 Pharmacology & Pharmacy, 2013, 4, 663-678 Published Online December 2013 (http://www.scirp.org/journal/pp) http://dx.doi.org/10.4236/pp.2013.49093 Open Access PP 663 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism* Yutaka Takeuchi1, Masayuki Namiki2, Yasumi Kitahara1, Setsuo Hasegawa3,4, Akihiro Ohnishi5, Nobuyuki Yasuda6, Takashi Inoue6, Richard Clark6, Kazuto Yamazaki6# 1Clinical Development, Japan/Asia Clinical Research PCU, Eisai Co., Ltd., Tokyo, Japan; 2Clinical Pharmacology, Clinical Science, SOCS CFU, Eisai Co., Ltd., Tokyo, Japan; 3Sekino Clinical Pharmacology Clinic, Tokyo, Japan; 4Present Address: Pharmaspur, Inc., Tokyo, Japan; 5Department of Laboratory Medicine, Daisan Hospital, Jikei University School of Medicine, Tokyo, Japan; 6Tsukuba Research Laboratories, Eisai Co., Ltd., Tsukuba, Japan. Email: #k5-yamazaki@hhc.eisai.co.jp Received October 16th, 2013; revised November 28th, 2013; accepted December 2nd, 2013 Copyright © 2013 Yutaka Takeuchi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT E3024 (3-but-2-ynyl-5-methyl-2-piperazin-1-yl-3,5-dihydro-4H-imidazo[4,5-d]pyridazin-4-one tosylate) is a dipeptidyl peptidase-IV (DPP-IV) inhibitor that was expected to be an antidiabetic agent. Its safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) were investigated in a randomized, double-blind, placebo-controlled, ascending single-dose study in 48 healthy Japanese male subjects. Fasted subjects were orally administered E3024 (5, 10, 20, 40, or 80 mg) or placebo. E3024 was rapidly absorbed, with tmax values ranging between 0.33 and 3 h after dosing. The mean t1/2 ranged from 5.34 to 11.68 h. AUC0-inf and Cmax increased dose-proportionately. PK-PD relationship of E3024 was evaluated by using an Imax model, indicating that plasma E3024 concentrations and inhibitory effects of plasma DPP-IV activity were well correlated. The IC50 value was calculated as 33.7 ng/mL, which was consistent with in vitro data. Thus, E3024 showed a good PK profile and inhibited DPP-IV dose-dependently. Of 30 subjects administered E3024, 12 (40%) experienced adverse events (AEs). Dose escalation to 160 mg was abandoned owing to undesired subjective/objective findings in 4 of 6 subjects receiving 40 mg and 5 of 6 subjects receiving 80 mg. The most promi- nent AE was rash, but there were no serious AEs or deaths. The maximum tolerated dose was considered to be 20 mg. We hypothesized that histamine was a cause of the rash induction, and examined blood histamine levels of normal Fischer rats treated with E3024. Blood histamine levels were increased significantly by E3024 at 500 mg/kg (p < 0.001), but not by vildagliptin or valine-pyrrolidide (DPP-IV inhibitors) at the same dose. No blood histamine increases were observed in genetically mast cell-deficient Ws/Ws rats treated with E3024 at 500 mg/kg. In in vitro assays, E3024 in- duced histamine release from normal rat peritoneal mast cells in a concentration-dependent manner, but not from baso- phils. The structure-activity relationship study suggested that a piperazine group N-linked to the 2-position of the 5,6-membered fused heterocyclic rings was a key structural element for triggering histamine release. Keywords: Dipeptidyl Peptidase-IV Inhibitor; Rash; Histamine; Structure-Activity Relationship 1. Introduction Dipeptidyl peptidase-IV (DPP-IV) inhibitors have been considered highly attractive for the treatment of type 2 diabetes, as the inhibition of DPP-IV results in an in- crease of the endogenous active glucagon-like peptide-1 (GLP-1) levels [1-5]. Of this class of drugs, sitagliptin (MK-0431) [6], vildagliptin (LAF237) [7], saxagliptin (BMS-477118) [8], alogliptin (SYR-322) [9], linagliptin *This clinical study was sponsored by Eisai Co., Ltd. Dr. Hasegawa was the director of the study site. Dr. Ohnishi was a paid consultant to Eisai and other pharmaceutical companies. #Corresponding author.  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 664 (BI-1356) [10] and anagliptin [11] have all been launched into the market for the treatment of type 2 diabetes. E3024 (3-but-2-ynyl-5-methyl-2-piperazin-1-yl-3,5- dihydro-4H-imidazo[4,5-d]pyridazin-4-one tosylate) (Fig- ure 1) with a molecular weight of 282.91 is a novel, highly selective and competitive DPP-IV inhibitor syn- thesized by Eisai Co., Ltd. [12-14]. E3024 inhibited the DPP-IV activity in human, mouse, rat and canine plasma with IC50 (concentration required for 50% of the maxi- mum inhibition) values of 0.14, 0.28, 0.40 and 0.36 mol/L, respectively. In an oral glucose tolerance test using Zucker fa/fa rats, E3024 dose-dependently in- creased plasma insulin levels and reduced the area under the curve (AUC) of delta blood glucose at doses of 1 and 3 mg/kg. E3024 had no effect on fasting blood glucose levels in normal rats at doses of 1 or 10 mg/kg. These non-clinical data had suggested that E3024 would be a novel antidiabetic agent in the treatment of postprandial hyperglycemia with a low risk of causing hypoglycemia. The objectives of the present studies were: 1) to evalu- ate the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) after single oral dose of E3024 in healthy Japanese male subjects, and 2) to examine possible mechanisms of rash development observed in this clinical trial, using normal and genetically mast cell- deficient rats. 2. Materials and Methods 2.1. Clinical Study This study was conducted at Sekino Clinical Pharmacol- ogy Clinic, Tokyo, Japan, in accordance with the ethical principles of the Declaration of Helsinki, good clinical practice in Japan, and International Conference on Har- monization guidelines. The clinical study protocol and informed consent documents were approved by the insti- tutional review board of Sekino Clinical Pharmacology Clinic. Informed consent was obtained from all subjects in writing before implementation of any study-related procedures. 2.1.1. Study Design This study was performed as a randomized, double-blind, placebo-controlled, escalating single-dose study. Differ- ent groups of eight subjects each were orally adminis- tered single doses of E3024 (5, 10, 20, 40, or 80 mg, n = 6) or placebo (n = 2 for 5, 10, 80 mg; n = 6 for 20, 40 mg) after an overnight fast of 10 h. E3024 was supplied by Eisai Co., Ltd. E3024 was ad- ministered in film-coated tablets containing 1, 10, or 40 mg of E3024. Placebo was administered in visually matching tablets. 2.1.2. Subj ects Healthy Japanese male subjects between 20 and 39 years of age and with body mass index (BMI) of 18.5 to 25.0 kg/m2 were eligible for participation in this study. Sub- jects were excluded if they had a known history of any significant drug or food allergy, a significant organ dys- function, or any clinically significant deviation from normal in medical history, physical examination findings, vital signs, electrocardiogram, or laboratory test results. Subjects with gastrointestinal, hepatic, renal, respiratory, or cardiovascular diseases; congenital metabolic disorder; Figure 1. Chemical structures of E3024, ER-319441-15, ER-319433-15, and ER-463809-15. Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 665 a positive test result for hepatitis B surface antigen, hepa- titis C antibody or human immunodeficiency virus; or alcohol or drug abuse (or a positive urine drug test result at screening) were excluded from participation. Subjects were excluded if they had a known history of any gas- trointestinal surgery that could impact upon absorption of the study drug. Subjects were also excluded if they had experienced a weight change >10% from screening to baseline. Furthermore, any subject was excluded who had received blood within three months or donated blood (400 mL within three months or 200 mL within 30 days of study start), or ingested any investigational medication within four months before study start. Subjects were pro- hibited from any prescription drugs and over-the-counter (OTC) acid controllers within 30 days prior to and during the study, and other OTC medications within seven days prior to and during the study. 