precipitated with the DNMT3A protein and analyzed by a qPCR protocol to Copyright © 2013 SciRes. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 Copyright © 2013 SciRes. 288 OPEN ACCESS determine the DNA levels of desired target genes in each ChIP reaction. 2.7. Western Blot Hybridization Western blots were performed using a previously de- scribed protocol [16] and 50 μg total protein was used in the western blots for each cell lysate. Antibodies against β-actin (C-2), DNMT1 (N-16), DNMT3A (64B814 and H-295), and DNMT3B (2280C3a), were purchased from the Santa Cruz Biotechnologies, Inc. and used in this study. 2.8. Statistical Analysis Results were expressed as the mean ±S.D. Statistically significant differences were determined using a student t-test with 95% confidence interval (CI). The data was obtained from at least three independent experiments. 3. RESULTS 3.1. The XPC Deficiency Caused an Increase for TSC-Induced DNA Hypermethylation at the Promoter Region CpG Island-Containing Sequences of the Brca1, Mlh1, and Xpc Genes in XPC Cells In order to determine the role of XPC protein attenuation and deficiency in DNA hypermethylation of important tumor suppressor genes observed in many lung cancer patients, we first studied the effects of XPC protein defi- ciency on tobacco smoke-induced DNA hypermethyla- tion in Brca1, Mlh1, and Xpc genes, which were fre- quently observed in lung cancer patients [21,24-28]. Both NF (GM00043) and XPC (GM16684) cells were treated with TSC (25 μg/ml) for two months to allow for DNA hypermethylation accumulation in genomic DNA of the treated cells. As a control, the XPG cells, which are deficient in the XPG protein required for a late step of the NER process, were also treated with TSC in a par- allel experiment; however, the XPG cells did not survive the two-month TSC treatment due to their high sensitiv- ity to DNA-damaging treatment. Therefore, genomic DNA was prepared only from untreated and TSC-treated NF and XPC cells for our DNA methylation study. The genomic DNA was then digested with both BbvI and Dpn1 restriction enzymes to release the 372 bp, 390 bp, and 413 bp DNA fragments containing the promoter re- gion CpG island sequences of the Brca1, Mlh1, and Xpc genes respectively. The methylated DNA fragments were enriched from the digested genomic DNA and quantified by a quantitative PCR (qPCR) protocol to determine the level of the promoter region CpG island-containing se- quences of the Brca1, Mlh1, and Xpc genes in the me- thylation-enriched DNA samples (Table 2). The results of our qPCR assay revealed that the TSC treatment caused some decrease in the level of DNA methylation for Brca1, Mlh1, and Xpc genes in NF cells (Table 2). In XPC cells, however, the TSC treatment caused 37.90 ± 12.80, 7.85 ± 2.62, and 15.40 ± 4.24 fold increase in DNA methylation for the Brc a1, Mlh1, and Xpc genes respectively (Table 2). These results suggest that the XPC deficiency causes an increase for TSC-induced DNA hypermethylation at the promoter region CpG is- land-containing sequences of the Brca1, Mlh1, and Xpc genes. 