 Open Journal of Genetics, 2013, 3, 262-269 OJGen http://dx.doi.org/10.4236/ojgen.2013.34029 Published Online December 2013 (http://www.scirp.org/journal/ojgen/) C-type lectins and human epithelial membrane protein1: Are they new proteins in keratin disorders? Nilüfer Karadeniz1*, Thomas Liehr2, Kristin Mrasek2, Ibrahim Aşık3, Zuleyha Aşık4, Nadezda Kosyakova2, Hasmik Mkrtchyan2 1Medical Genetics of Zubeyde Hanım Maternity Hospital, Ankara, Turkey 2Jena University Hospital, Institute of Human Genetics, Kollegiengasse, Jena, Germany 3Department of Anaesthesiology and Intensive Care, Faculty of Medicine, University of Ankara, Ankara, Turkey 4Dermatology of Dr Sami Ulus Children’s Hospital, Ankara, Turkey Email: *trkaradeniz@hotmail.com Received 16 August 2013; revised 26 September 2013; accepted 25 October 2013 Copyright © 2013 Nilüfer Karadeniz et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Here we report a family with a clinical spectrum of Pachyonychia Congenita Tarda (PCT) encompassing two generations via a balanced chromosomal trans- location between 4q26 and 12p12.3. We discuss the effects of chromosomal translocations on gene ex- pression through involved breakpoints and structural gene abnormalities detected by array CGH. We be- lieve that the family we present gives further insight to the better understanding of molecular and struc- tural basis of keratin disorders, and to the late onset and genetic basis of PCT through the possible role of C-type lectins and human epithelial membrane pro- tein1 (EMP1). Better understanding of the molecular basis of keratin disorders is the foundation for im- proved diagnosis, genetic counseling and novel thera- peutic approaches to overcome the current treatment limitations related to this disease. Keywords: Keratins; Palmoplantar Keratoderma; Pachyonychia Congenital Tarda; Lectins; Epithelial Membrane Protein1; Gene Expression and Chromosome Translocation 1. INTRODUCTION Humans encode keratins in 54 genes, and of these 28 are localized at 17q12-21 as acidic type I and as basic type II at 12q13.13. Mutations within those genes are associated with tissue specific “fragility disorders’’ which may manifest both on the skin and mucosa depending on their expressions. One of these is Pachyonychia congenita (PC) seen in three forms as PC-1, PC-2, and Pachyonychia Congenita Tarda (PCT). Although PC is well docu- mented both clinically and genetically, there are only few reports for PCT, and its genetic basis is still obscure [1,2]. Pachyonychia Congenita Tarda (PCT) is characterized by nail changes appearing during the second or third decades of life. More than 10 cases of PCT, both familial and sporadic, have been reported in the literature since it was first described in 1991 [3]. The age of onset ranges from teenage years to 44 years [4-9]. The reasons behind the late onset of PCT in some patients as well as the exclusive involvement of the nails are not yet fully understood (Bahhady et al., 2008). Here we report a family with PCT and its intra-famil- ial phenotypic variation associated with a balanced chromosomal translocation, t (4; 12) (q26; p12.3). Fur- thermore, we propose a modification for PC classifica- tion. The impacts of reported chromosomal breakpoints in human keratin disorders that lead to skin fragility are discussed in general for further insight. 2. CASE REPORTS An 18-year-old male patient (I-1) was referred to Medi- cal Genetics Department due to a request by his derma- tologist. Upon admission he was found to have been suf- fering from severe hyperhydrosis of his palms and soles since the beginning of his adolescence. Redness, blister and scaling were also detected in his detailed anamnesis. He had undergone different clinical treatments and took different medications due to these symptoms previously. On physical examination, he had focal palmoplantar keratoderma (PPK) with scaling due to blisters, slight onycholysis on palmar thumb nail and vasomotor altera- tions such as Raynaud’s phenomenon (see Figure 1). The results of palmar and plantar biopsies were compati- *Corresponding author. OPEN ACCESS 