 Journal of Biosciences and Medicines, 2013, 1, 26-30 JBM http://dx.doi.org/10.4236/jbm.2013.13006 Published Online December 2013 (http://www.scirp.org/journal/jbm/) OPEN ACCESS The genome of herpes simplex virus type 1 is prone to form short repeat seq uen c es Xiangyan Zhao1*, Xiaolong Wu1*, Lv Qin2, Zhongyang Tan1#, Shifang Li2, Qingjian Ouyang1, You Tian1 1College of Biology, State Key Laboratory for Chemo/Biosensing and Chemometrics, Hunan University, Changsha, China 2State Key Laboratory of Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China Email: #zhongyang@hnu.edu.cn Received September 2013 ABSTRACT Herein, we report a very high content of simple se- quence repeats (SSRs) covering 66.12% of the herpes simplex virus type 1 (HSV-1) genome when a low threshold is adopted to define SSRs, indicating that repeat sequence is a very important character of the HSV-1 genome. The repeats with two iterations ac- count for 68.33% of the total repeats. In reality, the genome of HSV-1 is prone to form shorter repeat se- quences. For mono-, di- and trinucleotide repeats, the repeat numbers decreased with the increase of re- peats iterations, implicating that the formation ten- dency of SSRs might be from low iterations to high iterations. The high iterations SSRs might have sub- jected to strong selected pressure and survived to perform different functions. The analysis suggested that the repeats formation may be an essential evolu- tionary driving force for the HSV-1 genome, and the results might be helpful for studying the genome structure, repeats genesis and genome evolution of HSV-1. Keywords: Simple Sequence Repeat; HSV-1 Genome; Microsatellite; SSR 1. INTRODUCTION Herpes simplex virus type 1 (HSV-1) is a member of the Simplexvirus genus in the Herpesviridae family and it is widely distributed in the human population. HSV-1 is the leading cause of viral induced blindness [1]; it gives rise to a variety of clinical disorders and is a major cause of morbidity and mortality worldwide [2]. The global pre- valence of HSV-1 is approximately 90%, including 65% more or less in the USA [3] and 52% - 67% in northern Europe [4]. Like other creatures, the HSV-1 genome contains amounts of repeats. What’s interesting is that the content of GC repeats is far high in the HSV-1 ge- nome, which may be related to some pathogenesis of HSV-1 [5]. To our knowledge, the small scope of muta- tion in repeated regions can cause many diseases [6]; in addition, expansion or contraction of repeats can change the sequence length [7]. Therefore, researching the re- petitive sequences we can learn something not only about the interrelationship between genome structure and pathogenesis, but about genome evolution of the HSV-1 from the perspective of molecular biology. In recent decades, increased attention has been paid to genomic repeat sequences. According to differential lengths