3.2. The XPC Deficiency Resulted in an Increased Binding of DNMT3A at the Promoter Region CpG Island-Containing Sequences of the Mlh1 and Xpc Genes after the TSC Treatment The results of our DNA methylation studies suggested an important role of the XPC protein deficiency for TSC-induced DNA hypermethylation of the Brca1, Mlh1, and Xpc genes. To define a mechanism by which the XPC deficiency led to more elevated DNA hypermethy- lation of these genes in the TSC-treated XPC cells, we determined the protein levels of DNMT1, DNMT3A, and Table 2. The promoter region CpG island-containing sequence DNA methylation levels of the Brca1, Mlh1, and Xpc genes in both untreated and TSC-treated NF and XPC cellsa. Brca1 Mlh1 Xpc NF (GM00043) 1.00 1.00 1.00 0.36 ± 0.06 0.76 ± 0.22 0.29 ± 0.13 NF (GM00043) + TSC p = 0.003 p = 0.204 p = 0.01 XPC (GM16684) 1.00 1.00 1.00 37.90 ± 12.80 7.85 ± 2.62 15.40 ± 4.24 XPC (GM16684) + TSC P = 0.038 P = 0.045 P = 0.028 aThe level of methylated DNA in the untreated NF and XPC cells was counted as 100% and the level of the methylated DNA in the TSC-treated NF and XPC cells was calculated at fold change to that of the untreated cells. The P value was calculated between the TSC-treated cells vs untreated cells for both NF and XPC cells in individual genes with 95% confidence interval (CI) using student’s t-test. 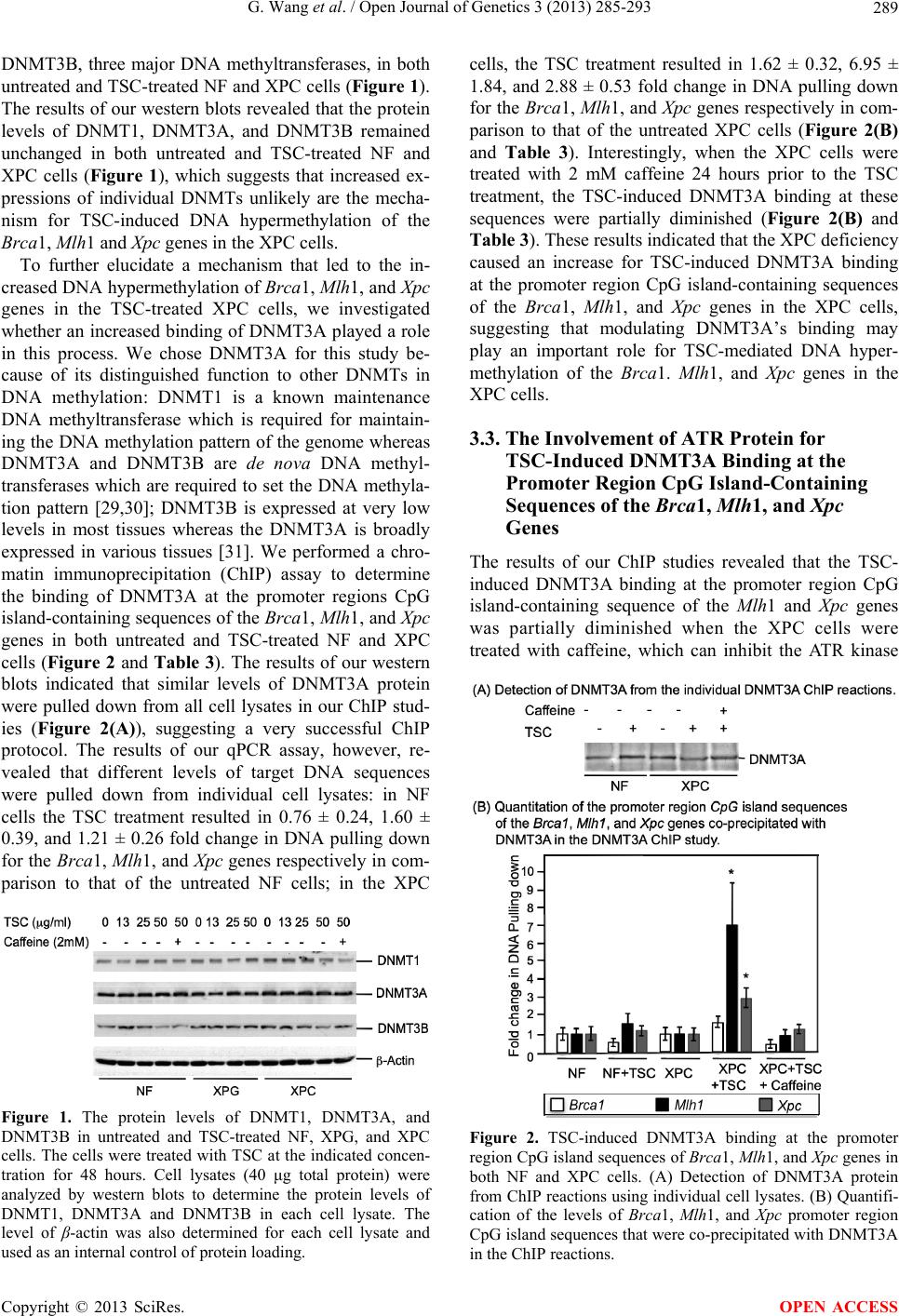 G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 289 DNMT3B, three major DNA methyltransferases, in both untreated and TSC-treated NF and XPC cells (Figure 1). The results of our western blots revealed that the protein levels of DNMT1, DNMT3A, and DNMT3B remained unchanged in both untreated and TSC-treated NF and XPC cells (Figure 1), which suggests that increased ex- pressions of individual DNMTs unlikely are the mecha- nism for TSC-induced DNA hypermethylation of the Brca1, Mlh1 and Xpc genes in the XPC cells. To further elucidate a mechanism that led to the in- creased DNA hypermethylation of Brca1, Mlh1, and Xp c genes in the TSC-treated XPC cells, we investigated whether an increased binding of DNMT3A played a role in this process. We chose DNMT3A for this study be- cause of its distinguished function to other DNMTs in DNA methylation: DNMT1 is a known maintenance DNA methyltransferase which is required for maintain- ing the DNA methylation pattern of the genome whereas DNMT3A and DNMT3B are de nova DNA methyl- transferases which are required to set the DNA methyla- tion pattern [29,30]; DNMT3B is expressed at very low levels in most tissues whereas the DNMT3A is broadly expressed in various tissues [31]. We performed a chro- matin immunoprecipitation (ChIP) assay to determine the binding of DNMT3A at the promoter regions CpG island-containing sequences of the Brca1, Mlh1, and Xpc genes in both untreated and TSC-treated NF and XPC cells (Figure 2 and Table 3). The results of our western blots indicated that similar levels of DNMT3A protein were pulled down from all cell lysates in our ChIP stud- ies (Figure 2(A)), suggesting a very successful ChIP protocol. The results of our qPCR assay, however, re- vealed that different levels of target DNA sequences were pulled down from individual cell lysates: in NF cells the TSC treatment resulted in 0.76 ± 0.24, 1.60 ± 0.39, and 1.21 ± 0.26 fold change in DNA pulling down for the Brca1, Mlh1, and Xpc genes respectively in com- parison to that of the untreated NF cells; in the XPC Figure 1. The protein levels of DNMT1, DNMT3A, and DNMT3B in untreated and TSC-treated NF, XPG, and XPC cells. The cells were treated with TSC at the indicated concen- tration for 48 hours. Cell lysates (40 μg total protein) were analyzed by western blots to determine the protein levels of DNMT1, DNMT3A and DNMT3B in each cell lysate. The level of β-actin was also determined for each cell lysate and used as an internal control of protein loading. cells, the TSC treatment resulted in 1.62 ± 0.32, 6.95 ± 1.84, and 2.88 ± 0.53 fold change in DNA pulling down for the Brca1, Mlh1, and Xpc genes respectively in com- parison to that of the untreated XPC cells (Figure 2(B) and Table 