2.1.3. Procedures Screening procedures, including medical history taking, physical examination, 12-lead electrocardiography (ECG), clinical laboratory evaluations, vital signs measurement, and urine drug screening, were performed from 30 days before study drug administration along with the assess- ment of inclusion/exclusion criteria. Eligible subjects were admitted to the study site on the day prior to dosing for base line evaluations. Subjects were required to ab- stain from food and beverages, except water, for at least 10 h prior to check-in. After the check-in evaluation was completed, subjects were provided with an appropriate meal(s); thereafter, they were required to fast (abstain from food and fluids, except water) overnight for at least 10 h prior to drug administration on the following day. Subjects took the study drug with 200 mL of water in a fasted state. Water was allowed ad libitum, except from 2 h before dosing to 1 h after dosing. Subjects were re- quired to abstain from food up to 4.5 h after dosing. Subjects received a standardized meal at 4.5 (lunch) and 10.5 (dinner) h after dosing to assess the pharmacody- namic effects of E3024 on GLP-1, insulin, C-peptide, glucagon, and glucose. The total energy of each meal was 800 kcal, with a nutrient breakdown of 25% fat, 15% protein, and 60% carbohydrate. Subsequent meals were provided as per the regular meal schedule at the site. Subjects were to maintain an upright (seated or standing) position for at least 4.5 h following administration of the study drug. 2.1.4. Ph ar ma c oki netic Assessments Blood samples were collected at 0 (pre-dose), 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, and 96 h after ad- ministration of the study drug for determination of E3024 in human plasma. Blood samples (3 mL each) were col- lected from a cutaneous vein in the forearm into a so- dium-heparinized tube. Samples were centrifuged (4˚C at 1500× g for 15 min) to obtain plasma. Urine samples were collected before dosing and at the following inter- vals: 0 to 6 h, 6 to 12 h, 12 to 24 h, 24 to 48 h, 48 to 72 h, and 72 to 96 h after dosing, for determination of E3024 in human urine. Plasma and urine samples were stored at −20˚C until sample analysis. Analysis was performed by Bioanalysis Section, Clinical Research Center at Eisai Co., Ltd. (Tokyo, Ja- pan). For quantitative determination of E3024, plasma and urine samples were analyzed by a validated liquid chromatographic-tandem mass spectrometry (LC/MS/MS) method. This method was based on solid-phase extrac- tion using Empore extraction disk plates (3M, St. Paul, MN) in a 96-well format, with 0.02 mL (plasma) or 0.005 mL (urine) eluent samples injected into the LC/ MS/MS. Pharmacokinetic parameters were calculated from plasma and urine concentrations of E3024 by model-in- dependent analysis using WinNonlin Professional ver- sion 4.1 (Pharsight Corp., Mountain View, CA). The dose-proportionality of maximum observed concentra- tion (Cmax) and area under the plasma concentration-time curve from 0 to infinity (AUC0-inf) obtained from model- independent analysis was assessed both visually and us- ing a power model (Y = αXβ; X, dose; Y, Cmax or AUC0-inf). Dose proportionality was assessed based on whether 95% confidence intervals (CIs) of β lay within the range from 0.7 to 1.3 [15]. 2.1.5. Pharmacodynamic Assessments For DPP-IV activity assay, blood samples were collected before dosing and at 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h after dosing. For active GLP-1 and glucagon, blood collection was performed before the meals provided at 4.5 and 10.5 h after dosing (lunch and dinner, respec- tively); and at 0.33, 0.67, 1, 1.5, 2, and 3 h after the meals provided at 4.5 and 10.5 h after dosing. Blood (2 mL) was withdrawn into tubes containing ethylenedia- minetetraacetic acid (EDTA) alone (plasma DPP-IV ac- tivity assay and plasma active GLP-1) or EDTA plus aprotinin (plasma glucagon). For serum insulin, C-pep- tide and glucose, blood (3 mL) was withdrawn into se- rum separator tubes. For active GLP-1 samples, 50 μL of DPP-IV inhibitor solution (Linco Research, Inc., St. Charles, MO) was added to each tube within 30 sec after collection, and the tubes were gently mixed and placed on ice water immediately. After centrifugation, plasma and serum samples were stored at −20˚C or below until assayed. Pharmacodynamic parameters were measured at Mi- tsubishi Kagaku Bio-clinical Laboratories, Inc. (now, Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 666 Mitsubishi Chemical Medience Corp., Tokyo, Japan). DPP-IV enzyme activity was determined via incubation of 20-μL EDTA-treated human plasma (5-fold dilution in assay) with the substrate, glycyl-L-proline 7-amido-4- methyl-coumarin hydrobromide (H-Gly-Pro-AMC·HBr; 0.08 mmol/L in assay) at room temperature for 10 min by measurement of the release of 7-amino-4-methyl-cou- marin with a multifunctional microplate reader (excita- tion 360 nm; emission 465 nm). Enzyme activity (1 mU/mL) was defined as the amount of enzyme required to degrade 1 nmole of substrate per min in 1 mL of reac- tion solution (mU/mL = nmol/mL·min). The range of reliable quantitation was 0.05 to 20.0 mU/mL. Active GLP-1 (GLP-1-[7-36]amide and GLP-1-[7-37]) was as- sayed with an enzyme-linked immunosorbent assay (ELISA) kit (Linco Research, Inc.). The lower limit of reliable quantitation was estimated to be 5.00 pmol/L. If concentrations could be calculated from measured fluo- rescence intensity, the values were used in the analysis even if less than 5.00 pmol/L. If concentrations could not be calculated, the measured values were defined as zero. Insulin, C-peptide, glucagon, and glucose concentrations were measured by standard methods in the laboratory, i.e. immunoradiometric assay for insulin, radioimmunoassay for glucagon and C-peptide, and enzymatic assay for glucose. For pharmacodynamic parameters, the values meas- ured and changes from baseline at each time point were summarized using descriptive statistics by dose. Percent inhibition of plasma DPP-IV activity for each subject was plotted against plasma E3024 concentration, and an Imax model (effect = Imax·C/(IC50 + C); where C is plasma E3024 concentration) was used to determine the IC50 values. 2.1.6. Sa f ety Assessmen t s The following data were collected during the study to assess safety: physical examination findings, vital signs (blood pressure, pulse rate, respiratory rate, and body temperature), body weight, 12-lead ECGs, and clinical laboratory parameters (hematology, biochemistry and urinalysis). In the case of a clinically significant abnor- mal value, the evaluation was to be repeated until the value was within an acceptable or normal range. AEs were to be followed to resolution. From subjects who had rash in the 40-mg group, blood samples were collected for measurement of non-specific immunoglobulin E (IgE) at 24 and 96 h, and drug-in- duced lymphocyte stimulation test (DLST) at 96 h after dosing. In the same way, from subjects who had rash in the 80-mg group, blood samples were collected for measurement of IgE, serotonin, histamine, and substance P at onset of rash (the nearest pharmacokinetic time point), 24 and 96 h after dosing. Blood samples, which were collected for clinical laboratory tests the day before dosing, were also used to obtain baseline data for IgE, histamine and substance P. These additional assays were performed at the study site for IgE, and at Mitsubishi Kagaku Bio-clinical Laboratories, Inc. for DLST, sero- tonin, histamine and substance P. The numbers of subjects with AEs were tabulated. For clinical laboratory parameters (except urinalysis), vital signs, body weight, and 12-lead ECG parameters, the values measured and changes from baseline at each time point were summarized using descriptive statistics by dose. For urinalysis, cross tables were prepared. 