of repetitive units, the repeat sequences are usu- ally divided into three basic types: microsatellites, mini- satellites and satellite DNA [8]. Mono-, di-, tri-, te- tra-, penta- and hexanucleotide repeats are usually classified as microsatellite-also referred to as short tandem repeats (STRs) or simple sequence repeats (SSRs) [9], and it widely studied in the genomes of prokaryotes and euka- ryotes. Repeats of longer units are referred to as minisa- tellites, the extreme long units, called satellite DNA [6]. Long tandem repeats are observed to be hypermutable, but are rare in exons and only occasionally related with diseases in human [7,10]. However, STRs are extremely common and scattered in both coding and non-coding regions in eukaryote [11], prokaryote [12] and also viral [13] genomes. Why are the SSRs so common? Do they perform some functions or are they just “junk DNA” sequences that should perhaps be regarded as “selfish DNA”? Exploring these questions is important to under- stand how genomes originated, organized and expanded. In previous studies, microsatellites were researched by using the statistics of relative abundance, relative density, composition and location, etc. In this paper, however, we mainly use repeat iterations to study repeats of short length. Different research ers used different thresholds *Xiaolong Wu a nd Xiangyan Zhao are the C o-First Author. #Corresponding author. 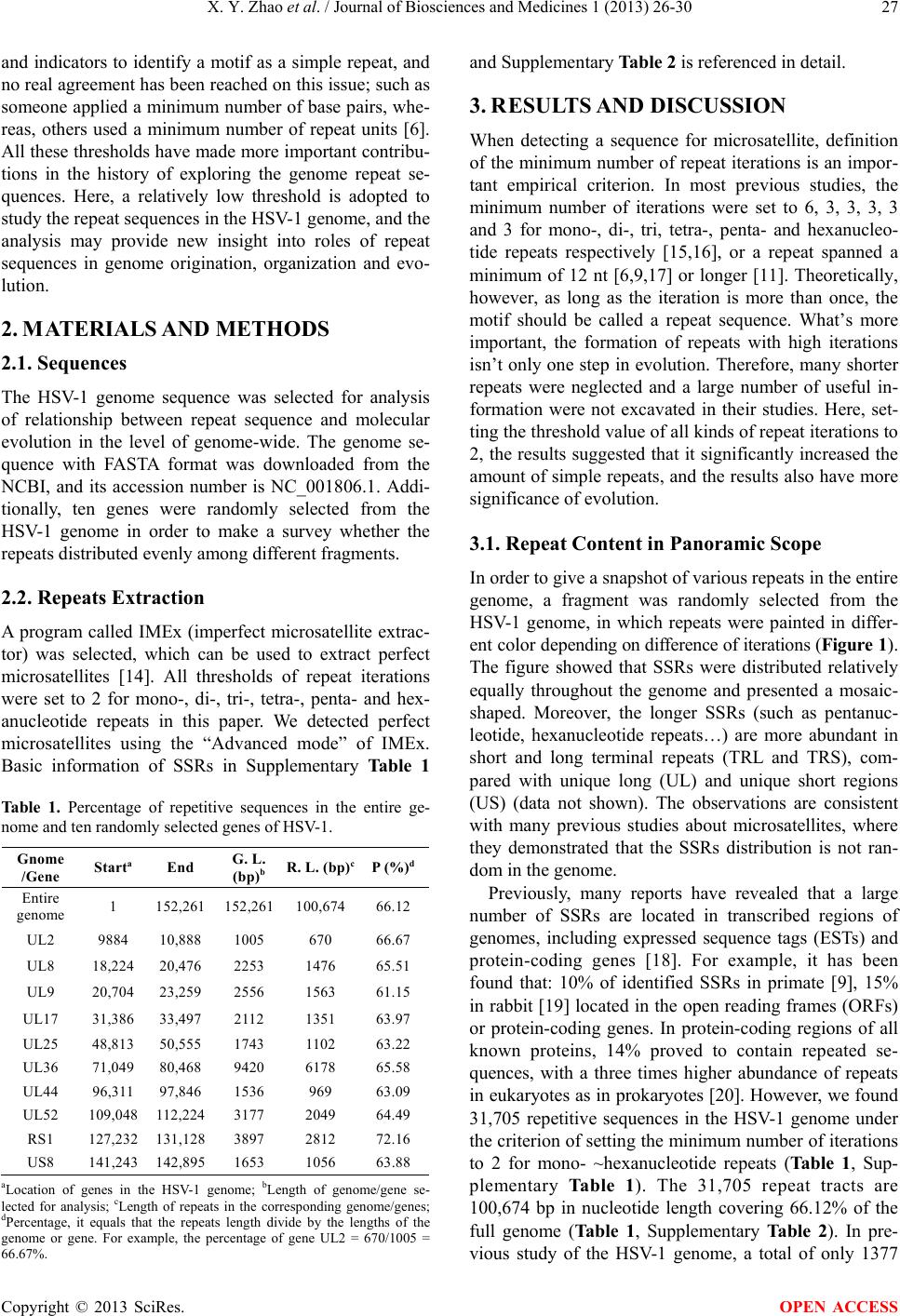 X. Y. Zhao et al. / Journal of Biosciences and Medicines 1 (2013) 26-30 Copyright © 2013 SciRes. OPEN ACCESS and indicators to identify a motif as a simple repeat, and no real agreement has been reached on this issue; such as someone applied a minimum number of base pairs, whe- reas, others used a minimum number of repeat units [6]. All these thresholds have made mor e important contribu - tions in the history of exploring the genome repeat se- quences. Here, a relatively low threshold is adopted to study the repeat sequences in the HSV-1 genome, and the analysis may provide new insight into roles of repeat sequences in genome origination, organization and evo- lution. 2. MATERIALS AND METHODS 2.1. Sequences The HSV-1 genome sequence was selected for analysis of relationship between repeat sequence and molecular evolution in the level of genome-wide. The genome se- quence with FASTA format was downloaded from the NCBI, and its accession number is NC_001806.1. Addi- tionally, ten genes were randomly selected from the HSV-1 genome in order to make a survey whether the repeats distributed evenly among different fragments. 