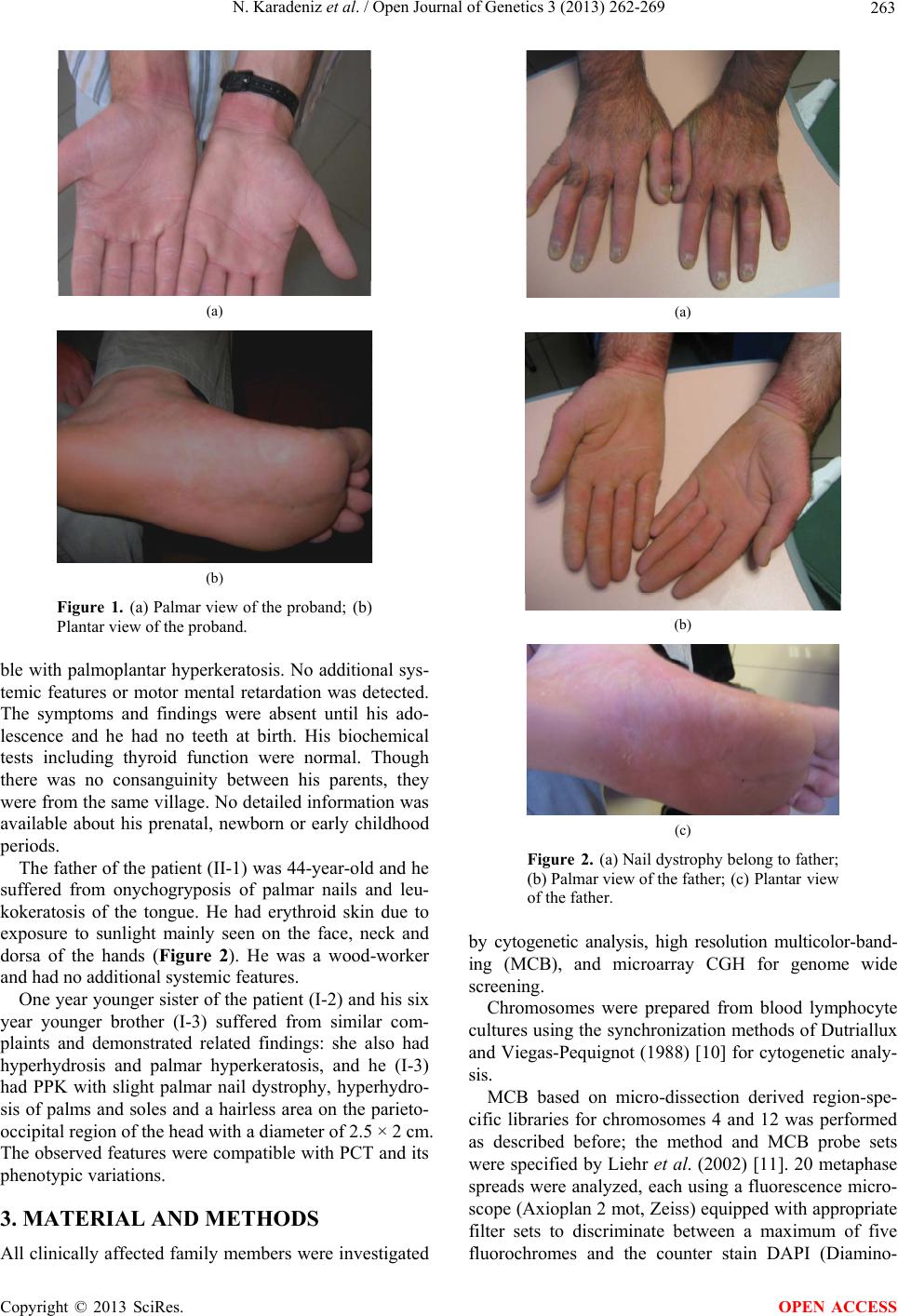 N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 263 (a) (b) Figure 1. (a) Palmar view of the proband; (b) Plantar view of the proband. ble with palmoplantar hyperkeratosis. No additional sys- temic features or motor mental retardation was detected. The symptoms and findings were absent until his ado- lescence and he had no teeth at birth. His biochemical tests including thyroid function were normal. Though there was no consanguinity between his parents, they were from the same village. No detailed information was available about his prenatal, newborn or early childhood periods. The father of the patient (II-1) was 44-year-old and he suffered from onychogryposis of palmar nails and leu- kokeratosis of the tongue. He had erythroid skin due to exposure to sunlight mainly seen on the face, neck and dorsa of the hands (Figure 2). He was a wood-worker and had no additional systemic features. One year younger sister of the patient (I-2) and his six year younger brother (I-3) suffered from similar com- plaints and demonstrated related findings: she also had hyperhydrosis and palmar hyperkeratosis, and he (I-3) had PPK with slight palmar nail dystrophy, hyperhydro- sis of palms and soles and a hairless area on the parieto- occipital region of the head with a diameter of 2.5 × 2 cm. The observed features were compatible with PCT and its phenotypic variations. 3. MATERIAL AND METHODS All clinically affected family members were investigated (a) (b) (c) Figure 2. (a) Nail dystrophy belong to father; (b) Palmar view of the father; (c) Plantar view of the father. by cytogenetic analysis, high resolution multicolor-band- ing (MCB), and microarray CGH for genome wide screening. Chromosomes were prepared from blood lymphocyte cultures using the synchronization methods of Dutriallux and Viegas-Pequignot (1988) [10] for cytogenetic analy- sis. MCB based on micro-dissection derived region-spe- cific libraries for chromosomes 4 and 12 was performed as described before; the method and MCB probe sets were specified by Liehr et al. (2002) [11]. 20 metaphase spreads were analyzed, each using a fluorescence micro- scope (Axioplan 2 mot, Zeiss) equipped with appropriate filter sets to discriminate between a maximum of five fluorochromes and the counter stain DAPI (Diamino- Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 264 phenylindol). Image capturing and processing were car- ried out using a mFISH imaging system (MetaSystems, Altlussheim, Germany) for the evaluation of MCB. MCB results were confirmed and refined by BAC-FISH using the following probes: RP11-18D18, RP11-320L7, RP11- 692M23 and RP11-12D15. The oligo-array method is the simultaneous hybridiza- tion of test-DNA (patient-DNA) and the genomic refer- ence DNA (human male/female genomic DNA, Promega) on a chip with 170.334 specific oligonucleotide sequen- ces (Agilent Human Genome CGH Microarray 180 K). The probes located on the array have an average distance of 17.665 kb between each other, so that changes that are smaller than 100 kb will be detectable. To reach maxi- mum statistical accuracy, changes only by a row of 5 probes (deletion; 88.3 kb) or of 10 probes (amplifica- tion; 176.6 kb) are to be further analyzed. Moreover, it was checked if there were CNV (Copy Number Varia- tions) by means of “Database of Genomic Variants”. For the interpretation of the results, we used the “UCSC Ge- nome Browser on Human Mar2006/ NCBI36/hg18” [12]. 4. RESULTS Cytogenetic analysis of the index patient (I-1) revealed 12p+ and 4q− pointing out to further investigation tech- niques to determine the related breakpoints on each of the chromosomes. It was transmitted from the father (II-1) to the patient. Figure 3 shows the intra-familial segrega- tion of the chromosomal abnormalities. Chromosomal analysis with MCB revealed a balanced translocation between chromosomes 4 and 12, t (4; 12) (q26; p12.3) for both the index patient and his father. The results were confirmed by BAC-FISH probes nar- rowing down to positions 112,485,135 and 117,524,000 at 4q25-q26 and positions 19,157,000 and 22,210,387 at 12p12.3-12p12 [13]. Figure 