2.2. Non-Clinical in Vivo and in Vitro Studies 2.2.1. Chem i c al s E3024, vildagliptin, valine-pyrrolidide (a DPP-IV in- hibitor [16]), ER-319441-15 (trifluoroacetate salt form of ER-319441 (2-(3-amino-piperidin-1-yl)-3-but-2-ynyl-5- methyl-3,5-dihydro-4H-imidazo[4,5-d]pyridazin-4-one)), ER-319433-15 (trifluoroacetate salt form of ER-319433 (2-{[7-(bu t-2-yn-1- yl)-1-[(4 -cyanoph enyl)methyl]-6 -oxo- 8-(piperazin-1-yl)-6,7-dihydro-1H-purin-2-yl]methyl} benzamide)), and ER-463809-15 (trifluoroacetate salt form of ER-463809 (2-({8-(3-aminopiperidin-1-yl)-7-(but-2- yn-1-yl)-1-[(4-cyanophenyl)methyl]-6-oxo-6,7-dihydro- 1H-purin-2-yl}methyl)benzamide)) were synthesized in our laboratories. Chemical structures of ER-319441-15, ER-319433-15 and ER-463809-15 are indicated in Fig- ure 1. A23187 (a calcium ionophore) and dimethyl sul- foxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Methylcellulose (MC) was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). 2.2.2. Animals Five-week-old normal male Fischer (F344/Jcl) rats were purchased from CLEA Japan, Inc. (Tokyo, Japan). Five- week-old male Slc:WsRC-Ws/Ws (Ws/Ws) and Slc: WsRC-+/+ (+/+; wild-type homozygous) rats were ob- tained from Japan SLC, Inc. (Hamamatsu, Japan). The rats were provided with a commercial diet (MF; Oriental Yeast, Tokyo, Japan) and water ad libitum, and were kept under conventional conditions of controlled tem- perature, humidity and lighting (22 2˚C, 55 5% and a 12-hr light/dark cycle with lights on at 07:00 a.m.). All procedures were conducted according to the Eisai Ani- mal Care Committee’s guideline. 2.2.3. Determination of Plasma Compound Concentrations in Rats Compounds were suspended in 0.5% MC, and adminis- tered to Fischer rats aged eight weeks orally (10 mL/kg). Open Access PP 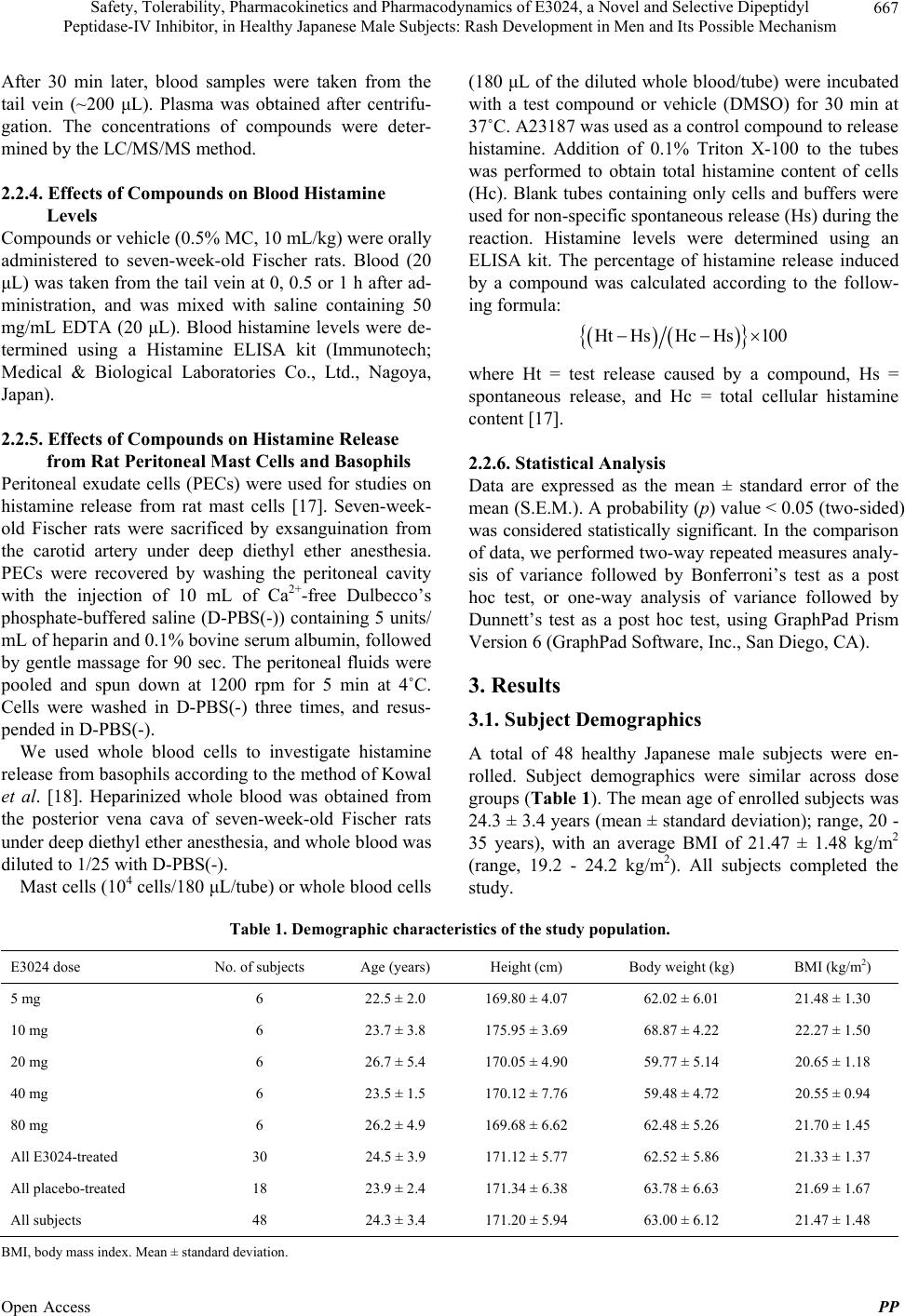 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism Open Access PP 667 (180 μL of the diluted whole blood/tube) were incubated with a test compound or vehicle (DMSO) for 30 min at 37˚C. A23187 was used as a control compound to release histamine. Addition of 0.1% Triton X-100 to the tubes was performed to obtain total histamine content of cells (Hc). Blank tubes containing only cells and buffers were used for non-specific spontaneous release (Hs) during the reaction. Histamine levels were determined using an ELISA kit. The percentage of histamine release induced by a compound was calculated according to the follow- ing formula: After 30 min later, blood samples were taken from the tail vein (~200 μL). Plasma was obtained after centrifu- gation. The concentrations of compounds were deter- mined by the LC/MS/MS method. 2.2.4. Effects of Compounds on Blood Histamine Levels Compounds or vehicle (0.5% MC, 10 mL/kg) were orally administered to seven-week-old Fischer rats. Blood (20 μL) was taken from the tail vein at 0, 0.5 or 1 h after ad- ministration, and was mixed with saline containing 50 mg/mL EDTA (20 μL). Blood histamine levels were de- termined using a Histamine ELISA kit (Immunotech; Medical & Biological Laboratories Co., Ltd., Nagoya, Japan). HtHsHc Hs100 where Ht = test release caused by a compound, Hs = spontaneous release, and Hc = total cellular histamine content [17]. 2.2.5. Effects of Compounds on Histamine Release from Rat Peritoneal Mast Cells and Basophils 2.2.6. Stati s tical Analysi s Peritoneal exudate cells (PECs) were used for studies on histamine release from rat mast cells [17]. Seven-week- old Fischer rats were sacrificed by exsanguination from the carotid artery under deep diethyl ether anesthesia. PECs were recovered by washing the peritoneal cavity with the injection of 10 mL of Ca2+-free Dulbecco’s phosphate-buffered saline (D-PBS(-)) containing 5 units/ mL of heparin and 0.1% bovine serum albumin, followed by gentle massage for 90 sec. The peritoneal fluids were pooled and spun down at 1200 rpm for 5 min at 4˚C. Cells were washed in D-PBS(-) three times, and resus- pended in D-PBS(-). Data are expressed as the mean ± standard error of the mean (S.E.M.). A probability (p) value < 0.05 (two-sided) was considered statistically significant. In the comparison of data, we performed two-way repeated measures analy- sis of variance followed by Bonferroni’s test as a post hoc test, or one-way analysis of variance followed by Dunnett’s test as a post hoc test, using GraphPad Prism Version 6 (GraphPad Software, Inc., San Diego, CA). 3. Results 3.1. Subject Demographics We used whole blood cells to investigate histamine release from basophils according to the method of Kowal et al. [18]. Heparinized whole blood was obtained from the posterior vena cava of seven-week-old Fischer rats under deep diethyl ether anesthesia, and whole blood was diluted to 1/25 with D-PBS(-). A total of 48 healthy Japanese male subjects were en- rolled. Subject demographics were similar across dose groups (Table 1). The mean age of enrolled subjects was 24.3 ± 3.4 years (mean ± standard deviation); range, 20 - 35 years), with an average BMI of 21.47 ± 1.48 kg/m2 (range, 19.2 - 24.2 kg/m2). All subjects completed the study. Mast cells (104 cells/180 μL/tube) or whole blood cells Table 1. Demographic characteristics of the study population. E3024 dose No. of subjects Age (years) Height (cm) Body weight (kg) BMI (kg/m2) 5 mg 6 22.5 ± 2.0 169.80 ± 4.07 62.02 ± 6.01 21.48 ± 1.30 10 mg 6 23.7 ± 3.8 175.95 ± 3.69 68.87 ± 4.22 22.27 ± 1.50 20 mg 6 26.7 ± 5.4 170.05 ± 4.90 59.77 ± 5.14 20.65 ± 1.18 40 mg 6 23.5 ± 1.5 170.12 ± 7.76 59.48 ± 4.72 20.55 ± 0.94 80 mg 6 26.2 ± 4.9 169.68 ± 6.62 62.48 ± 5.26 21.70 ± 1.45 All E3024-treated 30 24.5 ± 3.9 171.12 ± 5.77 62.52 ± 5.86 21.33 ± 1.37 All placebo-treated 18 23.9 ± 2.4 171.34 ± 6.38 63.78 ± 6.63 21.69 ± 1.67 All subjects 48 24.3 ± 3.4 171.20 ± 5.94 63.00 ± 6.12 21.47 ± 1.48 B MI, body mass index. Mean ± standard deviation. 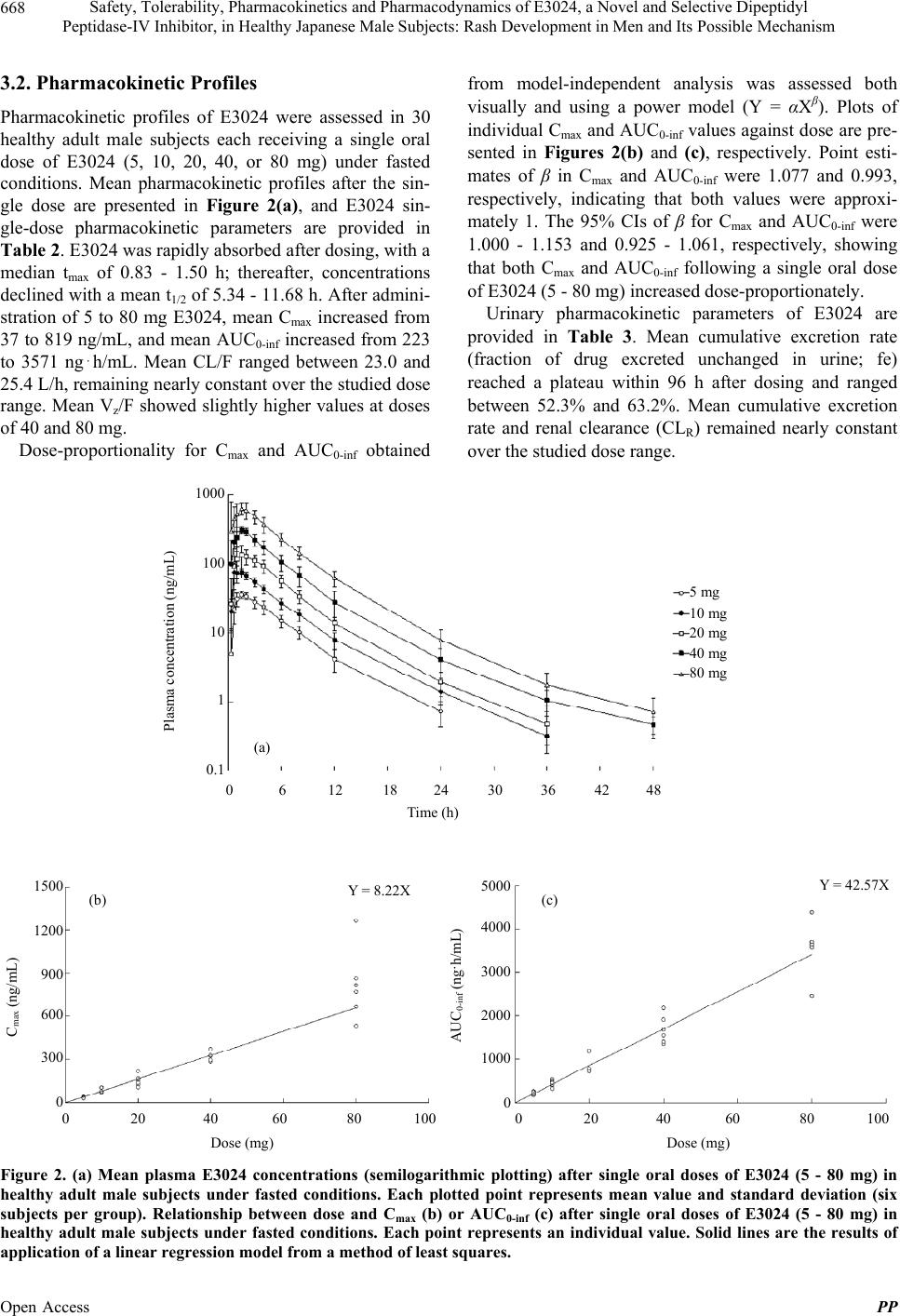 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 668 3.2. Pharmacokinetic Profiles Pharmacokinetic profiles of E3024 were assessed in 30 healthy adult male subjects each receiving a single oral dose of E3024 (5, 10, 20, 40, or 80 mg) under fasted conditions. Mean pharmacokinetic profiles after the sin- gle dose are presented in Figure 2(a), and E3024 sin- gle-dose pharmacokinetic parameters are provided in Table 2. E3024 was rapidly absorbed after dosing, with a median tmax of 0.83 - 1.50 h; thereafter, concentrations declined with a mean t1/2 of 5.34 - 11.68 h. After admini- stration of 5 to 80 mg E3024, mean Cmax increased from 37 to 819 ng/mL, and mean AUC0-inf increased from 223 to 3571 ng⋅h/mL. Mean CL/F ranged between 23.0 and 25.4 L/h, remaining nearly constant over the studied dose range. Mean Vz/F showed slightly higher values at doses of 40 and 80 mg. Dose-proportionality for Cma x and AUC0-inf obtained from model-independent analysis was assessed both visually and using a power model (Y = αXβ). Plots of individual Cmax and AUC0-inf values against dose are pre- sented in Figures 2(b) and (c), respectively. Point esti- mates of β in Cmax and AUC0-inf were 1.077 and 0.993, respectively, indicating that both values were approxi- mately 1. The 95% CIs of β for Cmax and AUC0-inf were 1.000 - 1.153 and 0.925 - 1.061, respectively, showing that both Cmax and AUC0-inf following a single oral dose of E3024 (5 - 80 mg) increased dose-proportionately. Urinary pharmacokinetic parameters of E3024 are provided in Table 3. Mean cumulative excretion rate (fraction of drug excreted unchanged in urine; fe) reached a plateau within 96 h after dosing and ranged between 52.3% and 63.2%. Mean cumulative excretion rate and renal clearance (CLR) remained nearly constant over the studied dose range. BC A Fig. 1000 100 10 1 0.1 06 12 18 24303642 48 5 mg 10 mg 20 mg 40 mg 80 mg Plasma concentration (ng/mL) (a) 0 Time (h) 20 40 60 80100020 4060 80100 Dose (mg) Dose (mg) (b) (c) 0 300 600 900 1200 1500 5000 4000 3000 2000 1000 0 Y = 8.22X Y = 42.57X C max (ng/mL) AUC 0-inf (ng·h/mL) Figure 2. (a) Mean plasma E3024 concentrations (semilogarithmic plotting) after single oral doses of E3024 (5 - 80 mg) in healthy adult male subjects under fasted conditions. Each plotted point represents mean value and standard deviation (six subjects per group). Relationship between dose and Cmax (b) or AUC0-inf (c) after single oral doses of E3024 (5 - 80 mg) in healthy adult male subjects under fasted conditions. Each point represents an individual value. Solid lines are the results of application of a linear regression model from a method of least squares. Open Access PP 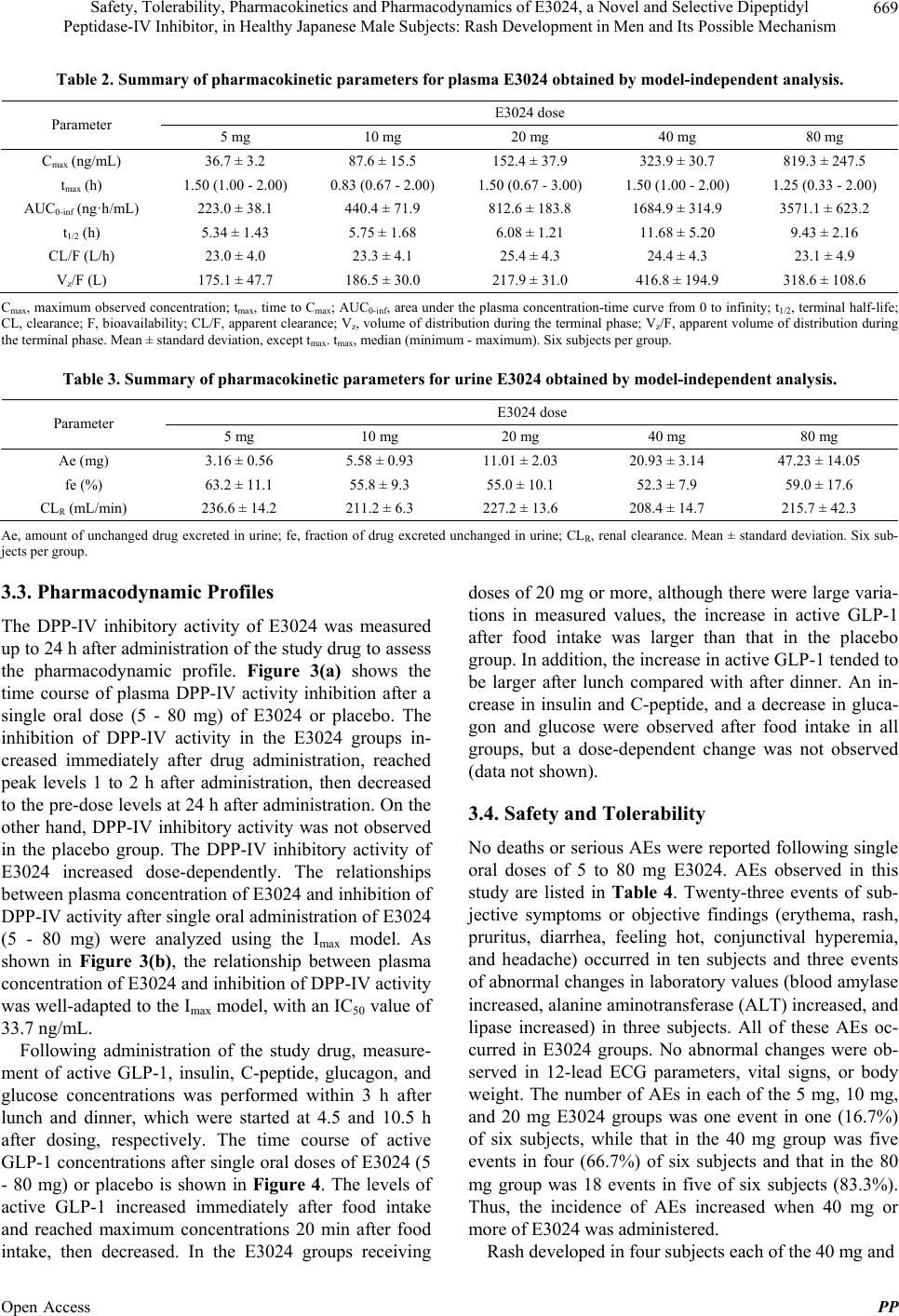 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 669 Table 2. Summary of pharmacokinetic parameters for plasma E3024 obtained by model-independent analysis. E3024 dose Parameter 5 mg 10 mg 20 mg 40 mg 80 mg Cmax (ng/mL) 36.7 ± 3.2 87.6 ± 15.5 152.4 ± 37.9 323.9 ± 30.7 819.3 ± 247.5 tmax (h) 1.50 (1.00 - 2.00) 0.83 (0.67 - 2.00) 1.50 (0.67 - 3.00) 1.50 (1.00 - 2.00) 1.25 (0.33 - 2.00) AUC0-inf (ng·h/mL) 223.0 ± 38.1 440.4 ± 71.9 812.6 ± 183.8 1684.9 ± 314.9 3571.1 ± 623.2 t1/2 (h) 5.34 ± 1.43 5.75 ± 1.68 6.08 ± 1.21 11.68 ± 5.20 9.43 ± 2.16 CL/F (L/h) 23.0 ± 4.0 23.3 ± 4.1 25.4 ± 4.3 24.4 ± 4.3 23.1 ± 4.9 Vz/F (L) 175.1 ± 47.7 186.5 ± 30.0 217.9 ± 31.0 416.8 ± 194.9 318.6 ± 108.6 Cmax, maximum observed concentration; tmax, time to Cmax; AUC0-inf, area under the plasma concentration-time curve from 0 to infinity; t1/2, terminal half-life; CL, clearance; F, bioavailability; CL/F, apparent clearance; Vz, volume of distribution during the terminal phase; Vz/F, apparent volume of distribution during the terminal phase. Mean ± standard deviation, except tmax. tmax, median (minimum - maximum). Six subjects per group. Table 3. Summary of pharmacokinetic parameters for urine E3024 obtained by model-independent analysis. E3024 dose Parameter 5 mg 10 mg 20 mg 40 mg 80 mg Ae (mg) 3.16 ± 0.56 5.58 ± 0.93 11.01 ± 2.03 20.93 ± 3.14 47.23 ± 14.05 fe (%) 63.2 ± 11.1 55.8 ± 9.3 55.0 ± 10.1 52.3 ± 7.9 59.0 ± 17.6 CLR (mL/min) 236.6 ± 14.2 211.2 ± 6.3 227.2 ± 13.6 208.4 ± 14.7 215.7 ± 42.3 Ae, amount of unchanged drug excreted in urine; fe, fraction of drug excreted unchanged in urine; CLR, renal clearance. Mean ± standard deviation. Six sub- jects per group. 3.3. Pharmacodynamic Profiles The DPP-IV inhibitory activity of E3024 was measured up to 24 h after administration of the study drug to assess the pharmacodynamic profile. Figure 3(a) shows the time course of plasma DPP-IV activity inhibition after a single oral dose (5 - 80 mg) of E3024 or placebo. The inhibition of DPP-IV activity in the E3024 groups in- creased immediately after drug administration, reached peak levels 1 to 2 h after administration, then decreased to the pre-dose levels at 24 h after administration. On the other hand, DPP-IV inhibitory activity was not observed in the placebo group. The DPP-IV inhibitory activity of E3024 increased dose-dependently. The relationships between plasma concentration of E3024 and inhibition of DPP-IV activity after single oral administration of E3024 (5 - 80 mg) were analyzed using the Imax model. As shown in Figure 3(b), the relationship between plasma concentration of E3024 and inhibition of DPP-IV activity was well-adapted to the Imax model, with an IC50 value of 33.7 ng/mL. Following administration of the study drug, measure- ment of active GLP-1, insulin, C-peptide, glucagon, and glucose concentrations was performed within 3 h after lunch and dinner, which were started at 4.5 and 10.5 h after dosing, respectively. The time course of active GLP-1 concentrations after single oral doses of E3024 (5 - 80 mg) or placebo is shown in Figure 4. The levels of active GLP-1 increased immediately after food intake and reached maximum concentrations 20 min after food intake, then decreased. In the E3024 groups receiving doses of 20 mg or more, although there were large varia- tions in measured values, the increase in active GLP-1 after food intake was larger than that in the placebo group. In addition, the increase in active GLP-1 tended to be larger after lunch compared with after dinner. An in- crease in insulin and C-peptide, and a decrease in gluca- gon and glucose were observed after food intake in all groups, but a dose-dependent change was not observed (data not shown). 3.4. Safety and Tolerability No deaths or serious AEs were reported following single oral doses of 5 to 80 mg E3024. AEs observed in this study are listed in Table 4. Twenty-three events of sub- jective symptoms or objective findings (erythema, rash, pruritus, diarrhea, feeling hot, conjunctival hyperemia, and headache) occurred in ten subjects and three events of abnormal changes in laboratory values (blood amylase increased, alanine aminotransferase (ALT) increased, and lipase increased) in three subjects. All of these AEs oc- curred in E3024 groups. No abnormal changes were ob- served in 12-lead ECG parameters, vital signs, or body weight. The number of AEs in each of the 5 mg, 10 mg, and 20 mg E3024 groups was one event in one (16.7%) of six subjects, while that in the 40 mg group was five events in four (66.7%) of six subjects and that in the 80 mg group was 18 events in five of six subjects (83.3%). Thus, the incidence of AEs increased when 40 mg or more of E3024 was administered. R ash developed in four subjects each of the 40 mg and Open Access PP 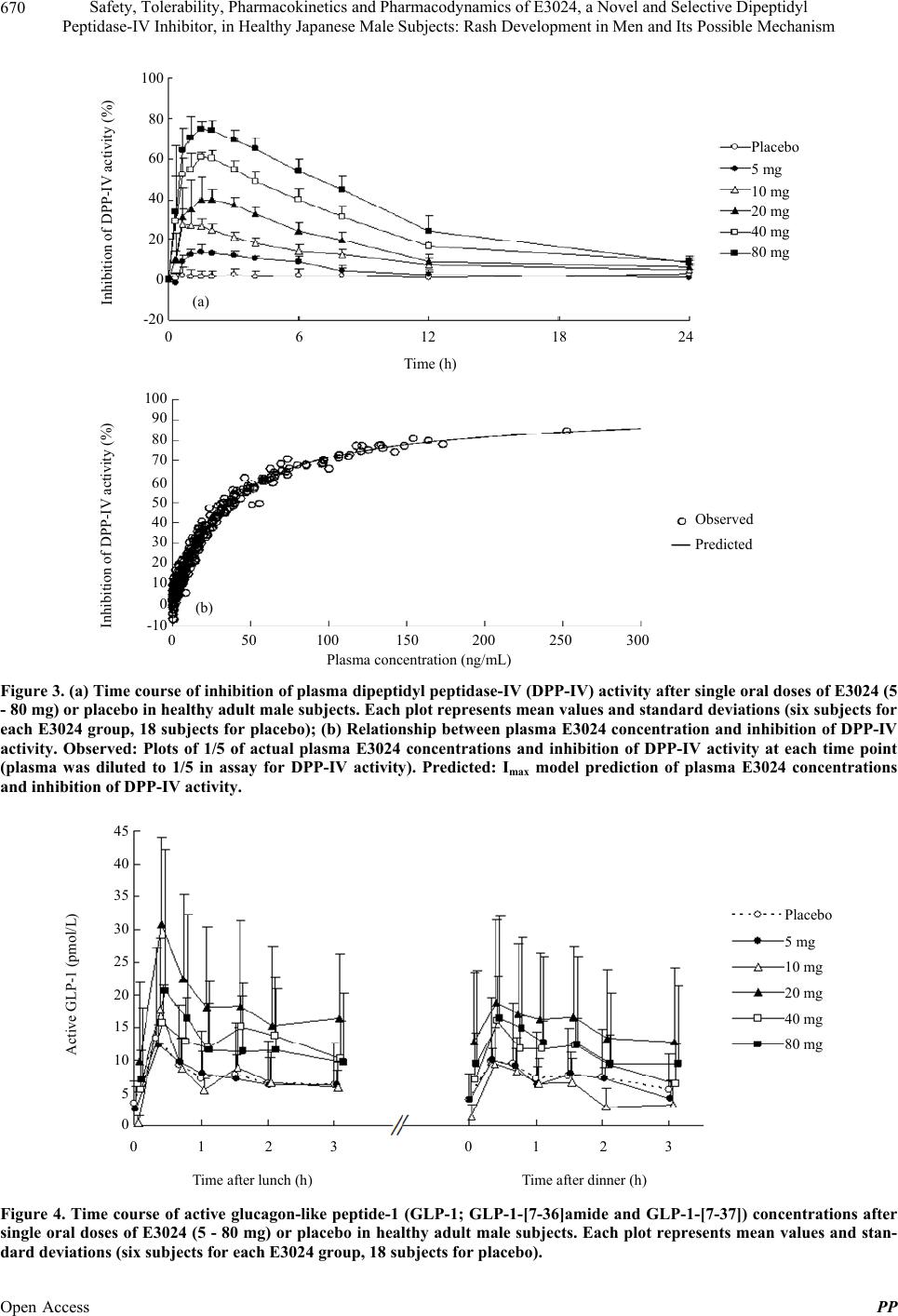 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 670 100 20 0 -20 0 6 121824 5 mg 10 mg 20 mg 40 mg 80 mg Inhibition of DPP-IV activity (%) (a) Time (h) 40 60 80 Placebo 100 20 0 -10 0 Inhibition of DPP-IV activity (%) (b) Plasma concentration (ng/mL) 40 60 80 Predicted 50 100 150 250200 300 90 70 50 30 10 Observed Figure 3. (a) Time course of inhibition of plasma dipeptidyl peptidase-IV (DPP-IV) activity after single oral doses of E3024 (5 - 80 mg) or placebo in healthy adult male subjects. Each plot represents mean values and standard deviations (six subjects for each E3024 group, 18 subjects for placebo); (b) Relationship between plasma E3024 concentration and inhibition of DPP-IV activity. Observed: Plots of 1/5 of actual plasma E3024 concentrations and inhibition of DPP-IV activity at each time point (plasma was diluted to 1/5 in assay for DPP-IV activity). Predicted: Imax model prediction of plasma E3024 concentrations and inhibition of DPP-IV activity. 10 5 0 0 5 mg 10 mg 20 mg 40 mg 80 mg Active GLP-1 (pmol/L) Time after lunch (h) 15 20 Placebo 25 35 45 30 40 1 2 3 012 3 Time after dinner (h) Figure 4. Time course of active glucagon-like peptide-1 (GLP-1; GLP-1-[7-36]amide and GLP-1-[7-37]) concentrations after single oral doses of E3024 (5 - 80 mg) or placebo in healthy adult male subjects. Each plot represents mean values and stan- ard deviations (six subjects for each E3024 group, 18 subjects for placebo). d Open Access PP 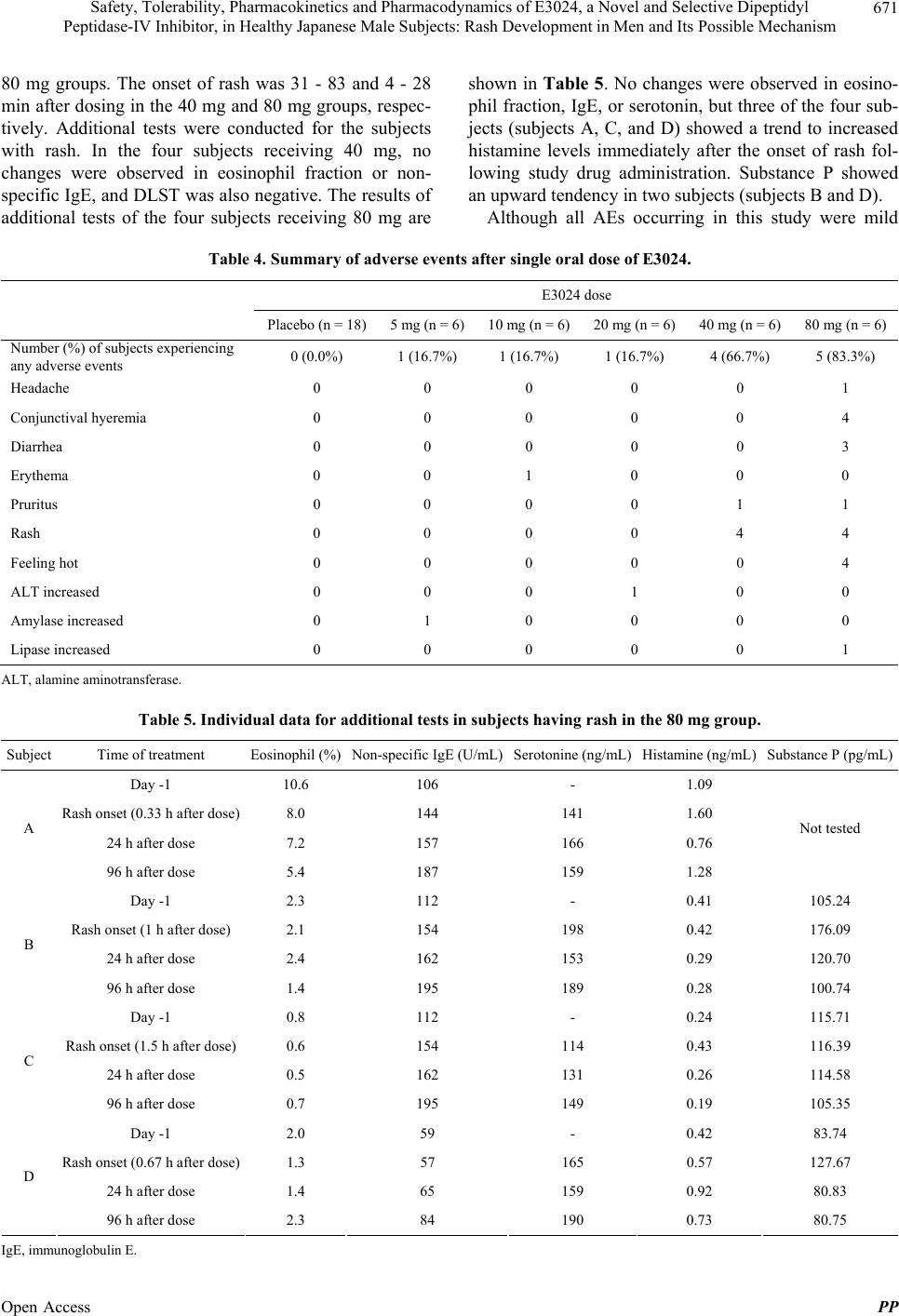 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 671 80 mg groups. The onset of rash was 31 - 83 and 4 - 28 min after dosing in the 40 mg and 80 mg groups, respec- tively. Additional tests were conducted for the subjects with rash. In the four subjects receiving 40 mg, no changes were observed in eosinophil fraction or non- specific IgE, and DLST was also negative. The results of additional tests of the four subjects receiving 80 mg are shown in Table 5. No changes were observed in eosino- phil fraction, IgE, or serotonin, but three of the four sub- jects (subjects A, C, and D) showed a trend to increased histamine levels immediately after the onset of rash fol- lowing study drug administration. Substance P showed an upward tendency in two subjects (subjects B and D). Although all AEs occurring in this study were mild Table 4. Summary of adverse events after single oral dose of E3024. E3024 dose Placebo (n = 18)5 mg (n = 6)10 mg (n = 6)20 mg (n = 6)40 mg (n = 6) 80 mg (n = 6) Number (%) of subjects experiencing any adverse events 0 (0.0%) 1 (16.7%) 1 (16.7%) 1 (16.7%) 4 (66.7%) 5 (83.3%) Headache 0 0 0 0 0 1 Conjunctival hyeremia 0 0 0 0 0 4 Diarrhea 0 0 0 0 0 