3). Interestingly, when the XPC cells were treated with 2 mM caffeine 24 hours prior to the TSC treatment, the TSC-induced DNMT3A binding at these sequences were partially diminished (Figure 2(B) and Table 3). These results indicated that the XPC deficiency caused an increase for TSC-induced DNMT3A binding at the promoter region CpG island-containing sequences of the Brca1, Mlh1, and Xpc genes in the XPC cells, suggesting that modulating DNMT3A’s binding may play an important role for TSC-mediated DNA hyper- methylation of the Brca1. Mlh1, and Xpc genes in the XPC cells. 3.3. The Involvement of ATR Protein for TSC-Induced DNMT3A Binding at the Promoter Region CpG Island-Containing Sequences of the Brca1, Mlh1, and Xpc Genes The results of our ChIP studies revealed that the TSC- induced DNMT3A binding at the promoter region CpG island-containing sequence of the Mlh1 and Xpc genes was partially diminished when the XPC cells were treated with caffeine, which can inhibit the ATR kinase Figure 2. TSC-induced DNMT3A binding at the promoter region CpG island sequences of Brca1, Ml h1, and Xpc genes in both NF and XPC cells. (A) Detection of DNMT3A protein from ChIP reactions using individual cell lysates. (B) Quantifi- cation of the levels of Brca1, Mlh1, and Xpc promoter region CpG island sequences that were co-precipitated with DNMT3A in the ChIP reactions. Copyright © 2013 SciRes. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 290 Table 3. Quantitative analysis of the TSC-induced DNMT3A binding at the promoter region CpG island-containing sequences of the Brca1, Mlh1, and Xpc genes in both NF and XPC cellsb. Brca1 Mlh1 Xpc NF (GM00043) 1.00 ± 0.11 1.00 ± 0.16 1.00 ± 0.27 NF + TSC 0.76 ± 0.24 1.60 ± 0.39 1.21 ± 0.26 XPC (GM16684) 1.00 ± 0.32 1.00 ± 0.34 1.00 ± 0.36 XPC + TSC 1.62 ± 0.32 6.95 ± 1.84 2.88 ± 0.53 XPC+ Caffeine + TSC 0.45 ± 0.18 0.93 ± 0.32 1.34 ± 0.25 bThe DNA levels of the Brca1, Mlh1, and Xpc gene sequences obtained from the untreated NF and XPC cells of the ChIP study were accounted as 100% and the DNA levels of the Brca1, Mlh1, and Xpc gene sequences obtained from the treated cells of the ChIP study were calculated as fold changes to that of the un- treated cells for both NF and XPC cells respectively. activity, prior to the TSC treatment, suggesting a possible role of ATR protein in the process. To further define the involvement of ATR protein in this process, we deter- mined the protein-protein interaction between ATR and DNMT3A proteins in both untreated and TSC-treated NF and XPC cells using an immuno-precipitation (IP) pro- tocol (Figure 3(A)). When ATR protein was pulled down from individual cell lysates in our IP study, DNMT3A protein was also co-precipitated with the ATR protein from the cell lysates prepared from both untreated and TSC-treated NF and XPC cells (Figure 3(A)), which sug- gests an interaction between the ATR and the DNMT3A protein. Interestingly, majority of the DNMT3A protein co-precipitated with the ATR protein in the IP study was from one of two DNMT3A protein bands as shown in the western blots (the lower band in Figure 3( A )). To further define the role of ATR protein in TSC-in- duced DNA hypermethylation of important target genes, we also determined the phosphorylation status of the ATR protein at serine 428, an indicator of ATR acti- vation [32-36], in both untreated and TSC-treated NF and XPC cells (Figure 3(B)). Our western blot results indicated that the TSC treatment caused a greater in- crease in the level of the ATR phosphorylation at serine 428 in the XPC cells than that of the NF cells (Figure 3(B) top panel lane 4 vs lane 2). This result suggests that activation of the ATR protein plays an important role for TSC-induced DNA hypermethylation of important target genes. 