2.2. Repeats Extraction A program called IMEx (imperfect microsatellite extrac- tor) was selected, which can be used to extract perfect microsatellites [14]. All thresholds of repeat iterations were set to 2 for mono-, di-, tri-, tetra-, penta- and hex- anucleotide repeats in this paper. We detected perfect microsatellites using the “Advanced mode” of IMEx. Basic information of SSRs in Supplementary Table 1 Table 1. Percentage of repetitive sequences in the entire ge- nome and ten randomly selected genes of HSV-1. Gnome /Gene Starta End G. L. (bp)b R. L. (bp)c P (%)d Entire genome 1 152,261 152,261 100,674 66.12 UL2 9884 10,888 1005 670 66.67 UL8 18,224 20,476 2253 1476 65.51 UL9 20,704 23,259 2556 1563 61.15 UL17 31,386 33,497 2112 1351 63.97 UL25 48,813 50,555 1743 1102 63.22 UL36 71,049 80,468 9420 6178 65.58 UL44 96,311 97,846 1536 969 63.09 UL52 109,048 112,224 3177 2049 64.49 RS1 127,232 131,128 3897 2812 72.16 US8 141,243 142,895 1653 1056 63.88 aLocation of genes in the HSV-1 genome; bLength of genome/gene se- lected for analysis; cLength of repeats in the corresponding genome/genes; dPercentage, it equals that the repeats length divide by the lengths of the genome or gene. For example, the percentage of gene UL2 = 670/1005 = 66.67%. and Supplementary Table 2 is referenced in detail. 3. RESULTS AND DISCUSSION When detecting a sequence for microsatellite, definition of the minimum number of repeat iterations is an impor- tant empirical criterion. In most previous studies, the minimum number of iterations were set to 6, 3, 3, 3, 3 and 3 for mono-, di-, tri, tetra-, penta- and hexanucleo- tide repeats respectively [15,16], or a repeat spanned a minimum of 12 nt [6,9,17] or longer [11]. Theoretically, however, as long as the iteration is more than once, the motif should be called a repeat sequence. What’s more important, the formation of repeats with high iterations isn’t only one step in evolution. Therefore, many shorter repeats were neglected and a large number of useful in- formation were not excavated in their studies. Here, set- ting the threshold value of all kinds of repeat iterations to 2, the results suggested that it significantly increased the amount of simple repeats, and the results also have more significance of evolution. 3.1. Repeat Content in Panoramic Scope In order to give a snapshot of various repeats in the entire genome, a fragment was randomly selected from the HSV-1 genome, in which repeats were painted in differ- ent color depending on difference of iterations (Figure 1). The figure showed that SSRs were distributed relatively equally throughout the genome and presented a mosaic- shaped. Moreover, the longer SSRs (such as pentanuc- leotide, hexanucleotide repeats…) are more abundant in short and long terminal repeats (TRL and TRS), com- pared with unique long (UL) and unique short regions (US) (data not shown). The observations are consistent with many previous studies about microsatellites, where they demonstrated that the SSRs distribution is not ran- dom in the genome. Previously, many reports have revealed that a large number of SSRs are located in transcribed regions of genomes, including expressed sequence tags (ESTs) and