4 shows the molecular cy- togenetic definition of each breakpoint. Although we studied all the clinically affected mem- + + Figure 3. Pedigree of the presented family. (a) (b) Figure 4. (a) Partial karyotype with MCB; (b) BAC-FISH results. bers of the family for aCGH analysis in the form of ge- nome wide screening, we could only get results from the two of these members (I-2 and I-3), not having the translocations. So, we do not have any results for the translocation carriers for CGH microarray. Otherwise, members with focal PK without translocations (I-2 and I-3) have a lot of known benign CNV. They also have deletions of 7q34 which is very small (minimum 0.065166 Mb to maximum 0.089971 Mb; NCBI36/ HG18). Case I-2 also has amplification in 7q11.21 span- ning from 0.262634 to 0.307307 Mb. 5. DISCUSSION Keratins, the major structural proteins of the epidermis, form intermediate filament networks providing the integ- rity of the skin via support to keratinocytes. In humans, there are around 30 keratin families divided into two groups, namely, acidic and basic keratins, which are ar- ranged in pairs. There are also at least 54 functional keratin genes which are expressed in tissue and differen- Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 265 tiation-specific manner. These keratins have roles in cell growth, apoptosis, tissue polarity, wound healing and tissue remodeling as well [14-16]. The expressions of specific keratin genes are regulated by the differentiation of epithelial cells within the stratifying squamous epithe- lium [2]. Over the last decade, keratin mutations have been iden- tified as the causes of a number of skin fragility disorders including quite different phenotypes such as epidermoly- sis bullosa simplex (EBS), bullous congenital ichthyosi- form erythroderma (BCIE), PC, steatoma multiplex, ich- thyosis bullosa of Siemens (IBS), and white sponge ne- vus (WSN) of the orogenital mucosa depending on the expression patterns of the keratins involved [1,17]. Re- cently, Shetty and Gokul (2012) [2] well documented the structure of keratin, the various types of keratins and their distributions as well as the diseases associated with keratinization. The exact phenotype of each disease reflects the spa- tial level of expression and types of the mutated keratin genes, positions of the mutations as well as their conse- quences at subcellular levels [16]. In general, the muta- tions are either heterozygous missense mutations or small-in frame deletion/insertion mutations [18]. Over 90% of the pathogenic mutations in keratinopathies are missense mutations with a small number of small in frame insertion vs. deletion mutations and a few intronic splice site defects leading to larger in-frame deletions. At the protein level, the consequences of mutant polypep- tides are expressions at normal or near normal levels with substitutions, deletions or insertions of a different amino acid [16]. The molecular classification of keratin disorders was recently based on mutations involving keratin, loricin, desmosemes, connexins, and catepsins [19]. How these mutations in the keratins cause hyperkeratosis of the nail is not entirely clear, presumably the fragility of kerati- nocytes in the nail bed leads to the release of cytokines upon proliferative cells of the nail matrix thereby causing overgrowth of the nail [20]. A number of in vitro studies have demonstrated that keratin mutations can cause dis- ruptions to filament assembly and network formation, the severity of which can vary with the position of, or the actual mutation. At the clinical level, these mutations present skin fragility disorders such as PC, PCT, EBS, and so on depending on the expression patterns of the keratins involved [21]. On the other hand, the co-distri- bution of certain structural proteins such as desmoplakin, connexin, and plakoglobin with keratins onto other tis- sues may explain some genetic diseases associated with PPK: desmoplakin and plakoglobin mutations with car- diocutaneous syndromes and connexin mutations with PPK, deafness, and neuropathy [22]. A correct diagnosis based on molecular genetic