4. DISCUSSION In this work we determined the role of XPC protein defi- ciency in tobacco smoke-caused DNA hypermethylation of the Brca1, Mlh 1, and Xp c genes and further defined the involving mechanism. The results of our DNA me- thylation studies demonstrated that the XPC deficiency resulted in a greater increase for TSC-caused DNA me- thylation at the promoter region CpG island-containing sequences of the Brca1, Mlh1, and Xpc genes in XPC cells. The results of our ChIP studies revealed that the XPC deficiency caused a significant more increase for TSC-induced DNMT3A binding at the promoter region CpG island-containing sequences of the Brca1, Mlh1 and Xpc genes in XPC cells; however, this increase in DNMT3A binding was partially diminished when XPC cells were treated with caffeine prior to the TSC treat- ment. The results of our IP study further demonstrated that the ATR protein interacts with the DNMT3A. In addition, the results of our western blot studies also in- dicated that the XPC deficiency resulted in an increase for TSC-induced ATR phosphorylation at serine 428. Given the important roles of XPC and ATR proteins in DNA damage response (DDR) and the fact that attenu- ated levels of XPC protein are frequently observed in lung cancer patients, these results suggest that the XPC protein attenuation and deficiency play an important role for tobacco smoke-caused DNA hypermethylation of im- portant tumor suppressor genes. The results of our DNA methylation studies revealed that the XPC deficiency caused an increase for tobacco smoke-induced DNA hypermethylation of the Brca1, Mlh1, and Xpc genes in the XPC cells. Considering the important functions of the BRCA1 and MLH1 proteins in DNA repair and other DNA damage response, it is pos- sible that silence of these genes is necessary for cells surviving the tobacco smoke-caused DNA damage; however, silence of these genes clearly could lead to some foreseeable consequence, such as uncontrolled cell proliferation and transformation from normal cells to tumor cells. Therefore, XPC protein attenuation and de- ficiency may lead to high risk of lung cancer develop- ment under tobacco smoking through enhancing tobacco smoke-induced DNA hypermethylation of important tumor suppressor genes and transformation from normal lung epithelial cells to lung tumor cells. In addition, al- though our studies only determined the DNA methyla- tion status of the Brca1, Mlh1, and Xpc genes, it was likely that the DNA methylation status of many other tumor suppressors and cancer-related gene were also affected by XPC deficiency under the TSC exposure. Further identification of those genes, therefore, would provide a better understanding of the molecular mecha- Copyright © 2013 SciRes. OPEN ACCESS 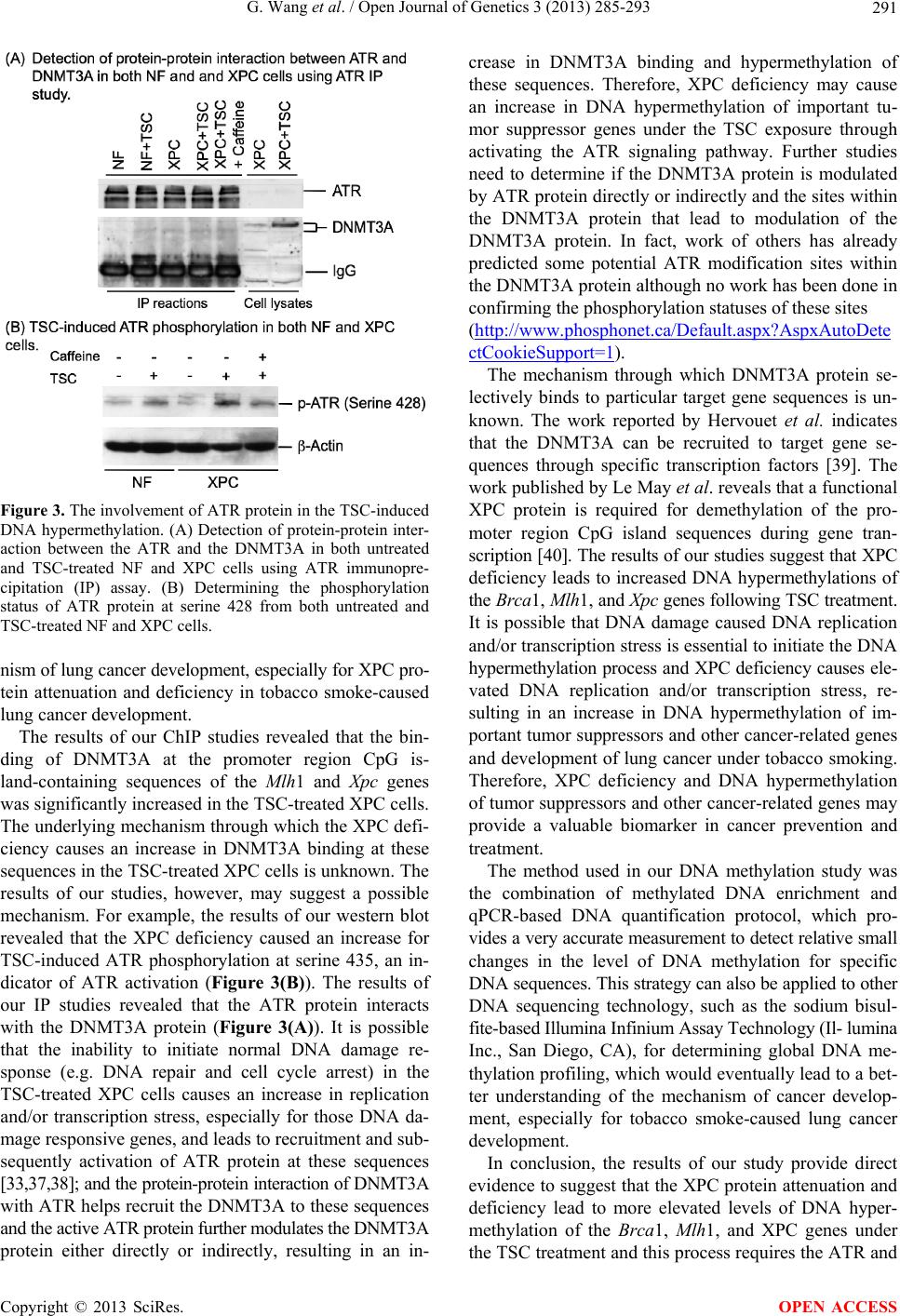 G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 291 Figure 3. The involvement of ATR protein in the TSC-induced DNA hypermethylation. (A) Detection of protein-protein inter- action between the ATR and the DNMT3A in both untreated and TSC-treated NF and XPC cells using ATR immunopre- cipitation (IP) assay. (B) Determining the phosphorylation status of ATR protein at serine 428 from both untreated and TSC-treated NF and XPC cells. nism of lung cancer development, especially for XPC pro- tein attenuation and deficiency in tobacco smoke-caused lung cancer development. The results of our ChIP studies revealed that the bin- ding of DNMT3A at