3 Erythema 0 0 1 0 0 0 Pruritus 0 0 0 0 1 1 Rash 0 0 0 0 4 4 Feeling hot 0 0 0 0 0 4 ALT increased 0 0 0 1 0 0 Amylase increased 0 1 0 0 0 0 Lipase increased 0 0 0 0 0 1 ALT, alamine aminotransferase. Table 5. Individual data for additional tests in subjects having rash in the 80 mg group. Subject Time of treatment Eosinophil (%) Non-specific IgE (U/mL)Serotonine (ng/mL)Histamine (ng/mL) Substance P (pg/mL) Day -1 10.6 106 - 1.09 Rash onset (0.33 h after dose) 8.0 144 141 1.60 24 h after dose 7.2 157 166 0.76 A 96 h after dose 5.4 187 159 1.28 Not tested Day -1 2.3 112 - 0.41 105.24 Rash onset (1 h after dose)2.1 154 198 0.42 176.09 24 h after dose 2.4 162 153 0.29 120.70 B 96 h after dose 1.4 195 189 0.28 100.74 Day -1 0.8 112 - 0.24 115.71 Rash onset (1.5 h after dose)0.6 154 114 0.43 116.39 24 h after dose 0.5 162 131 0.26 114.58 C 96 h after dose 0.7 195 149 0.19 105.35 Day -1 2.0 59 - 0.42 83.74 Rash onset (0.67 h after dose) 1.3 57 165 0.57 127.67 24 h after dose 1.4 65 159 0.92 80.83 D 96 h after dose 2.3 84 190 0.73 80.75 I gE, immunoglobulin E. Open Access PP 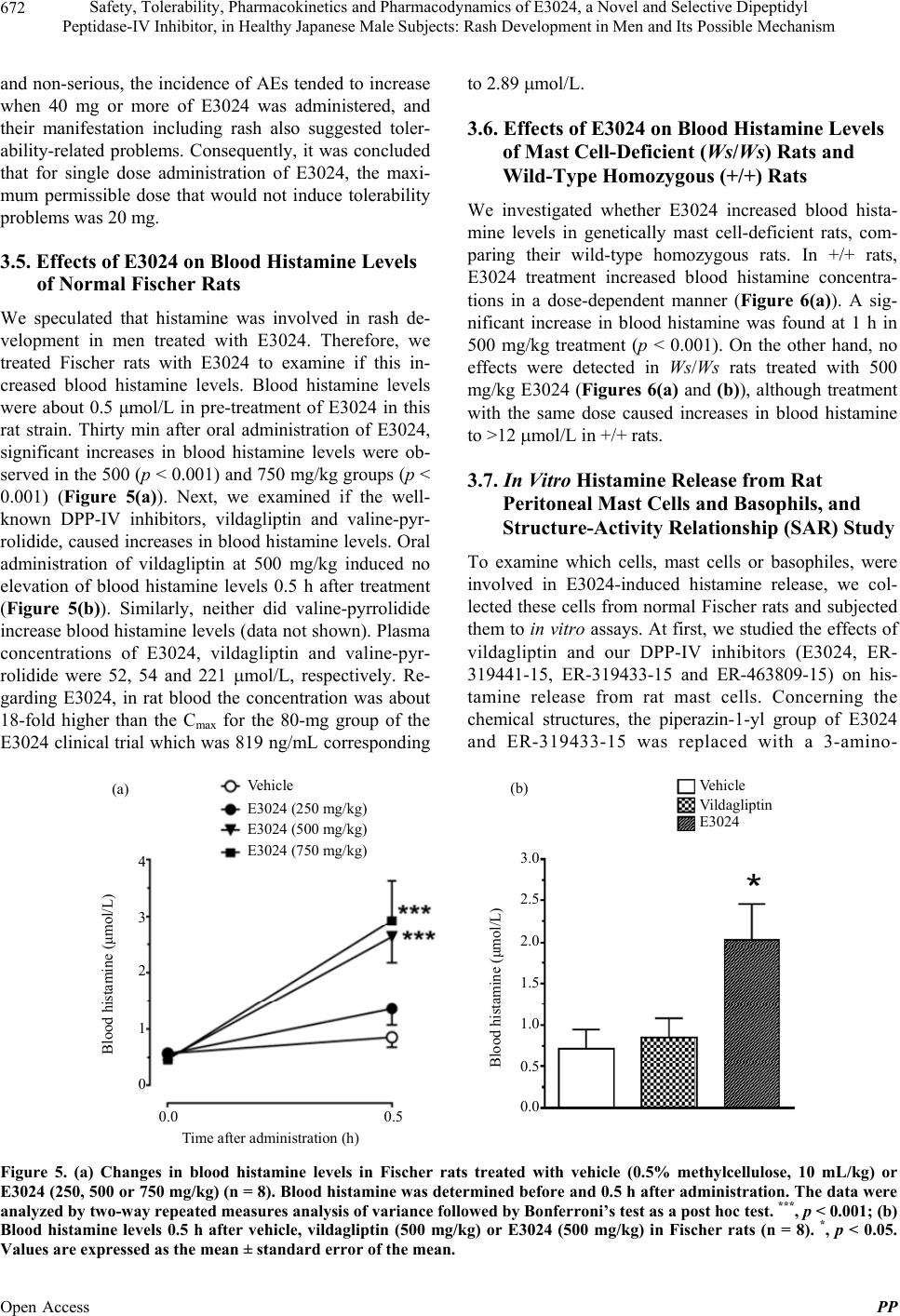 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 672 and non-serious, the incidence of AEs tended to increase when 40 mg or more of E3024 was administered, and their manifestation including rash also suggested toler- ability-related problems. Consequently, it was concluded that for single dose administration of E3024, the maxi- mum permissible dose that would not induce tolerability problems was 20 mg. 3.5. Effects of E3024 on Blood Histamine Levels of Normal Fischer Rats We speculated that histamine was involved in rash de- velopment in men treated with E3024. Therefore, we treated Fischer rats with E3024 to examine if this in- creased blood histamine levels. Blood histamine levels were about 0.5 μmol/L in pre-treatment of E3024 in this rat strain. Thirty min after oral administration of E3024, significant increases in blood histamine levels were ob- served in the 500 (p < 0.001) and 750 mg/kg groups (p < 0.001) (Figure 5(a)). Next, we examined if the well- known DPP-IV inhibitors, vildagliptin and valine-pyr- rolidide, caused increases in blood histamine levels. Oral administration of vildagliptin at 500 mg/kg induced no elevation of blood histamine levels 0.5 h after treatment (Figure 5(b)). Similarly, neither did valine-pyrrolidide increase blood histamine levels (data not shown). Plasma concentrations of E3024, vildagliptin and valine-pyr- rolidide were 52, 54 and 221 mol/L, respectively. Re- garding E3024, in rat blood the concentration was about 18-fold higher than the Cmax for the 80-mg group of the E3024 clinical trial which was 819 ng/mL corresponding to 2.89 mol/L. 3.6. Effects of E3024 on Blood Histamine Levels of Mast Cell-Deficient (Ws/Ws) Rats and Wild-Type Homozygous (+/+) Rats We investigated whether E3024 increased blood hista- mine levels in genetically mast cell-deficient rats, com- paring their wild-type homozygous rats. In +/+ rats, E3024 treatment increased blood histamine concentra- tions in a dose-dependent manner (Figure 6(a)). A sig- nificant increase in blood histamine was found at 1 h in 500 mg/kg treatment (p < 0.001). On the other hand, no effects were detected in Ws/Ws rats treated with 500 mg/kg E3024 (Figures 6(a) and (b)), although treatment with the same dose caused increases in blood histamine to >12 mol/L in +/+ rats. 3.7. In Vitro Histamine Release from Rat Peritoneal Mast Cells and Basophils, and Structure-Activity Relationship (SAR) Study To examine which cells, mast cells or basophiles, were involved in E3024-induced histamine release, we col- lected these cells from normal Fischer rats and subjected them to in vitro assays. At first, we studied the effects of vildagliptin and our DPP-IV inhibitors (E3024, ER- 319441-15, ER-319433-15 and ER-463809-15) on his- tamine release from rat mast cells. Concerning the chemical structures, the piperazin-1-yl group of E3024 and ER-319433-15 was replaced with a 3-amino- AB 0 0.0 Blood histamine ( mol/L) (a) Time after administration (h) 0.5 0.0 0.5 1.0 2.0 3.0 1.5 2.5 1 2 3 4 (b) Blood histamine (μmol/L) Veh ic le E3024 (250 mg/kg) E3024 (500 mg/kg) E3024 (750 mg/kg) Veh i cle Vildagliptin E3024 Figure 5. (a) Changes in blood histamine levels in Fischer rats treated with vehicle (0.5% methylcellulose, 10 mL/kg) or E3024 (250, 500 or 750 mg/kg) (n = 8). Blood histamine was determined before and 0.5 h after administration. The data were analyzed by two-way repeated measures analysis of variance followed by Bonferroni’s test as a post hoc test. ***, p < 0.001; (b) Blood histamine levels 0.5 h after vehicle, vildagliptin (500 mg/kg) or E3024 (500 mg/kg) in Fischer rats (n = 8). *, p < 0.05. Values are expressed as the mean ± standard error of the mean. Open Access PP 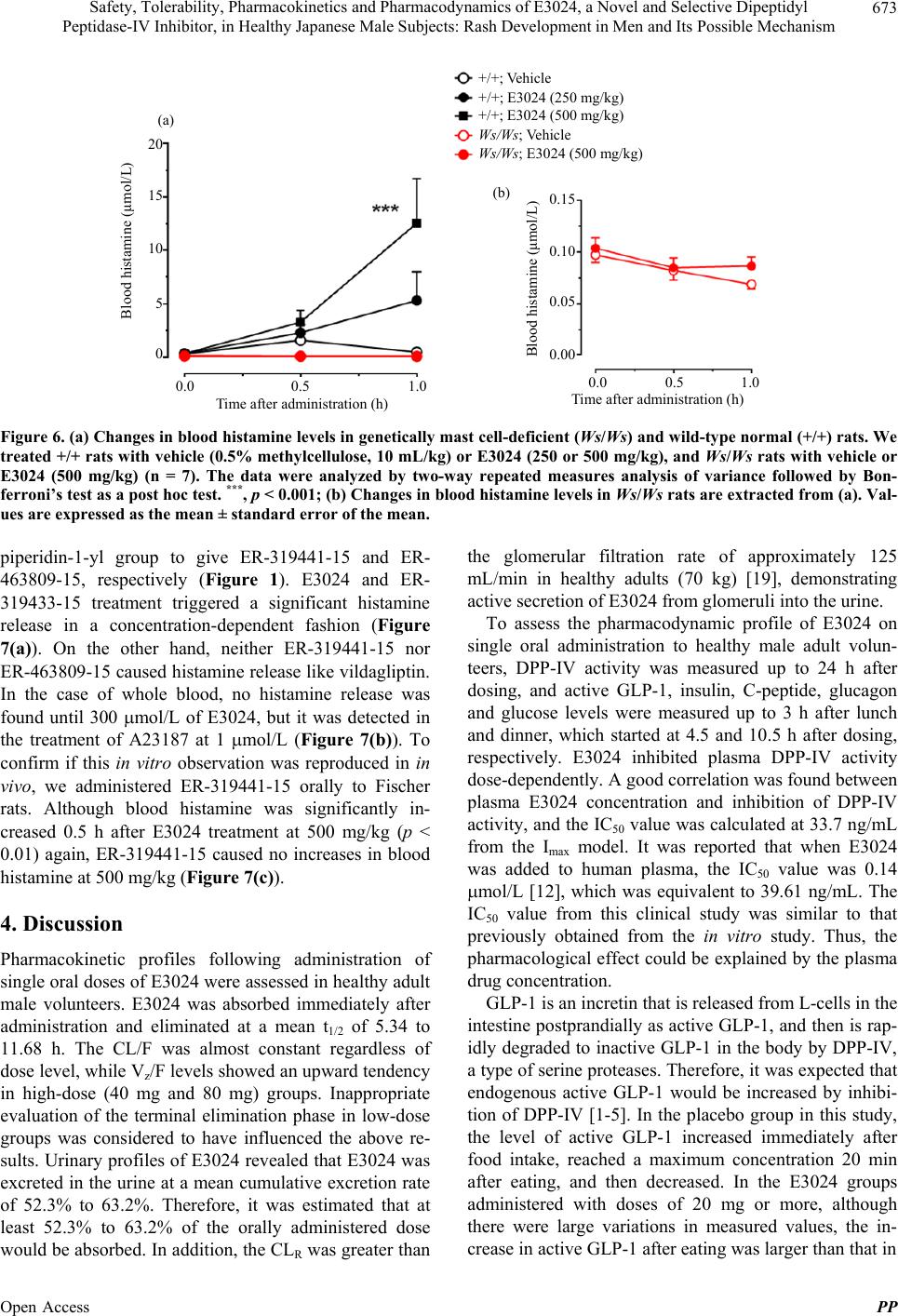 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 673 A B 0 0.0 Blood histamine (μmol/L) Time after administration (h) 0.5 5 +/+; Vehicle +/+; E3024 (250 mg/kg) +/+; E3024 (500 mg/kg) 1.0 +/+; E3024 (250 mg/kg) Ws/Ws; Vehicle Ws/Ws; E3024 (500 mg/kg) 10 15 20 (a) (b) 0.00.5 1.0 Time after administration (h) 0.00 0.05 0.10 0.15 Blood histamine (µmol/L) Figure 6. (a) Changes in blood histamine levels in genetically mast cell-deficient (Ws/Ws) and wild-type normal (+/+) rats. We treated +/+ rats with vehicle (0.5% methylcellulose, 10 mL/kg) or E3024 (250 or 500 mg/kg), and Ws/Ws rats with vehicle or E3024 (500 mg/kg) (n = 7). The data were analyzed by two-way repeated measures analysis of variance followed by Bon- ferroni’s test as a post hoc test. ***, p < 0.001; (b) Changes in blood histamine levels in Ws/Ws rats are extracted from (a). Val- ues are expressed as the mean ± standard error of the mean. piperidin-1-yl group to give ER-319441-15 and ER- 463809-15, respectively (Figure 1). E3024 and ER- 319433-15 treatment triggered a significant histamine release in a concentration-dependent fashion (Figure 7(a)). On the other hand, neither ER-319441-15 nor ER-463809-15 caused histamine release like vildagliptin. In the case of whole blood, no histamine release was found until 300 mol/L of E3024, but it was detected in the treatment of A23187 at 1 mol/L (Figure 7(b)). To confirm if this in vitro observation was reproduced in in vivo, we administered ER-319441-15 orally to Fischer rats. Although blood histamine was significantly in- creased 0.5 h after E3024 treatment at 500 mg/kg (p < 0.01) again, ER-319441-15 caused no increases in blood histamine at 500 mg/kg (Figure 7(c)). 4. Discussion Pharmacokinetic profiles following administration of single oral doses of E3024 were assessed in healthy adult male volunteers. E3024 was absorbed immediately after administration and eliminated at a mean t1/2 of 5.34 to 11.68 h. The CL/F was almost constant regardless of dose level, while Vz/F levels showed an upward tendency in high-dose (40 mg and 80 mg) groups. Inappropriate evaluation of the terminal elimination phase in low-dose groups was considered to have influenced the above re- sults. Urinary profiles of E3024 revealed that E3024 was excreted in the urine at a mean cumulative excretion rate of 52.3% to 63.2%. Therefore, it was estimated that at least 52.3% to 63.2% of the orally administered dose would be absorbed. In addition, the CLR was greater than the glomerular filtration rate of approximately 125 mL/min in healthy adults (70 kg) [19], demonstrating active secretion of E3024 from glomeruli into the urine. To assess the pharmacodynamic profile of E3024 on single oral administration to healthy male adult volun- teers, DPP-IV activity was measured up to 24 h after dosing, and active GLP-1, insulin, C-peptide, glucagon and glucose levels were measured up to 3 h after lunch and dinner, which started at 4.5 and 10.5 h after dosing, respectively. E3024 inhibited plasma DPP-IV activity dose-dependently. A good correlation was found between plasma E3024 concentration and inhibition of DPP-IV activity, and the