analysis is mandatory, al- though a causal therapy is still not available [19]. PC refers to a group of rare disorders characterized by transverse over curvature and onycholysis of the nail developing in infancy together with thick skin and other ectodermal features. The condition is separated into four different types by the presence of associated features and different keratin abnormalities have been described in the two main subtypes. It is usually inherited as an auto- somal dominant trait. However, autosomal recessive and sporadic cases have also been described [3,23,24]. In PC-1, hypertrophic nail dystrophy is accompanied by severe focal keratoderma especially on the soles of the feet and often with white plaques in the mouth (oral leu- kokeratosis) as seen in the presented case II-1. In PC-2, nail dystrophy is accompanied by mild palmoplantar keratoderma and multiple pilosebaceous cysts that typi- cally develop after puberty. Natal teeth and hair abnor- malities as twisted hair are associated features but are not fully penetrating [25]. Milder phenotypic variations of PC-1 and PC-2 are now being recognized as FNEPPK and steatocytoma multiplex respectively [26]. Type 3 PC may have features of both types 1 and 2, in addition to corneal leukokeratosis and cataracts. Laryngeal lesions with hoarseness, hair abnormalities, and mental retarda- tion are characteristics of type 4 [4]. The clinical features of PC are closely related to allelic disorders like FNEPPK and steatocytoma multiplex. Mu- tations in keratins K6a or K16 cause PC-1 or FNEPPK phenotypes, whereas K6b or K17 defects lead to PC2 or steatocystoma multiplex phenotypes [27-29]. We pro- pose that fifth type of PC in which the typical nail changes begin in the second or third decades of life has previously been described as PCT [3]. In PC-1, the mutations in the gene for K6a and K16, which are normally regarded as stress response keratins induced during wound healing, have been identified. The tissue distribution of this keratin in the nail bed, nail fold, as well as in palmoplantar skin and oral mucosa matches well with the epithelial phenotypes [27]. The majority of the mutations in PC are missense changes with smaller numbers of in-frame insertions and deletions. In PC-1, two-thirds of all known mutations occur in the KRTA 6A gene, whereas one-third occurs in KRT16 [26]. Smith et al. (2005) [30] reported 30 new mutations in PC. All the identified mutations were heterozygous amino acid sub- stitutions or small in-frame deletion mutations. All the novel PC-1 mutations reported by them were in the helix boundary motifs of either K6a or K16, consistent with previous findings. Although the genetic basis and the clinical manifesta- tions of PC are well documented as explained above, there is scarce information for PCT. Few reports that were documented clinically and genetically [4] since it was first described in 1991 [3]. Hannaford and Stapleton Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 266 (2000) [23] reviewed 15 cases of the PCT variety. They found that the most common features were nail changes and palmoplantar keratoderma. The nail changes were reported to be present in all the fingers and the toes, unlike their case where there was sparing of some fingers. Although there has been some debate concerning whe- ther these late-onset cases represent a separate genetic syndrome or a variant form of PC-1, generally it was ac- cepted that PCT can be thought to represent a delayed form of type 1 PC and may have similar underlying ge- netic defects affecting the keratin 6a/i6 pair [31]. The main symptom of the family presented here is hyperhydrosis. Hyperhydrosis is the most commonly seen clinical feature on PC of the report by Leachman et al. (2005) [32]. Because of this, when the patient with PPK is being evaluated, hyperhydrosis should also be considered. All affected members have the PPK with variable degrees and with increasing severity with ageing. Phenotypic variations of PPK in the presented family are features of both PC-1 and PCT. In the presented family, due to the late onset of clinical features and