the promoter region CpG is- land-containing sequences of the Mlh1 and Xpc genes was significantly increased in the TSC-treated XPC cells. The underlying mechanism through which the XPC defi- ciency causes an increase in DNMT3A binding at these sequences in the TSC-treated XPC cells is unknown. The results of our studies, however, may suggest a possible mechanism. For example, the results of our western blot revealed that the XPC deficiency caused an increase for TSC-induced ATR phosphorylation at serine 435, an in- dicator of ATR activation (Figure 3(B)). The results of our IP studies revealed that the ATR protein interacts with the DNMT3A protein (Figure 3(A)). It is possible that the inability to initiate normal DNA damage re- sponse (e.g. DNA repair and cell cycle arrest) in the TSC-treated XPC cells causes an increase in replication and/or transcription stress, especially for those DNA da- mage responsive genes, and leads to recruitment and sub- sequently activation of ATR protein at these sequences [33,37,38]; and the protein-protein interaction of DNMT3A with ATR helps recruit the DNMT3A to these sequences and the active ATR protein further modulates the DNMT3A protein either directly or indirectly, resulting in an in- crease in DNMT3A binding and hypermethylation of these sequences. Therefore, XPC deficiency may cause an increase in DNA hypermethylation of important tu- mor suppressor genes under the TSC exposure through activating the ATR signaling pathway. Further studies need to determine if the DNMT3A protein is modulated by ATR protein directly or indirectly and the sites within the DNMT3A protein that lead to modulation of the DNMT3A protein. In fact, work of others has already predicted some potential ATR modification sites within the DNMT3A protein although no work has been done in confirming the phosphorylation statuses of these sites (http://www.phospho net.ca/Default.aspx?Asp xAutoDete ctCookieSupport=1 ). The mechanism through which DNMT3A protein se- lectively binds to particular target gene sequences is un- known. The work reported by Hervouet et al. indicates that the DNMT3A can be recruited to target gene se- quences through specific transcription factors [39]. The work published by Le May et al. reveals that a functional XPC protein is required for demethylation of the pro- moter region CpG island sequences during gene tran- scription [40]. The results of our studies suggest that XPC deficiency leads to increased DNA hypermethylations of the Brca1, Mlh1, and Xpc genes following TSC treatment. It is possible that DNA damage caused DNA replication and/or transcription stress is essential to initiate the DNA hypermethylation process and XPC deficiency causes ele- vated DNA replication and/or transcription stress, re- sulting in an increase in DNA hypermethylation of im- portant tumor suppressors and other cancer-related genes and development of lung cancer under tobacco smoking. Therefore, XPC deficiency and DNA hypermethylation of tumor suppressors and other cancer-related genes may provide a valuable biomarker in cancer prevention and treatment. The method used in our DNA methylation study was the combination of methylated DNA enrichment and qPCR-based DNA quantification protocol, which pro- vides a very accurate measurement to detect relative small changes in the level of DNA methylation for specific DNA sequences. This strategy can also be applied to other DNA sequencing technology, such as the sodium bisul- fite-based Illumina Infinium Assay Technology (Il- lumina Inc., San Diego, CA), for determining global DNA me- thylation profiling, which would eventually lead to a bet- ter understanding of the mechanism of cancer develop- ment, especially for tobacco smoke-caused lung cancer development. In conclusion, the results of our study provide direct evidence to suggest that the XPC protein attenuation and deficiency lead to more elevated levels of DNA hyper- methylation of the Brca1, Mlh 1, and XPC genes under the TSC treatment and this process requires the ATR and Copyright © 2013 SciRes. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 292 DNMT3A proteins. Therefore, the knowledge obtained from this study provides some valuable insight into the mechanism of lung cancer development, especially the role of XPC protein attenuation and deficiency in tobacco smoke-caused lung cancer development. This knowledge also will have important implications in detection, pre- vention, and treatment for many types of cancer in the future. 5. ACKNOWLEDGEMENTS We thank Dr. Douglas Ruden for his encouraging discussion on the project. Performance of this work was facilitated by the Cell Culture Core, the Imaging and Flow Cytometry Core, and the Microarray and Bioinformatic Core of the Institute of Environmental Health Sciences at Wayne State University (WSU). REFERENCES [1] Kerr, K.M., Galler, J.S., Hagen, J.A., Laird, P.W. and Laird-Offringa, I.A. (2007) The role of DNA methylation in the development and progression of lung adenocarci- noma. Disease Markers, 23, 5-30. http://dx.doi.org/10.1155/2007/985474 [2] Anglim, P.P., Alonzo, T.A. and Laird-Offringa, I.A. (2008) DNA methylation-based biomarkers for early detection of non-small cell lung cancer: An update. Molecular Cancer, 7, 81. http://dx.doi.org/10.1186/1476-4598-7-81 [3] Risch, A. and Plass, C. (2008) Lung cancer epigenetics and genetics. International Journal of Cancer, 123, 1-7. http://dx.doi.org/10.1002/ijc.23605 [4] Pfeifer, G.P. and Rauch, T.A. (2009) DNA methylation patterns in lung carcinomas. Seminars in Cancer Biology, 19, 181-187. http://dx.doi.org/10.1016/j.semcancer.2009.02.008 [5] Van Den Broeck, A., Ozenne, P., Eymin, B. and Gazzeri, S. (2010) Lung cancer: A modified epigenome. Cell Ad- hesion & Migration, 4, 107-113. http://dx.doi.org/10.4161/cam.4.1.10885 [6] Holland-Frei (2006) Cancer medicine 7. American Asso- ciation for Cancer Research. [7] Akhavan-Niaki, H. and Samadani, A.A. (2013) DNA methylation and cancer development: Molecular mecha- nism. Cell Biochemistry and Biophysics. http://link.springer.com/article/10.1007%2Fs12013-013-9 555-2 [8] Das, P.M. and Singal, R. (2004) DNA methylation and cancer. Journal of Clinical Oncology, 22, 4632-4642. http://dx.doi.org/10.1200/JCO.2004.07.151 [9] Sancar, A., Lindsey-Boltz, L.A., Kang, T.H., Reardon, J.T., Lee, J.H. and Ozturk, N. (2010) Circadian clock control of the cellular response to DNA damage. FEBS Letters, 584.2618-2625. http://dx.doi.org/10.1016/j.febslet.2010.03.017 [10] Friedberg, E.C., Walker, G.C., Siede, W., Wood, R.D., Schultz, R.A. and Ellenberger, T. (2006) DNA repair and mutagenesis. ASM Press, Washington DC. [11] Hanawalt, P. (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene, 21, 8949-8956. http://dx.doi.org/10.1038/sj.onc.1206096 [12] Sarker, A.H., Tsutakawa, S.E., Kostek, S., Ng, C., Shin, D.S., Peris, M., Campeau, E., Tainer, J.A., Nogales, E., and Cooper, P.K. (2005) Recognition of RNA poly- merase II and transcription bubbles by XPG, CSB, and TFIIH: Insights for transcription-coupled repair and Cockayne Syndrome. Molecular Cell, 20, 187-198. http://dx.doi.org/10.1016/j.molcel.2005.09.022 [13] Lainé, J.P. and Egly, J.M. (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. The