protein-coding genes [18]. For example, it has been found that: 10% of identified SSRs in primate [9], 15% in rabbit [19] located in the open reading frames (ORFs) or protein-coding genes. In protein-coding regions of all known proteins, 14% proved to contain repeated se- quences, with a three times higher abundance of repeats in eukaryotes as in prokaryotes [20]. However, w e found 31,705 repetitive sequences in the HSV-1 genome under the criterion of setting the minimum number of iterations to 2 for mono- ~hexanucleotide repeats (Table 1, Sup- plementary Table 1). The 31,705 repeat tracts are 100,674 bp in nucleotide length covering 66.12% of the full genome (Table 1, Supplementary Table 2). In pre- vious study of the HSV-1 genome, a total of only 1377 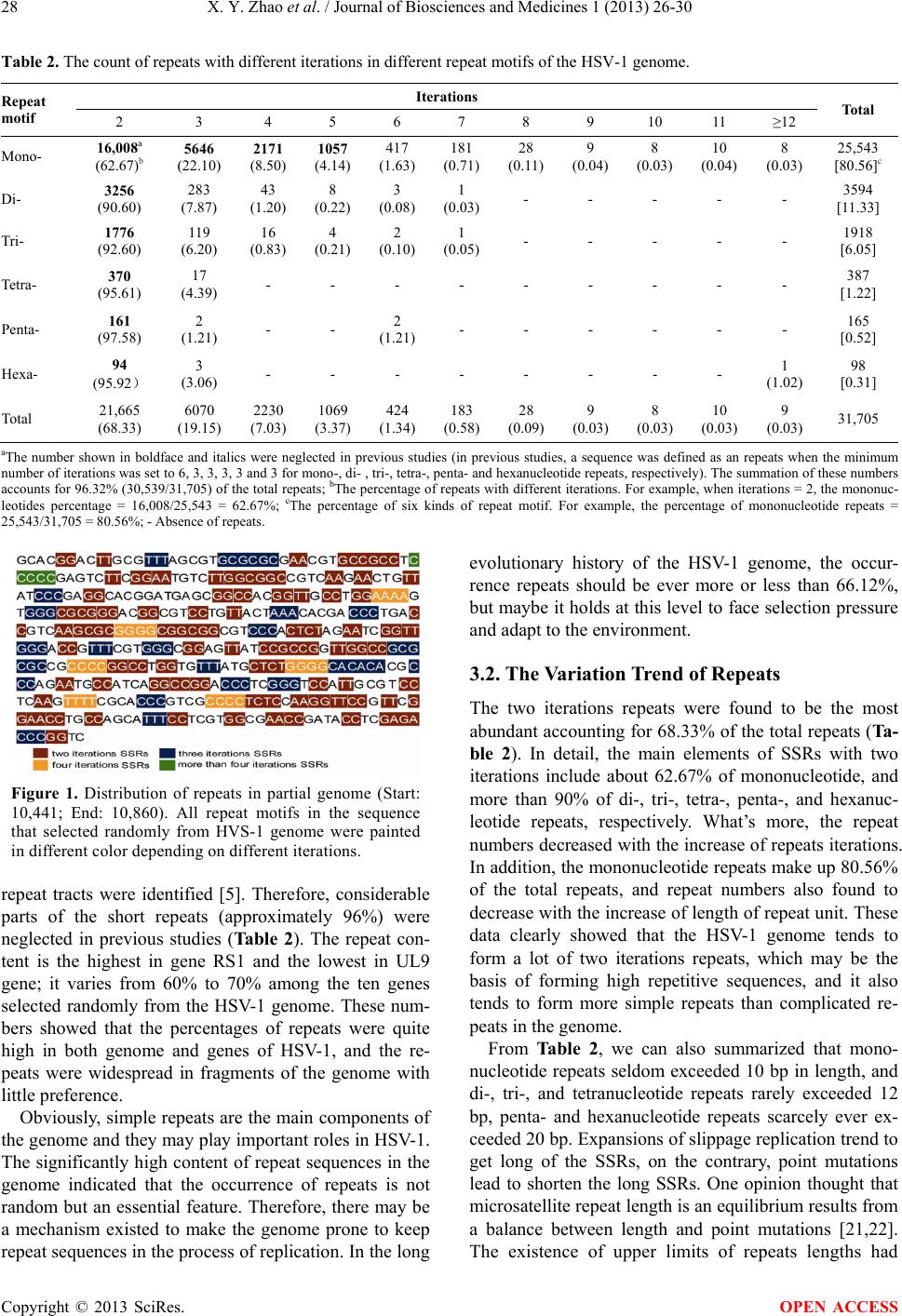 X. Y. Zhao et al. / Journal of Biosciences and Medicines 1 (2013) 26-30 Copyright © 2013 SciRes. OPEN ACCESS Table 2. The count of repeats with different iterations in different repeat motifs of the HSV-1 genome. Repeat motif Iterations Total 2 3 4 5 6 7 8 9 10 11 ≥12 Mono- 16,008a (62.67)b 5646 (22.10) 2171 (8.50) 1057 (4.14) 417 (1.63) 181 (0.71) 28 (0.11) 9 (0.04) 8 (0.03) 10 (0.04) 8 (0.03) 25,543 [80.56]c Di- 3256 (90.60) 283 (7.87) 43 (1.20) 8 (0.22) 3 (0.08) 1 (0.03) - - - - - 3594 [11.33] Tr i- 1776 (92.60) 119 (6.20) 16 (0.83) 4 (0.21) 2 (0.10) 1 (0.05) - - - - - 1918 [6.05] Tetra- 370 (95.61) 17 (4.39) - - - - - - - - - 387 [1.22] Penta- 161 (97.58) 2 (1.21) - - 2 (1.21) - - - - - - 165 [0.52] Hexa- 94 (95.92) 3 (3.06) - - - - - - - - 1 (1.02) 98 [0.31] Total 21,665 (68.33) 6070 (19.15) 2230 (7.03) 1069 (3.37) 424 (1.34) 183 (0.58) 28 (0.09) 9 (0.03) 8 (0.03) 10 (0.03) 9 (0.03) 31,705 aThe number shown in boldface and italics were neglected in previous studies (in previous studies, a sequence was defined as an repeats when the minimum number of iterations was set to 6, 3, 3, 3, 3 and 3 for mono-, di- , t ri-, tetra-, penta- and hex anucl eotid e repeat s, res pecti vely). The summation of these numbers accounts for 96.32% (3 0,539/31,705) of the total r epeats; bThe per centage of r epeats with different i terations. Fo r example, when iteration s = 2, the mononuc- leotides percentage = 16,008/25,543 = 62.67%; cThe percentage of six kinds of repeat motif. For example, the percentage of mononucleotide repeats = 25,543/31,705 = 80.56%; - Absence of repeats. Figure 1. Distribution of repeats in partial genome (Start: 10,441; End: 10,860). All repeat motifs in the sequence that selected randomly from HVS-1 genome were painted in different c olor d epe ndi n g on d i fferent it er at ion s. repeat tracts were identified [5]. Therefore, considerable parts of the short repeats (approximately 96%) were neglected in previous studies (Ta b l e 2 ). The repeat con- tent is the highest in gene RS1 and the lowest in UL9 gene; it varies from 60% to 70% among the