slight mal- formations on nails, we arrive at the diagnosis of PCT. Only in one patient of ours (I-3) the scalp was slightly and limitedly affected while in their report it was dif- fusely affected in one patient. The nails of the case II-1, the oldest member of the family, was more affected than the others indicating age dependent severity and the ef- fects of anticipation and/or environmental factors on clinical features. He also has white plaques on the tongue which is one of the main features of PC1, indicating in- tra-familial phenotypic variation. We suggest that it can result from the activation of the affected allele leading to this progressive event, and some environmental or im- munological factors may also be relevant for this situa- tion as described by Hannaford and Stapleton (2000) [23]. On the other hand, while we respect the results be- longing to two of four patients with balanced transloca- tions, this association could be coincidental as well. But, it requires significantly more attention to each of the breakpoints. Trembath et al. (2004) [33] investigated two patients with translocations in Rieger’s syndrome. They described how they sequenced the breakpoint regions on each chromosome breakpoint and how they cloned the translocation segments themselves using pandle PCR (PHPCR) together with characterization of enhancer and silencer functions in fragments near the breakpoints. Their results demonstrated that PTIX2 was rich in repeti- tive elements, but no novel genes were identified. FISH demonstrated that PTIX2 was intact, and transfection studies revealed a slight enhancer affected by chromo- some 4 sequences and a strong silencer effect on other affected chromosome sequences (chromosome 11). They concluded that due to absence of novel genes near either of the breakpoints, changes in the potential regulatory elements may be the best model to explain the loss of PTIX2 expression in their patients and hence the Rieger’s syndrome phenotype. But we cannot cite any information in the literature about the relationship between the pre- sented breakpoints and keratin disorders either clinically or genetically. So, to the best of our knowledge, ours is a first time description. Moreover, the patients with normal karyotypes (I-2 and I-3) present with a deletion on 7q34 detected by aCGH. Seven CNVs are located in this region; three of them span a large part of the deleted region. In the lit- erature, no cases have been described and in the ISCA (The International Standards for Cytogenomic Arrays Consortium) database, there are no registrations of pa- tients with such a deletion. In the Decipher database there is one patient (254,949) with a clinical phenotype. But this patient has three additional deletions. So these are not comparable with each other. There are two genes in this region: PIP (Prolactin Inducible Protein, OMIM 17,620) is expressed in benign and malignant breast tu- mor tissues and in some normal exocrine organs such as sweat, salivary and lacrimal glands, and TAS2R39 (Taste Receptor, type 2, member 39). So this deletion can be the possible cause of hyperhydrosis seen in the patients. If someone reevaluates his patients with PCT, it can be determined as a further clinical and/or laboratory finding related to 7q34. Otherwise, there are no genes located on 7q11.21 which are amplified on case I-2, and therefore it is not potentially pathogenical. Indeed, in this region, there can be possible genetic-regulatory elements that influence other genes. In most cases, the genetic defect can be explained as a disruption of normal cis regulation of transcription, without clear evidence of altered chro- matin organization, although mechanisms are difficult to assess in human diseases, in which access to the affected tissues is often impossible [34]. But, unfortunately we couldn’t get any results using aCGH on each of these translocation carriers. We can say that chromosomal rearrangements can come into play through two mechanisms: 1) by disturbing the interac- tions of the promoter and transcription units with its cis-acting regulators, either by mutation or by physical