EMBO Iournal, 25, 387-397. http://dx.doi.org/10.1038/sj.emboj.7600933 [14] Wood, R.D. (1999) DNA damage recognition during nu- cleotide excision repair in mammalian cells. Biochimie, 81, 39-44. http://dx.doi.org/10.1016/S0300-9084(99)80036-4 [15] Sugasawa, K., Okamoto, T., Shimizu, Y., Masutani, C., Iwai, S. and Hanaoka, F. (2001) A multistep damage re- cognition mechanism for global genomic nucleotide ex- cision repair. Genes & Development, 15, 507-521. http://dx.doi.org/10.1101/gad.866301 [16] Wang, G., Chuang, L., Zhang, X., Colton, S., Domb- kowski, A., Reiners, J., Diakiw, A. and Xu, X.S. (2004) The initiative role of XPC protein in cisplatin DNA da- maging treatment-mediated cell cycle regulation. Nucleic Acids Research, 32, 2231-2240. http://dx.doi.org/10.1093/nar/gkh541 [17] Colton, S.L., Xu, X., Wang, Y.A. and Wang, G. (2006) The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. The Journal of Biologi- cal Chemistry, 281, 27117-27125. http://dx.doi.org/10.1074/jbc.M602826200 [18] Lomonaco, S.L., Xu, X. and Wang, G. (2009) The role of Bcl-x(L) protein in nucleotide excision repair-facilitated cell protection against cisplatin-induced apoptosis. DNA and Cell Biology, 28, 285-294. http://dx.doi.org/10.1089/dna.2008.0815 [19] Hollander, M.C., Philburn, R.T., Patterson, A.D., Velasco- Miguel, S., Friedberg, E.C., Linnoila, R.I. and Fornace Jr., A.J. (2005) Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung car- cinogenesis. Proceedings of the National Academy of Sci- ences of the United States of America, 102, 13200- 13205. http://dx.doi.org/10.1073/pnas.0503133102 [20] Chen, Z., Yang, J., Wang, G., Song, B., Li, J. and Xu, Z. (2007) Attenuated expression of xeroderma pigmentosum group C is associated with critical events in human blad- der cancer carcinogenesis and progression. Cancer Re- search, 67, 4578-4585. http://dx.doi.org/10.1158/0008-5472.CAN-06-0877 [21] Wu, Y.H., Tsai Chang, J.H., Cheng, Y.W., Wu, T.C., Chen, C.Y. and Lee, H. (2007) Xeroderma pigmentosum group C gene expression is predominantly regulated by promoter hypermethylation and contributes to p53 muta- tion in lung cancers. Oncogene, 26, 4761-4773. http://dx.doi.org/10.1038/sj.onc.1210284 Copyright © 2013 SciRes. OPEN ACCESS  G. Wang et al. / Open Journal of Genetics 3 (2013) 285-293 Copyright © 2013 SciRes. 293 OPEN ACCESS [22] Cheo, D.L., Burns, D.K., Meira, L.B., Houle, J.F. and Friedberg, E.C. (1999) Mutational inactivation of the xe- roderma pigmentosum group C gene confers predisposi- tion to 2-acetylaminofluorene-induced liver and lung cancer and to spontaneous testicular cancer in Trp53-/- mice. Cancer Research, 59, 771-775. [23] Friedberg, E.C., Bond, J.P., Burns, D.K., Cheo, D.L., Greenblatt, M.S., Meira, L.B., Nahari, D. and Reis, A.M. (2000) Defective nucleotide excision repair in Xpc mutant mice and its association with cancer predisposition. Mu- tation Research/DNA Repair, 459, 99-108. http://dx.doi.org/10.1016/S0921-8777(99)00068-3 [24] Wang, Y.C., Lu, Y.P., Tseng, R.C., Lin, R.K., Chang, J.W., Chen, J.T., Shih, C.M. and Chen, C.Y. (2003) Inac- tivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched spu- tum samples. The Journal of Clinical Investigation, 111, 887-895. [25] Okuda, T., Kawakami, K., Ishiguro, K., Oda, M., Omura, K. and Watanabe, G. (2005) The profile of hMLH1 me- thylation and microsatellite instability in colorectal and non-small cell lung cancer. International Journal of Mo- lecular Medicine, 15, 85-90. [26] Lee, M.N., Tsen, R.C., Hsu, H.S., Chen, J.Y., Tzao, C., Ho, W.L. and Wang, Y.C. (2007) Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clinical