ten genes selected randomly from the HSV-1 genome. These num- bers showed that the percentages of repeats were quite high in both genome and genes of HSV-1, and the re- peats were widespread in fragments of the genome with little preference. Obviously, simple repeats are the main components of the genome and they may play important roles in HSV-1. The significantly high content of repeat sequences in the genome indicated that the occurrence of repeats is not random but an essential feature. Therefore, there may be a mechanism existed to make the genome prone to keep repeat sequences in the process of replication. In the long evolutionary history of the HSV-1 genome, the occur- rence repeats should be ever more or less than 66.12%, but maybe it holds at this level to f ace selection pressure and adapt to the environment. 3.2. The Variation Trend of Repeats The two iterations repeats were found to be the most abundant accounting for 68.33% of the total repeats (Ta- ble 2). In detail, the main elements of SSRs with two iterations include about 62.67% of mononucleotide, and more than 90% of di-, tri-, tetra-, penta-, and hexanuc- leotide repeats, respectively. What’s more, the repeat numbers decreased with the increase of repeats iterations. In addition, the mononucleotide repeats make up 80.56% of the total repeats, and repeat numbers also found to decrease with the increase of length of repeat unit. These data clearly showed that the HSV-1 genome tends to form a lot of two iterations repeats, which may be the basis of forming high repetitive sequences, and it also tends to form more simple repeats than complicated re- peats in the genome. From Table 2, we can also summarized that mono- nucleotide repeats seldom exceeded 10 bp in length, and di-, tri-, and tetranucleotide repeats rarely exceeded 12 bp, penta- and hexanucleotide repeats scarcely ever ex- ceeded 20 bp. Expansions of slippage replication trend to get long of the SSRs, on the contrary, point mutations lead to shorten the long SSRs. One opinion thought that microsatellite repeat length is an equilibrium results from a balance between length and point mutations [21,22]. The existence of upper limits of repeats lengths had  X. Y. Zhao et al. / Journal of Biosciences and Medicines 1 (2013) 26-30 Copyright © 2013 SciRes. OPEN ACCESS another explanation that the tendency for repeat length at a locus to rise via mutation is counteracted by selection, and such selection might function through an unchara- cterized mechanism on the length of the repeat sequence itself or on gene expression as affected by the SSR se- quence at issue [23]. Microsatellite length might be an important factor in affecting mutation rate in HSV-1 genome. The count of SSRs reduces fast with the increase of iterations, and this indicated that it is relatively hard to form repeats with high iterations and long-unit, and these repeats seem to be more instability in HSV-1 genome. However, tetra-, penta- and hexanucleotide repeats were not seriously analyzed in the paper, on account of the low content, compared to mono-, di- and trinucleotide repeats. 