dissociation of the transcribed gene from its full set regulator elements; and/or 2) alteration of the local or global regulation of chromatin structure [35-38]. When genes that normally reside in euchromatic domains were transposed close to a heterochromatic region, mosaic pattern of gene expression is observed [39]. On the other hand, Chen et al. (1997) [40] and Liehr et al. (1999) [13] have mapped human epithelialmembrane protein CL-20 gene (EMP1) to chromosome 12p12-13 by fluorescence in situ hybridization. Previously, Marvin et al. (1995) [41] showed that high CL-20 expression cor- Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 267 relates with squamous differentiation in vivo and in vitro using Northern Blot and in situ hybridization. They also showed that retinoids repress the induction of CL-20. It is likely to play important roles in the regulation of cell proliferation, differentiation, and cell death. As it is known, the in vitro and in vivo effects of retinoids on keratins are paradoxical. In vitro, retinoids reduce the expressions of KRT5, KRT14, KRT6, KRT1 and KRT10 in cultured keratinocytes, whereas KRT7, KRT13, KRT15 and KRT19 are induced [42]. Subsequently, Cambi and Figdor (2009) [43] showed that C-type lectins, a family of surface receptors, are known to recognize microbial carbohydrate moieties as well as sense products from cells dying by necrosis with immunity and transduce inflammatory signals that modulate the immune system. We suggest that human epithelial membrane protein1 and/or C-type lectins, lo- calized 12p12-13, can also play a role for pathological pathways of keratins though flexible responses of the skin immune system during the wound healing and apoptosis as seen in the genes for K6a and K16. Both of them or one of them can also help to explain the under- lying pathology of late onset, increased severity of clini- cal features with ageing and slightly affected nails in the family. In conclusion, we suggest that the combination of whole-genome genetic association studies and measure- ments of global gene expression can shed light to the understanding of the differences in the gene expressions in human genetic diseases. Also, we believe that the family we present here will help us to better understand the molecular pathology of keratin disorders by giving us new insights and making it possible to develop new therapeutic approaches. REFERENCES [1] Lane, E.B. and McLean, W.H.I. (2004) Keratins and skin disorders. Journal of Pathology, 204, 355-366. http://dx.doi.org/10.1002/path.1643 [2] Shetty, S. and Gokul, S. (2012) Keratinization and its disorders. Oman Medical Journal, 27, 348-357. http://dx.doi.org/10.5001/omj.2012.90 [3] Paller, A.S., Moore, J.A. and Scher, R. (1991) Pachyon- ychia congenita tarda. A late-onset form of pachyonychia congenita. Archives of Dermatology, 127, 701-703. http://dx.doi.org/10.1001/archderm.1991.0168004010901 3 [4] Bahhady, R., Abbas, O. and Dahdah, M. (2008) Pachyo- nychia congenita tarda: Very late onset. International Journal of Dermatology, l47, 1172-1173. http ://d x.do i.o rg/10. 1111/ j. 1365-4632.2008.03830.x [5] Iraci, S., Bianchi, L., Gatti, S., Carrozzo, A.M., Bettini, D. and Nini, G. (1993) Pachyonychia congenita with late onset of nail dystrophy—A new clinical entity? Clinical and Experimental Dermatology, 18, 478-480. http ://d x.do i.o rg/10. 1111/ j. 1365-2230.1993.tb02257.x [6] Chang, A., Lucker, G.P., van de Kerkhof, P.C. and Stei- jlen, P.M. (1994) Pachyonychia congenita in the absence ofother syndrome abnormalities. Journal of the American Academy of Dermatology, 30, 1017-1018. http://dx.doi.org/10.1016/S0190-9622(09)80144-8 [7] Moon, S.E., Lee, Y.S. and Youn, J.I. (1994) Eruptive vellus hair cyst and steatocystoma multiplex in a patient with pachyonychia congenita. Journal of the American Academy of Dermatology, 30, 275-276. http://dx.doi.org/10.1016/S0190-9622(08)81928-7 [8] Haber, R.M. and Rose, T.H. (1986) Autosomal recessive pachyonychia congenital. Archives of Dermatology, 122, 919-923. http://dx.doi.org/10.1001/archderm.1986.0166020009102 3 [9] Mawhinney, H., Creswell, S. and Beare, J.M. (1981) Pa- chyonychia congenita with candidiasis. Clinical and Ex- perimental Dermatology, 6, 145-149. http ://d x.do i.o rg/10. 