Can- cer Research, 13, 832-838. http://dx.doi.org/10.1158/1078-0432.CCR-05-2694 [27] Seng, T.J., Currey, N., Cooper, W.A., Lee, C.S., Chan, C., Horvath, L., Sutherland, R.L., Kennedy, C., McCaughan, B. and Kohonen-Corish, M.R. (2008) DLEC1 and MLH1 promoter methylation are associated with poor prognosis in non-small cell lung carcinoma. British Journal of Can- cer, 99, 375-382. http://dx.doi.org/10.1038/sj.bjc.6604452 [28] Geng, X., Wang, F., Zhang, L. and Zhang, W.M. (2009) Loss of heterozygosity combined with promoter hyper- methylation, the main mechanism of human MutL Ho- molog (hMLH1) gene inactivation in non-small cell lung cancer in a Chinese population. Tumori, 95, 488-494. [29] Okano, M., Bell, D.W., Haber, D.A. and Li, E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essen- tial for de novo methylation and mammalian development. Cell, 99, 247-257. http://dx.doi.org/10.1016/S0092-8674(00)81656-6 [30] Chen, T., Ueda, Y., Dodge, J.E., Wang, Z. and Li, E. Es- tablishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Molecular and Cellular Biology, 2003, 23, 5594- 5605. http://dx.doi.org/10.1128/MCB.23.16.5594-5605.2003 [31] Watanabe, D., Suetake, I., Tada, T. and Tajima, S. (2002) Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis, Mechanisms of Development, 118, 187-190. http://dx.doi.org/10.1016/S0925-4773(02)00242-3 [32] Tibbetts, R.S., Brumbaugh, K.M., Williams, J.M., Sark- aria, J.N., Cliby, W.A., Shieh, S.Y., Taya, Y., Prives, C. and Abraham, R.T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes & De- velopment, 13, 152-157. http://dx.doi.org/10.1101/gad.13.2.152 [33] Heffernan, T.P., Simpson, D.A., Frank, A.R., Heinloth, A.N., Paules, R.S., Cordeiro-Stone, M. and Kaufmann, W.K. (2002) An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Molecular and Cellular Biology, 22, 8552-8561. http://dx.doi.org/10.1128/MCB.22.24.8552-8561.2002 [34] Helt, C.E., Cliby, W.A., Keng, P.C., Bambara, R.A. and O’Reilly, M.A. (2005) Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selec- tive target specificities in response to different forms of DNA damage. The Journal of Biological Chemistry, 280, 1186-1192. http://dx.doi.org/10.1074/jbc.M410873200 [35] Cimprich, K.A. and Cortez, D. (2008) ATR: An essential regulator of genome integrity. Nature Reviews Molecular Cell Biology, 9, 616-627. http://dx.doi.org/10.1038/nrm2450 [36] Despras, E., Daboussi, F., Hyrien, O., Marheineke, K. and Kannouche, P.L. (2010) ATR/Chk1 pathway is es- sential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Human Molecular Ge- netics, 19, 1690-1701. http://dx.doi.org/10.1093/hmg/ddq046 [37] Stiff, T., Walker, S.A., Cerosaletti, K., Goodarzi, A.A., Petermann, E., Concannon, P., O'Driscoll, M., Jeggo, P.A. (2006) ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. The EMBO Journal, 25, 5775-5782. http://dx.doi.org/10.1038/sj.emboj.7601446 [38] Ward, I.M., Minn, K. and Chen, J. (2004) UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) acti- vation requires replication stress. The Journal of Bio- logical Chemistry, 279, 9677-9680. http://dx.doi.org/10.1074/jbc.C300554200 [39] Hervouet, E., Vallette, F.M. and Cartron, P.F. (2009) Dnmt3/transcription factor interactions as crucial players in targted DNA methylation. Epigenetics, 4, 487-499. http://dx.doi.org/10.4161/epi.4.7.9883 [40] Le, May, N., Mota-Fernandes, D., Velez-Cruz, R., Iltis, I., Biard, D. and Egly, J.M. (2010) NER factors are recruited to active promoters and fa- cilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Molecular and Cellular Biology, 38, 54-66.
|