3.3. Short Repeats and Genome Evo lutio n The number of repeats with two iterations w as 21,665 in the entire genome, accounting for 68.33% of the total repeats, and they absolutely predominate in the HSV-1 genome (Table 2). In addition, each percentage of two iterations repeats was significantly higher than 90% ex- cept mononucleotides (62.67%) in the six kinds of repeat motifs, and the summation of repeats with iterations less than 5 accounts for 94.15% of the total repeats. The two iterations repeats should be subjected to weaker selective pressure in the HSV-1 evolutionary process. High per- centage of two times repeats suggested that two times repeats may be the basis of forming repeats with high iterations in the HSV-1 genome. The genome of HSV-1 is prone to form shorter repeat sequences, and these re- peats may provide a molecular bias for fast adapting to environmental change in the HSV-1 genome. 3.4. Long Repeats of Low Frequency The exceptions were that there were only two pentanu- cleotide repeats found with six iterations and one hexa- nucleotide repeat with more than 12 iterations. The ob- servation of low frequency of longer, complicated re- peats possibly result from those repeat sequences are faced with stronger selection pressure than short simple repeats. In the long evolutionary history, the occurrence of long repeat sequences may be lost resulting from harmful or lethal to genome, this may make the genome unable to observe growing longer very quickly. Some short repeats may be neutral to the genomes and been fixed by DNA replication. The remains of long repeat sequences may be benefit to the genomes and have sur- vived from selection pressure to maintain different re- ported functions [6]. And this repeats prone mechanism possible lead the genome growing longer and longer. This mechanism may explain the observation of the mi- crosatellite polymorphisms deriving mainly from vari- ability in length rather than in the primary sequence [24] and an experimental variant of citrus exocortis viroid (CEVd) expanding its genome with a 96 nucleotide re- peat sequences [25]. This is possibly an evidence for the repeat prone mechanism with relation to genome ex- panding. 4. CONCLUSION By setting low thresholds, the frequency of repeats in- creased a lot. SSRs were distributed relatively equally throughout the genome and presented a mosaic-shaped; and repeats were quite high in both genome and genes of HSV-1. Simple repeats are the main components of the genome and they may play important roles in HSV-1. HSV-1 genome tends to form a lot of two iterations re- peats, which may be the basis of forming high repetitive sequences, and it also tends to form more simple repeats than complicated repeats in the genome. Formation of repeat may be essential for evolution of HSV-1 genome, and the results might be helpful for studying the genome structure, repeats genesis and evolution of HSV-1. 