1111/ j. 1365-2230.1981.tb02281.x [10] Dutrillaux, B. and Viegas-Pequignot, E. (1981) High resolution R- and G-banding on the same preparation. Human Genetics, 57, 93-95. http://dx.doi.org/10.1007/BF00271176 [11] Liehr, T., Weise, A., Heller, A., Starke, H., Mrasek, K., Kuechler, A., Weier, H.U. and Claussen, U. (2002) Mul- ticolor chromosome banding (MCB) with YAC/BAC- based probes andregion-specific microdissection DNA libraries. Cytogenetic and Genome Research, 97, 43-50. http://dx.doi.org/10.1159/000064043 [12] UCSC Genome Browser on Human Mar2006/NCBI36/ hg18. [13] Liehr, T., Kuhlenbäumer, G., Wulf, P., Taylor, V., Suter, U., Van Broeckhoven, C., Lupski, J.R., Claussen, U. and Rautenstrauss, B. (1999) Regional localization of the human epithelial membrane protein genes 1, 2, and 3 (EMP1, EMP2, EMP3) to 12p12.3, 16p13.2, and 19q13.3. Genomics, 58, 106-108. http://dx.doi.org/10.1006/geno.1999.5803 [14] Schweizer, J., Bowden, P.E., Coulombe, P.A., Langbein, L., Lane, E.B., Magin, T.M., Maltais, L., Omary, M.B., Parry, D.A., Rogers, M.A. and Wright, M.W. (2006) New consensus nomenclature for mammalian keratins. The Journal of Cell Biology, 174, 169-174. http://dx.doi.org/10.1083/jcb.200603161 [15] Hesse, M., Zimek, A., Weber, K. and Magin, T.M. (2004) Comprehensive analysis of keratin gene clusters inhu- mans and rodents. The Journal of Cell Biology, 83, 19-26. http://dx.doi.org/10.1078/0171-9335-00354 [16] Chamcheu, J.C., Siddiqui, I.A., Syed, D.N., Adhami, V.M., Liovic, M. and Mukhtar, H. (2011) Keratin gene mutations in disorders of human skin and its appendages. Archives of Biochemistry and Biophysics, 508, 123-137. http://dx.doi.org/10.1016/j.abb.2010.12.019 [17] Rugg, E.L. and Leigh, I.M. (2004) The keratins and their disorders. American Journal of Medical Genetics, Part C, 131C, 4-11. http://dx.doi.org/10.1002/ajmg.c.30029 [18] Steinert, P.M., Yang, J.M., Bale, S.J. and Compton, J.G. (1993) Concurrence between the molecular overlap re- Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 268 gions in keratin intermediate filaments and the locations of keratin mutations in genodermatoses. Biochemical and Biophysical Research Communications, 197, 840-848. http://dx.doi.org/10.1006/bbrc.1993.2555 [19] Braun-Falco, M. (2009) Hereditary palmoplantar kerato- dermas. J DDG, 7, 971-984. [20] De Berker, D., Wojnarowska, F., Sviland, L., Westgate, G.E., Dawber, R.P. and Leigh, I.M. (2000) Keratin ex- pression in the normal nail unit: markers of regional dif- ferentiation. British Journal of Dermatology, 142, 89-96. http://dx.doi.org/10.1046/j.1365-2133.2000.03246.x [21] Smith, F. (2003) The molecular genetics of keratin disor- ders. American Journal of Clinical Dermatology, 4, 347- 464. http://dx.doi.org/10.2165/00128071-200304050-00005 [22] Kelsell, D.P. and Leigh, I.M. (2008) Inheridet kerato- dermas of palms and soles. In: Wolf, K., Goldsmith, L.A., Katz, S.I., Gilchrest, B.A., Paller, A. and Leffell, D.J., Eds., Fitzpatrick’s Dermatology in General Medicine, Mc Graw Hill Medical, NewYork, 224. [23] Hannaford, R.S. and Stapleton, K. (2000) Pachyonychia congenita tarda. Australasian Journal of Dermatology, 41, 175-177. http://dx.doi.org/10.1046/j.1440-0960.2000.00425.x [24] Su, W.P., Chun, S.I., Hammond, D.E. and Gordon, H. (1990) Pachyonychia congenita: A clinical study of 12 cases and review of the literature. Pediatric Dermatology, 7, 33-38. http ://d x.do i.o rg/10. 