IC50 value was calculated at 33.7 ng/mL from the Imax model. It was reported that when E3024 was added to human plasma, the IC50 value was 0.14 mol/L [12], which was equivalent to 39.61 ng/mL. The IC50 value from this clinical study was similar to that previously obtained from the in vitro study. Thus, the pharmacological effect could be explained by the plasma drug concentration. GLP-1 is an incretin that is released from L-cells in the intestine postprandially as active GLP-1, and then is rap- idly degraded to inactive GLP-1 in the body by DPP-IV, a type of serine proteases. Therefore, it was expected that endogenous active GLP-1 would be increased by inhibi- tion of DPP-IV [1-5]. In the placebo group in this study, the level of active GLP-1 increased immediately after food intake, reached a maximum concentration 20 min after eating, and then decreased. In the E3024 groups administered with doses of 20 mg or more, although there were large variations in measured values, the in- crease in active GLP-1 after eating was larger than that in Open Access PP 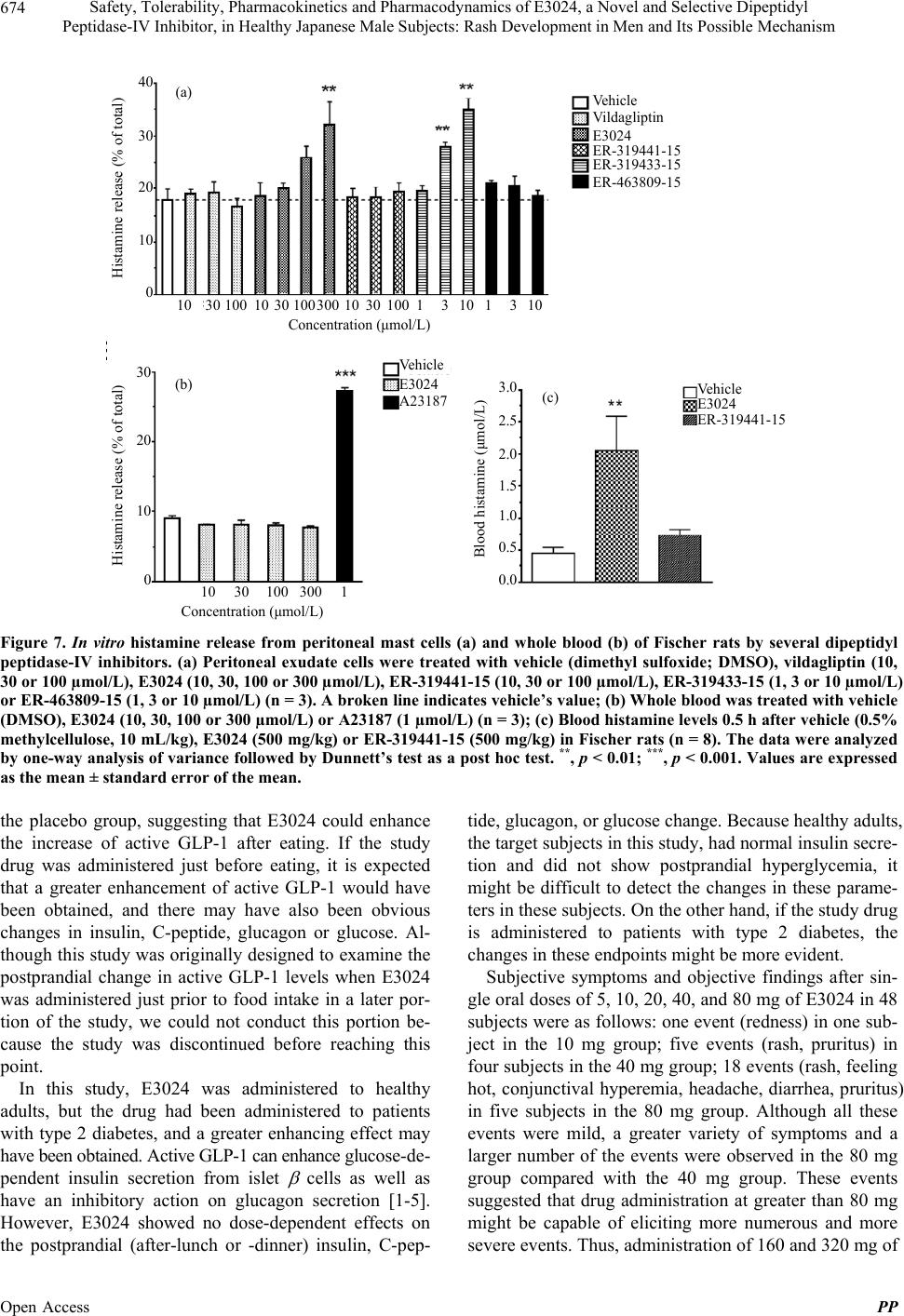 Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 674 C 0.0 0.5 1.0 2.0 3.0 1.5 2.5 (b) Blood histamine (μmol/L) Vehicle ER-319441-15 E3024 Vehicle Vildagliptin E3024 (c) (a) 40 Histamine release (% of total) 30 20 10 0 10 30 100 10 30 100 300 103010013 101310 Ve hi c le E3024 A23187 ER-319441-15 ER-463809-15 ER-319433-15 30 20 10 0 Histamine release (% of total) 10 100 300 30 1 Concentration (μmol/L) Concentration (μmol/L) Figure 7. In vitro histamine release from peritoneal mast cells (a) and whole blood (b) of Fischer rats by several dipeptidyl peptidase-IV inhibitors. (a) Peritoneal exudate cells were treated with vehicle (dimethyl sulfoxide; DMSO), vildagliptin (10, 30 or 100 µmol/L), E3024 (10, 30, 100 or 300 µmol/L), ER-319441-15 (10, 30 or 100 µmol/L), ER-319433-15 (1, 3 or 10 µmol/L) or ER-463809-15 (1, 3 or 10 µmol/L) (n = 3). A broken line indicates vehicle’s value; (b) Whole blood was treated with vehicle (DMSO), E3024 (10, 30, 100 or 300 µmol/L) or A23187 (1 µmol/L) (n = 3); (c) Blood histamine levels 0.5 h after vehicle (0.5% methylcellulose, 10 mL/kg), E3024 (500 mg/kg) or ER-319441-15 (500 mg/kg) in Fischer rats (n = 8). The data were analyzed by one-way analysis of variance followed by Dunnett’s test as a post hoc test. **, p < 0.01; ***, p < 0.001. Values are expressed as the mean ± standard error of the mean. the placebo group, suggesting that E3024 could enhance the increase of active GLP-1 after eating. If the study drug was administered just before eating, it is expected that a greater enhancement of active GLP-1 would have been obtained, and there may have also been obvious changes in insulin, C-peptide, glucagon or glucose. Al- though this study was originally designed to examine the postprandial change in active GLP-1 levels when E3024 was administered just prior to food intake in a later por- tion of the study, we could not conduct this portion be- cause the study was discontinued before reaching this point. In this study, E3024 was administered to healthy adults, but the drug had been administered to patients with type 2 diabetes, and a greater enhancing effect may have been obtained. Active GLP-1 can enhance glucose-de- pendent insulin secretion from islet cells as well as have an inhibitory action on glucagon secretion [1-5]. However, E3024 showed no dose-dependent effects on the postprandial (after-lunch or -dinner) insulin, C-pep- tide, glucagon, or glucose change. Because healthy adults, the target subjects in this study, had normal insulin secre- tion and did not show postprandial hyperglycemia, it might be difficult to detect the changes in these parame- ters in these subjects. On the other hand, if the study drug is administered to patients with type 2 diabetes, the changes in these endpoints might be more evident. Subjective symptoms and objective findings after sin- gle oral doses of 5, 10, 20, 40, and 80 mg of E3024 in 48 subjects were as follows: one event (redness) in one sub- ject in the 10 mg group; five events (rash, pruritus) in four subjects in the 40 mg group; 18 events (rash, feeling hot, conjunctival hyperemia, headache, diarrhea, pruritus) in five subjects in the 80 mg group. Although all these events were mild, a greater variety of symptoms and a larger number of the events were observed in the 80 mg group compared with the 40 mg group. These events suggested that drug administration at greater than 80 mg might be capable of eliciting more numerous and more severe events. Thus, administration of 160 and 320 mg of Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 675 E3024 was not conducted. In eight subjects reporting rash, no changes in eosino- phil fraction or non-specific IgE, the parameters for type 1 (immediate) allergy, were observed. DLST was per- formed on four subjects to examine for the possibility of drug hypersensitivity due to delayed allergy, also with negative results. In three subjects from the 80 mg group, an increasing tendency in histamine level was observed immediately after the onset of rash following study-drug administration. In view of the extremely early onset of rash, the dose-dependency in the rash development pro- file, and the increase in blood histamine levels, histamine release due to the direct action of E3024 on mast cells was considered most likely to have caused the rash. Be- cause there has been no report of such a frequent devel- opment of rash associated with other DPP-IV inhibitors, it is considered unlikely that the rash following E3024 administration had been induced by DPP-IV inhibition. It has been suggested that assessment of the selectivity of DPP-IV inhibition over dipeptidyl peptidase-8 (DPP-8) and dipeptidyl peptidase-9 (DPP-9) is important for ob- taining an optimal safety profile of DPP-IV inhibitors in the treatment of type 2 diabetes [20]. The Food and Drug Administration (FDA) requested conduct of skin lesion assessments in monkeys for all DPP-IV inhibitors, based on findings of necrotizing skin lesions due to some DPP-IV inhibitors. The FDA considers the skin lesion a result of off-target inhibition of DPP-8 or DPP-9. Be- cause E3024 was shown to be a highly selective DPP-IV inhibitor which did not inhibit DPP-8 or DPP-9 activity [12], we speculate that inhibition of DPP-8 or DPP-9 is not the cause of the rashes observed with E3024. We inferred that rash in the clinical trial was related to histamine release which was caused by E3024 directly. Then, we examined if E3024 increased histamine levels in rats. The reasons why we chose rats were: 1) we had pharmacokinetic and pharmacodynamics data of E3024 in rats, and 2) there is a mutant rat strain in which mast cells are deficient. We found that E3024 increased blood histamine levels in normal rats. On the other hand, valine- pyrrolidide and vildagliptin had no effects on blood his- tamine levels, even in the presence of sufficient plasma concentrations. However, there is a large difference in the concentrations of E3024 causing rash in men and increasing blood histamine in the rat. This may be due to different sensitivity between the species: men may be more sensitive to E3024 than rats. Ws/Ws rats are deficient in both mucosal-type and connective tissue-type mast cells [21]. The defected gene of Ws is c-kit, receptor-type tyrosine kinase [22]. c-kit is a receptor of stem cell factor essential for migration, dif- ferentiation, and proliferation of cells, such as hemato- poietic stem cells, neural crest-derived melanocytes. Thus, Ws/Ws mutant rats manifest depigmentation, ane- mia, and mast cell deficiency [21]. Histamine is pro- duced in mast cells, basophils, and entero-chromaffin- like (ECL) cells. Basophils of Ws/Ws rats are not differ- ent from wild-type rats in number, and produce histamine [23]. In addition, histamine is synthesized and stored in ECL cells of Ws/Ws rats [24]. In our study, E3024 in- creased blood histamine in wild-type