5. ACKNO WLEDGEMENTS This work was supported by the Special project for biodiversity 2012, 2013 of the Chinese ministry of environ mental protection and Ministry of Science and Technology torch plan [12C26214304703]. REFERENCES [1] Karimi, A. and MacLean, A. (2005) Replication charac- teristics of herpes simplex virus type 1 (HSV-1) recom- binants in 3 types of tissue cultures. Iranian Biomedical Journal, 9, 95-101. [2] Arduino, P.G. and Porter, S.R. (2006) Oral and perioral herpes simplex virus type 1 (HSV-1) infection: Review of its management. Oral Diseases, 12, 254-270. http://dx.doi.org/10.1111/j.1601-0825.2006.01202.x [3] Xu, F., Schillinger, J.A., Sternberg, M.R., Johnson, R.E., Lee, F.K., et al. (2002) Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988-1994. The Journal of Infectious Diseases, 185, 1019-1024. http://dx.doi.org/10.1086/340041 [4] Pebody, R.G., Andrews, N., Brown, D., Gopal, R., Melk- er, H.D., et al. (2004) The seroepidemiology of herpes simplex virus type 1 and 2 in Europe. Sexually Transmit- ted Infections, 80, 185-191. http://dx.doi.org/10.1136/sti.2003.005850 [5] Ouyang, Q., Zhao, X., Feng, H., Tian, Y., Li, D., et al. (2012) High GC content of simple sequence repeats in Herpes simplex virus type 1 genome. Gene, 499, 37-40. http://dx.doi.org/10.1016/j.gene.2012.02.049 [6] Ellegren, H. (2004) Microsatellites: Simple sequences with complex evolution. Nature Reviews Genetics, 5, 435-445. http://dx.doi.org/10.1038/nrg1348 [7] Mirkin, S.M. (2007) Expandable DNA repeats and hu-  X. Y. Zhao et al. / Journal of Biosciences and Medicines 1 (2013) 26-30 Copyright © 2013 SciRes. OPEN ACCESS man disease. Nature, 447, 932-940. http://dx.doi.org/10.1038/nature05977 [8] Ramel, C. (1997) Mini- and Microsatellites. Environ- mental Health Perspectives, 5, 781-789. [9] Jurka, J. and Pethiyagoda, C. (1995) Simple repetitive DNA sequences from primates: Compilation and analy- sis. Journal of Molecular Evolution, 40, 120-126. http://dx.doi.org/10.1007/BF00167107 [10] Sutherland, G.R. and Richard, R.L. (1995) Simple tan- dem DNA repeat and human genetic disease. Proceed- ings of the National Academy of Sciences, 92, 3636-3641. http://dx.doi.org/10.1073/pnas.92.9.3636 [11] Toth, G., Gaspari, Z. and Jurka, J. (2000) Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Research, 10, 967-981. http://dx.doi.org/10.1101/gr.10.7.967 [12] Mrazek, J., Guo, X.X. and Shah, A. (2007) Simple se- quence repeats in prokaryotic genomes. Proceedings of the National Academy of Sciences, 104, 8472-8477. http://dx.doi.org/10.1073/pnas.0702412104 [13] Zhao, X., Tian, Y., Yang, R., Feng, H., Ouyang, Q., et