1111/ j. 1525-1470.1990.tb01070.x [25] Itin, P.H. and Fistarol, S.K. (2005) Palmoplantar kerato- dermas. Clinics in Dermatology, 23, 15-22. http://dx.doi.org/10.1016/j.clindermatol.2004.09.005 [26] Arin, M.J. (2009) The molecular basis of human keratin disorders. Human Genetics, 1254, 355-373. http://dx.doi.org/10.1007/s00439-009-0646-5 [27] Bowden, P.E., Haley, J.L., Kansky, A., Rothnagel, J.A., Jones, D.O. and Turner, R.J. (1995) Mutation of a type II keratin gene (K6a) in pachyonychia congenita. Nature Genetics, 10, 363-365. http://dx.doi.org/10.1038/ng0795-363 [28] McLean, W.H.I., Rugg, E.L., Lunny, D.P., Morley, S.M., Lane, E.B., Swensson, O., Dopping-Hepenstal, P.J.C., Griffiths, W.A.D., Eady, R.A.J., Higgins, C., Navsaria, H.A., Leigh, I.M., Strachan, T., Kunkeler, L. and Munro, C.S. (1995) Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nature Geneti cs, 9, 273-278. http://dx.doi.org/10.1038/ng0395-273 [29] Smith, F.J.D., Jonkman, M.F., van Goor, H., Coleman, C.M., Covello, S.P., Uitto, J. and McLean, W.H. (1998) A mutation in human keratin K6b produces a phenocopy of the K17 disorder pachyonychia congenita type2. Human Molecular Genetics, 7, 1143-1148. http://dx.doi.org/10.1093/hmg/7.7.1143 [30] Smith, F.J., Liao, H., Cassidy, A.J., Stewart, A., Hamill, K.J., Wood, P., Joval, I., van Steensel, M.A., Björck, E., CallifDaley, F., Pals, G., Collins, P., Leachman, S.A., Munro, C.S. and McLean, W.H. (2005) The genetic basis of pachyonychia congenita. Journal of Investigative Der- matology Symposium, 10, 21-30. http ://d x.do i.o rg/10. 1111/ j. 1087-0024.2005.10204.x [31] Baran, R. and Haneke, E. (2006) The nail in differential diagnosis. Informa Healthcare, 51-60. [32] Leachman, S.A., Kaspar, R.L., Fleckman, P., Florell, S.R., Smith, F.J., McLean, W.H., Lunny, D.P., Milstone, L.M., van Steensel, M.A., Munro, C.S., O’Toole, E.A., Celebi, J.T., Kansky, A. and Lane, E.B. (2005) Clinical and pathological features of pachyonychia congenita. Journal of Investigative Dermatology Symposium, 10, 3-17. http ://d x.do i.o rg/10. 1111/ j. 1087-0024.2005.10202.x [33] Trembath, D.G., Semina, E.V., Jones, D.H., Patil, S.R., Qian, Q., Amendt, B.A., Russo, A.F. and Murray, J.C. (2004) Analysis of two translocation breakpoints and identification of anegative regulatory element in patients with Rieger’s syndrome. Birth Defects Research Part A: Clinical and Molecular Teratology, 70, 82-91. http://dx.doi.org/10.1002/bdra.10154 [34] Kleinjan, D.A. and van Heyningen, V. (2005) Long-range control of gene expression: Emerging mechanisms and disruption in disease. The American Journal of Human Genetics, 76, 8-32. http://dx.doi.org/10.1086/426833 [35] Kleinjan, D.A. and van Heyningen, V. (1998) Position effects in human genetic disease. Human Molecular Genetics, 7, 1611-1618. http://dx.doi.org/10.1093/hmg/7.10.1611 [36] Jiang, G., Yang, F., van Overveld, P.G., Vedanarayanan, V., van der Maarel, S. and Ehrlich, M. (2003) Testing the position-effect variegation hypothesis for facioscapulo- humeral muscular dystrophy by analysis of histone modi- fication and gene expression in subtelomeric 4q. Human Molecular Genetics, 15, 2909-2921. http://dx.doi.org/10.1093/hmg/ddg323 [37] Saveliev, A., Everett, C., Sharpe, T., Webster, Z. and Fes- tenstein, R. (2003). DNA triplet repeats mediate hetero- chromatin-protein-1-sensitive variegated gene silencing. Nature, 24, 909-913. http://dx.doi.org/10.1038/nature01596 [38] Tufarelli, C., Stanley, J.A., Garrick, D., Sharpe, J.A., Ayyub, H., Wood, W.G. and Higgs, D.R. (2003) Tran- scription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nature Genetics, 34, 157-165. http://dx.doi.org/10.1038/ng1157 [39] Wallrath, L.L. and Elgin, S.C. (1995) Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes & Development, 9, 1263- 1277. [40] Chen, Y., Medvedev, A., Ruzanov, P., Marvin, K.W. and Jetten, A.M. (1997) cDNA cloning, genomic structure, and chromosome mapping of the human epithelial membrane protein CL-20 gene (EMP1), a member ofthe PMP22 family. Genomics, 41, 40-48. http://dx.doi.org/10.1006/geno.1997.4524 [41] Marvin, K.W., Fujimoto, W. and Jenetten, A. (1995) Identification and characterization of anovel Squamous- cell-associated gene related to PMP22. The Journal of Biological Chemistry, 270, 28910-28916. http://dx.doi.org/10.1074/jbc.270.48.28910 [42] Törmä, H. (2011)Regulation of keratin expression by Copyright © 2013 SciRes. OPEN ACCESS  N. Karadeniz et al. / Open Journal of Genetics 3 (2013) 262-269 Copyright © 2013 SciRes. 269 OPEN ACCESS retinoids. Dermatoendocrinol, 3, 136-140. [43] Cambi, A. and Figdor, C. (2009) Necrosis: C-type lectins sense cell death. Current Biology, 19, R375-378. http://dx.doi.org/10.1016/j.cub.2009.03.032
|