homozygous (+/+) rats, while no response was observed in Ws/Ws rats. In view of the difference in histamine-producing cells be- tween Ws/Ws and wild-type rats, we concluded that E3024 acted specifically on mast cells to release hista- mine. The cell-specific effect was also confirmed by the observation that histamine was released from normal rat mast cells, but not from basophils in vitro. From the in vitro study, we obtained the following SAR: comparing between E3024 and ER-319441-15, the presence of a piperazin-1-yl group on position 2 of the imidazo[4,5-d]pyridazine may be a causal structure for induction of histamine release in rats. Similarly, com- parison of ER-319433-15 and ER-463809-15 revealed that the piperazin-1-yl group on position 8 of purine is important in determining whether histamine release oc- curs. These results showed that a key structure causing histamine release is piperazine linked to a 5,6-membered fused heterocyclic core, namely, either a pyrimidine ring fused to an imidazole ring, or a pyridazine ring fused to an imidazole ring. More interestingly, this piperazine- associated histamine release can be avoided by substitu- tion with 3-amino-piperidine, while maintaining DPP-IV inhibitory activity (data not shown). Unfortunately, we cannot demonstrate whether or not the 3-amino-piperidin-1-yl compounds do not induce rash clinically. However, among marketed DPP-IV in- hibitors, linagliptin (8-[(3R)-3-amino-piperidin-1-yl]-7- (but-2-yn-1-yl)-3-methyl-1-[(4-methylquinazolin-2-yl) methyl]-3,7-dihydro-1H-purine-2,6-dione) [10] has a structure very similar to our compounds and contains a 3-amino-piperidin-1-yl group, not a piperazin-1-yl group, on position 8 of the purine (Figure 8). High incidence of rash as observed in the Phase I trial of E3024 has not been reported in clinical trials of linagliptin: the first- in-man study was performed as a randomized, doubled- blind, placebo-controlled Phase I trial in which 63 healthy, male, Caucasian volunteers received the treat- ment (47 received linagliptin; 16 received placebo) [25]. Once-daily oral doses of linagliptin were 2.5, 5, 25, 50, 100, 200, 400 and 600 mg. No rash was observed in this trial. There was a report of a randomized, double-blind, placebo-controlled Phase I trial enrolling eight healthy Japanese male subjects (six received linagliptin and two received placebo per group). Linagliptin was adminis- tered as single escalating doses of 1, 2.5, 5 and 10 mg, or Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 676 N Linagliptin N N N N N N O O CH 3 CH 3 NH 2 CH 3 Figure 8. Chemical structure of linagliptin. multiple escalating doses of 2.5, 5 and 10 mg once daily for 12 days [26]. One subject in the 5 mg group showed an increase in histamine concentration. No clinical signs or symptoms attributed to this event were observed, and then they considered unrelated to linagliptin. In addition, no rash was reported in 2523 patients receiving lina- gliptin 5 mg once daily in eight randomed, double-blind, placebo-controlled Phase III trials lasting 12 to 24 weeks [27]. These facts may support that our speculation that rash could be avoided by the combination of 5- and 6-membered fused heterocyclic rings and 3-amino- piperidine. In conclusion, we found that E3024 was a compound with favorable pharmacokinetic profiles that inhibited plasma DPP-IV activity dose-dependently with a good correlation between its plasma concentrations and inhibi- tion of DPP-IV activity. Because AEs including rash occurred frequently when 40 mg or more of E3024 was administered, the maximum permissible single dose of E3024 that would not induce tolerability problems was considered to be 20 mg. Since superior safety is required for antidiabetic therapy, we decided not to continue fur- ther development of E3024. The results of in vivo and in vitro pre-clinical examinations using rats indicated that histamine release from mast cells by E3024 was involved in rash development in men. 5. Acknowledgements The authors wish to express our thanks to the volunteers who participated in this first-in-man study, and staff at Sekino Clinical Pharmacology Clinic. The authors also thank the Eisai study team for E3024 clinical study: Ta- kashi Sato and Hideo Takahira for the measurement of plasma and urine concentrations of E3024; Hiroshi Kuroiwa for clinical data management; Hideaki Miyagi- shi for statistical analysis; and Hiroshi Arakawa and Shohei Nishimoto for study monitoring. The authors highly appreciated Professor Tetsuo Shiohara, Depart- ment of Dermatology, Kyorin University School of Medicine, Tokyo, Japan, for his medical advice on inter- pretation of rash developing in this study. In addition, we are grateful to Tadashi Nagakura, Katsuhiro Moriya, Yoshihisa Sano, Osamu Takenaka, and Junichi Naga- kawa of Eisai Co., Ltd. for their invaluable advice and support of pre-clinical studies. REFERENCES [1] J. J. Holst and C. F. Deacon, “Inhibition of the Activity of Dipeptidyl-Peptidase IV as a Treatment for Type 2 Dia- betes,” Diabetes, Vol. 47, No. 11, 1998, pp. 1663-1670. http://dx.doi.org/10.2337/diabetes.47.11.1663 [2] D. J. Drucker, “Enhancing Incretin Action for the Treat- ment of Type 2 Diabetes,” Diabetes Care, Vol. 26, No. 10, 2003, pp. 2929-2940. http://dx.doi.org/10.2337/diacare.26.10.2929 [3] C. F. Deacon, “Therapeutic Strategies Based on Gluca- gon-Like Peptide 1,” Diabetes, Vol. 53, No. 9, 2004, pp. 2181-2189. http://dx.doi.org/10.2337/diabetes.53.9.2181 [4] J. J. Holst and C. Ørskov, “The Incretin Approach for Diabetes Treatment. Modulation of Islet Hormone Re- lease by GLP-1 Agonism,” Diabetes, Vol. 53, Suppl. 3, 2004, pp. S197-S204. http://dx.doi.org/10.2337/diabetes.53.suppl_3.S197 [5] B. Ahrén, “Dipeptidyl Peptidase-4 Inhibitors. Clinical Data and Clinical Implications,” Diabetes Care, Vol. 30, No. 6, 2007, pp. 1344-1350. http://dx.doi.org/10.2337/dc07-0233 [6] D. Kim, L. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. S. Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry and A. E. Weber, “(2R)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1, 2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl) butan-2-amine: A Potent, Orally Active Dipeptidyl Pep- tidase IV Inhibitor for the Treatment of Type 2 Diabetes,” Journal of Medicinal Chemistry, Vol. 48, No. 1, 2005, pp. 141-151. http://dx.doi.org/10.1021/jm0493156 [7] E. B. Villhauer, J. A. Brinkman, G. B. Naderi, B. F. Bur- key, B. E. Dunning, K. Prasad, B. L. Mangold, M. E. Russell and T. E. Hughes, “1-[[(3-Hydroxy-1-adamantyl) amino]acetyl]-2-cyano-(S)-pyrrolidine: A Potent, Selec- tive, and Orally Bioavailable Dipeptidyl Peptidase IV In- hibitor with Antihyperglycemic Properties,” Journal of Medicinal Chemistry, Vol. 46, No. 13, 2003, pp. 2774- 2789. http://dx.doi.org/10.1021/jm030091l [8] D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S. P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Mar- cinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, “Discovery and Preclinical Profile of Saxagliptin (BMS-477118): A Highly Potent, Long-Acting, Orally Active Dipeptidyl Peptidase IV In- hibitor for the Treatment of Type 2 Diabetes,” Journal of Medicinal Chemistry, Vol. 48, No. 15, 2005, pp. 5025- 5037. http://dx.doi.org/10.1021/jm050261p [9] J. Feng, Z. Zhang, M. B. Wallace, J. A. Stafford, S. W. Kaldor, D. B. Kassel, M. Navre, L. Shi, R. J. Skene, T. Asakawa, K. Takeuchi, R. Xu, D. R. Webb and S. L. Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism 677 Gwaltney, II, “Discovery of Alogliptin: A Potent, Selec- tive, Bioavailable, and Efficacious Inhibitor of Dipeptidyl Peptidase IV,” Journal of Medicinal Chemistry, Vol. 50, No. 10, 2007, pp. 2297-2300. http://dx.doi.org/10.1021/jm070104l [10] M. Eckhardt, E. Langkopf, M. Mark, M. Tadayyon, L. Thomas, H. Nar, W. Pfrengle, B. Guth, R. Lotz, P. Sieger, H. Fuchs and F. Himmelsbach, “8-(3-(R)-Amino- piperidin-1-yl)-7 -but-2-y ny l-3-methy l -1-(4-methy l-quinaz- olin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a Highly Potent, Selective, Long-Acting, and Orally Bioavailable DPP-4 Inhibitor for the Treatment of Type 2 Diabetes,” Journal of Medicinal Chemistry, Vol. 50, No. 26, 2007, pp. 6450-6453. http://dx.doi.org/10.1021/jm701280z [11] N. Kato, M. Oka, T. Murase, M. Yoshida, M. Sakairi, S. Yamashita, Y. Yasuda, A. Yoshikawa, Y. Hayashi, M. Makino, M. Takeda, Y. Mirensha and T. Kakigami, “Dis- covery and Pharmacological Characterization of N-[2-({2- [(2S)-2-cyanopyrrolidin-1-yl]-2-oxoethyl}amino)-2-methyl- propyl-2-methy lpy razolo[1,5-a]pyrimidine-6-car-boxamide Hydrochloride (Anagliptin Hydrochloride Salt) as a Po- tent and Selective DPP-IV Inhibitor,” Bioorganicand Medicinal Chemistry, Vol. 19, No. 23, 2011, pp. 7221- 7227. http://dx.doi.org/10.1016/j.bmc.2011.09.043 [12] N. Yasuda, T. Nagakura, T. Inoue, K. Yamazaki, N. Ka- tsutani, O. Takenaka, R. Clark, F. Matsuura, E. Emori, S. Yoshikawa, K. Kira, H. Ikuta, T. Okada, T. Saeki, O. Asano and I. Tanaka, “E3024, 3-But-2-ynyl-5-methyl- 2-piperazin-1-yl-3,5-dihy dro-4H-imidazo[4,5-d]pyridazin- 4-one Tosylate, Is a Novel, Selective and Competitive Dipeptidyl Peptidase-IV Inhibitor,” European Journal of Pharmacology, Vol. 548, No. 1-3, 2006, pp. 181-187. http://dx.doi.org/10.1016/j.ejphar.2006.08.011 [13] K. Yamazaki, T. Inoue, N. Yasuda, Y. Sato, T. Nagakura, O. Takenaka, R. Clark, T. Saeki and I. Tanaka, “Com- parison of Efficacies of a Dipeptidyl Peptidase IV Inhibi- tor and α-Glucosidase Inhibitors in Oral Carbohydrate and Meal Tolerance Tests and the Effects of Their Com- bination in Mice,” Journal of Pharmacological Sciences, Vol. 104, No. 1, 2007, pp. 29-38. http://dx.doi.org/10.1254/jphs.FP0061376 [14] K. Yamazaki, N. Yasuda, T. Inoue, E. Yamamoto, Y. Sugaya, T. Nagakura, M. Shinoda, R. Clark, T. Saeki and I. Tanaka, “Effects of the Combination of a Dipeptidyl Peptidase IV Inhibitor and an Insulin Secretagogue on Glucose and Insulin Levels in Mice and Rats,” The Jour- nal of Pharmacological and Experimental Therapeutics, Vol. 320, No. 2, 2007, pp. 738-746. http://dx.doi.org/10.1124/jpet.106.112011 [15] T. Hashimoto, H. Kasai, M. Yamada, M. Yamada, H. Sakaki, J. Handa, T. Takizawa and A. Hirata, “Statistical Assessment of Linear Pharmacokinetics in Clinical Phar- macological Studies,” Xenobiotic Metabolism and Dispo- sition, Vol. 16, No. 3, 2001, pp. 244-252. [16] C. F. Deacon, T. E. Hughes and J. J. Holst, “Dipeptidyl Peptidase IV Inhibition Potentiates the Insulinotropic Ef- fect of Glucagon-Like Peptide 1 in the Anesthetized Pig,” Diabetes, Vol. 47, No. 5, 1998, pp. 764-769. http://dx.doi.org/10.2337/diabetes.47.5.764 [17] S. N. Pramod, Y. P. Venkatesh and P. A. Mahesh, “Potato Lectin Activates Basophils and Mast Cells of Atopic Sub- jects by Its Interaction with Core Chitobiose of Cell- Bound Non-Specific Immunoglobulin E,” Clinical and Experimental Immunology, Vol. 148, No. 3, 2007, pp. 391-401. http://dx.doi.org/10.1111/j.1365-2249.2007.03368.x [18] K. Kowal, H. Nolte, P. S. Skov and L. M. DuBuske, “Ef- fect of Allergen-Specific Immunotherapy on Recombi- nant Human Interleukin 3-Mediated Amplification of Al- lergen-Induced Basophil Histamine Release,” Allergy and Asthma Proceedings, Vol. 26, No. 6, 2005, pp. 456-462. [19] B. Davies and T. Morris, “Physiological Parameters in Laboratory Animals and Humans,” Pharmaceutical Re- search, Vol. 10, No. 7, 1993, pp. 1093-1095. http://dx.doi.org/10.1023/A:1018943613122 [20] G. R. Lankas, B. Leiting, R. S. Roy, G. J. Eiermann, M. G. Beconi, T. Biftu, C. C. Chan, S. Edmondson, W. P. Feeney, H. He, D. E. Ippolito, D. Kim, K. A. Lyons, H. O. Ok, R. A. Patel, A. N. Petrov, K. A. Pryor, X. Qian, L. Reigle, A. Woods, J. K. Wu, D. Zaller, X. Zhang, L. Zhu, A. E. Weber and N. A. Thornberry, “Dipeptidyl Peptidase IV Inhibition for the Treatment of Type 2 Diabetes: Po- tential Importance of Selectivity over Dipeptidyl Pepti- dases 8 and 9,” Diabetes, Vol. 54, No. 10, 2005, pp. 2988-2994. http://dx.doi.org/10.2337/diabetes.54.10.2988 [21] Y. Niwa, T. Kasugai, K. Ohno, M. Morimoto, M. Yama- zaki, K. Dohmae, Y. Nishimune, K. Kondo and Y. Kita- mura, “Anemia and Mast Cell Depletion in Mutant Rats that Are Homozygous at ‘White Spotting (Ws)’ Locus,” Blood, Vol. 78, No. 8, 1991, pp. 1936-1941. [22] T. Tsujimura, S. Hirota, S. Nomura, Y. Niwa, M. Yama- zaki, T. Tono, E. Morii, H.-M. Kim, K. Kondo, Y. Ni- shimune and Y. Kitamura, “Characterization of Ws Mu- tant Allele of Rats: A 12-Base Deletion in Tyrosine Kinase Domain of c-kit Gene,” Blood, Vol. 78, No. 8, 1991, pp. 1942-1946. [23] T. Kasugai, M. Okada, M. Morimoto, N. Arizono, K. Maeyama, M. Yamada, H. Tei, K. Dohmae, H. Onoue, G. F. Newlands, T. Watanabe, Y. Nishimune, H. R. P. Miller and Y. Kitamura, “Infection of Nippostrongylus brasil- iensis Induces Normal Increase of Basophils in Mast Cell-Deficient Ws/Ws Rats with a Small Deletion at the Kinase Domain of c-kit,” Blood, Vol. 81, No. 10, 1993, pp. 2521-2529. [24] S. Nakamura, H. Watanabe, T. Yokota, H. Matsui, M. Onji and K. Maeyama, “Effect of Rabeprazole on Hista- mine Synthesis in Enterochromaffin-Like Cells of Mast Cell-Deficient (Ws/Ws) Rats,” Euro pean Journal of Phar- macology, Vol. 394, No. 1, 2000, pp. 9-16. http://dx.doi.org/10.1016/S0014-2999(00)00080-7 [25] S. Hüttner, E. U. Graefe-Mody, B. Withopf, A. Ring and K. A. Dugi, “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Single Oral Doses of BI 1356, an Inhibitor of Dipeptidyl Peptidase 4, in Healthy Male Vol- unteers,” The Journal of Clinical Pharmacology, Vol. 48, No. 10, 2008, pp. 1171-1178. http://dx.doi.org/10.1177/0091270008323753 Open Access PP  Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E3024, a Novel and Selective Dipeptidyl Peptidase-IV Inhibitor, in Healthy Japanese Male Subjects: Rash Development in Men and Its Possible Mechanism Open Access PP 678 [26] A. Sarashina, S. Sesoko, M. Nakashima, N. Hayashi, A. Taniguchi, Y. Horie, E. U. Graefe-Mody, H.-J. Woerle and K. A. Dugi, “Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor in Development for the Treatment of Type 2 Diabetes Mellitus: A Phase I, Randomized, Double-Blind, Placebo-Controlled Trial of Single and Multiple Escalat- ing Doses in Healthy Adult Male Japanese Subjects,” Clinical Therapeutics, Vol. 32, No. 6, 2010, pp. 1188- 1204. http://dx.doi.org/10.1016/j.clinthera.2010.06.004 [27] G. Schernthaner, A. H. Barnett, A. Emser, S. Patel, J. Troost, H.-J. Woerle and M. von Eynatten, “Safety and Tolerability of Linagliptin: A Pooled Analysis of Data from Randomized Controlled Trials in 3572 Patients with Type 2 Diabetes Mellitus,” Diabetes, Obesity and Me- tabolism, Vol. 14, No. 5, 2012, pp. 470-478. http://dx.doi.org/10.1111/j.1463-1326.2012.01565.x Abbreviations Ae, amount of unchanged drug excreted in urine; AE, adverse event; ALT, alanine aminotransferase; AUC, area under the curve; AUC0-inf, area under the plasma concentration-time curve from 0 to infinity; BMI, body mass index; CI, confidence interval; CL, clearance; CL/F, apparent clearance; CLR, renal clearance; Cmax, maxi- mum observed concentration; DLST, drug-induced lym- phocyte stimulation test; DMSO, dimethyl sulfoxide; D-PBS, Dulbecco’s phosphate-buffered saline; DPP-IV, dipeptidyl peptidase-IV; DPP-8, dipeptidyl peptidase-8; DPP-9, dipeptidyl peptidase-9; ECG, electrocardiography; ECL, entero-chromaffin-like; EDTA, ethylenediamine- tetraacetic acid; ELISA, enzyme-linked immunosorbent assay; F, bioavailability; FDA, Food and Drug Admini- stration; fe, fraction of drug excreted unchanged in urine; GLP-1, glucagon-like peptide-1; IC50, concentration re- quired for 50% of the maximum inhibition; IgE, immu- noglobulin E; LC/MS/MS, liquid chromatographic-tandem mass spectrometry; MC, methycellulose; OTC, over-the- counter; PD, pharmacodynamics; PEC, peritoneal exu- date cell; PK, pharmacokinetics; SAR, structure-activity relationship; t1/2, terminal half-life; tmax, time to Cmax; Vz, volume of distribution during the terminal phase; Vz/F, apparent volume of distribution during the terminal phase
|