al. (2012) Coevolution between simple sequence repeats (SSRs) and virus genome size. BMC Genomics, 13, 435. http://dx.doi.org/10.1186/1471-2164-13-435 [14] Mudunuri, S.B. and Nagarajaram, H.A. (2007) IMEx: Imperfect microsatellite extractor. Bioinformatics, 23, 1181-1187. http://dx.doi.org/10.1093/bioinformatics/btm097 [15] Rajendrakumar, P., Biswal, A.K., Balachandran, S.M., Srinivasarao, K., Sundaram, R.M., et al. (2007) Simple sequence repeats in organellar genomes of rice: Frequen- cy and distribution in genic and intergenic regions. Bio- informatics, 23, 1-4. http://dx.doi.org/10.1093/bioinformatics/btl547 [16] Zhao, X., Tan, Z., Feng, H., Yang, R., Li, M., et al. (2011) Microsatellites in different Potyvirus genomes: Survey and analysis. Gene, 488, 52-56. http://dx.doi.org/10.1016/j.gene.2011.08.016 [17] Chen, M., Zeng, G., Tan, Z., Jiang, M., Zhang, J., et al. (2011) Compound microsatellites in complete Escheri- chia coli genomes. FEBS Letters, 585, 1072-1076. http://dx.doi.org/10.1016/j.febslet.2011.03.005 [18] Morgante, M., Hanafey, M. and Powell, W. (2002) Mi - crosatellitesare preferentially associated with nonrepeti- tive DNA in plant genomes. Nature Genetics, 30, 194- 200. http://dx.doi.org/10.1038/ng822 [19] van Lith, H.A. and van Zutphen, L.F. (1996) Characteri- zation of rabbit DNA microsatellites extracted from the EMBL nucleotide sequence database. Animal Genetics, 27, 387-395. http://dx.doi.org/10.1111/j.1365-2052.1996.tb00505.x [20] Marcotte, E.M., Pellegrini, M., Yeates, T.O. and Eisen- berg, D. (1999) A census of protein repeats. Journal of Molecular Biology, 293, 151-160. http://dx.doi.org/10.1006/jmbi.1999.3136 [21] Bell, G.I. and Jurka, J. (1997) The length distribution of perfect dimer repetitive DNA is consistent with its evolu- tion by an unbiased single-step mutation process. Journal of Molecular Evolution, 44, 414-421. http://dx.doi.org/10.1007/PL00006161 [22] Kruglyak, S., Durrett, R.T., Schug, M.D. and Aquadro, C.F. (1998) Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations. Proceedings of the National Academy of Sciences, 95, 10774-10778. http://dx.doi.org/10.1073/pnas.95.18.10774 [23] Gur-Arie, R., Cohen, C.J., Eitan, Y., Shelef, L., Haller- man, E.M. et al. (2000) Simple sequence repeats in Es- cherichia coli: Abundance, distribution, composition, and polymorphism. Genome Research, 10, 62-71. [24] Mrazek, J. (2006) Analysis of distribution indicates di- verse functions of simple sequence repeats in mycoplas- ma genomes. Molecular Biology and Evolution, 23, 1370- 1385. http://dx.doi.org/10.1093/molbev/msk023 [25] Fadda, Z., Daros, J.A., Flores, R. and Duran-Vila, N. (2003) Identification in eggplant of a variant of citrus exocortis viroid (CEVd) with a 96 nucleotide duplication in the right terminal region of the rod-like secondary structure. Virus Resear ch , 97, 145-149. http://dx.doi.org/10.1016/j.virusres.2003.08.002
|