 Journal of Modern Physics, 2013, 4, 27-42 Published Online November 2013 (http://www.scirp.org/journal/jmp) http://dx.doi.org/10.4236/jmp.2013.411A2002 Open Access JMP Single Molecule Applications of Quantum Dots Thomas E. Rasmussen1, Liselotte Jauffred2, Jonathan Brewer1, Stefan Vogel1, Esben R. Torbensen1, B. Christoffer Lagerholm1,3, Lene Oddershede2, Eva C. Arnspang1,4,5 1Departments of Physics, Chemistry, Pharmacy, Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark 2Niels Bohr Institute, University of Copenhagen, Copenhagen, Denmark 3Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK 4National Institutes of Health, Bethesda, USA 5Department of Molecular Biology and Genetics and Interdisciplinary Nanoscience Center, Aarhus University, Aarhus, Denmark Email: arnspang@mb.au.dk Received September 25, 2013; revised October 23, 2013; accepted November 22, 2013 Copyright © 2013 Thomas E. Rasmussen et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Fluorescent nanocrystals composed of semiconductor materials were first introduced for biological applications in the late 1990s. The focus of this review is to give a brief survey of biological applications of quantum dots (QDs) at the single QD sensitivity level. These are described as follows: 1) QD blinking and bleaching statistics, 2) the use of QDs in high speed single particle tracking with a special focus on how to design the biofunctional coatings of QDs which en- able specific targeting to single proteins or lipids of interest, 3) a hybrid lipid-DNA analogue binding QDs which allows for tracking single lipids in lipid bilayers, 4) two-photon fluorescence correlation spectroscopy of QDs and 5) optical trapping and excitation of single QDs. In all of these applications, the focus is on the single particle sensitivity level of QDs. The high applicability of QDs in live cell imaging experiments held together with the prospects in localization microscopy and single molecule manipulation experiments gave QDs a promising future in single molecule research. Keywords: Quantum Dots; Single Particle Tracking; Fluorescence Correlation Spectroscopy; Optical Tweezers 1. Introduction Fluorescent nanocrystals composed of semiconductor materials are regularly referred to as quantum dots (QDs) because of their optical properties. Quantum confinement within the core material leads to the fluorescence emis- sion wavelength being dependent on the core size and material composition. These days QDs are regularly be- ing used in a variety of biological applications both in vitro and in vivo (References). Almost 15 years after their initial application in biology [1], QDs have found use in a multitude of different applications due to their superior optical properties (brightness and resistance to bleaching) as compared to conventional fluorophores. In this review we present basic concepts of QDs with a fo- cus on the optical properties that make QDs so special and further give a survey of single QD applications in biology. QDs have a higher brightness because of their very high absorption excitation coefficients and high quantum yield. QDs also display a higher resistance to- wards photobleaching since their inorganic nature makes them less prone to degradation resulting from photo- induced bond cleavage. The resulting fluorescence life- time (20 - 50 ns) [2] and high signal to noise ratio make them ideal for time-resolved microscopy studies. Section 2 is an introduction to the material composition, quantum physics and optical properties of QDs. In Section 3, there are examples of two single particle applications of QDs: QDs targeted to a protein of interest followed by high speed single particle tracking (SPT) of the QD signal in live cells, and QDs targeted to a DNA-lipid analogue and SPT in lipid bilayers. In Section 4, there is an example of the use of QDs as probe in two-photon fluorescence cor- relation spectroscopy in which the size of the QD is found. In Section 5 we present how the use of QDs as a force-handle as well as a visualization probe using an optical tweezers assay. In all the applications mentioned in this review, the focus is on the use of QDs in single particle detection experiments.  T. E. RASMUSSEN ET AL. 28 2. Quantum Dots—Fluorescent Semiconductor Nanocrystals Quantum dots (QDs) are inorganic fluorescent nanoscale crystals with considerable enhanced optical properties in terms of brightness, photostability, blinking and bleach- ing compared to conventional organic and protein fluo- rophores. QD core sizes are in the range 1 - 10 nm and the individual nanocrystals contain from a few hundred to several thousands of atoms each. The core of the QD nanocrystals is typically composed of binary mixtures of semiconductor materials (ZnS, CdS, CdSe, InP, CdTe, PbS, PbTe). Early QDs were composed of only a naked core but this led to inherent problems with fluorescence output due to interaction of the exciton with the sur- roundings. Increasing QD yield and efficiency was achieved by adding a second shell layer semiconducting material to the core resulting in a so-called core/shell QD [3,4]. Core-shell QDs is the main type used today in bio- logical applications. QDs preserve some bulk properties from the material they are made of, but because of their small size they also retain new unique physical, chemical and electronic properties due to quantum confinement effects. Probably the most amazing feature of the confinement effects is that the emission bands of QDs are dependent of the composition and size. If semiconductors are exposed to light excitation, the mobility of electrons in the material increases. Characteristic for semiconductor materials is that they have an unoccupied energy band called the conduction band and an occupied energy band called the valence band. When irradiated an electron from the va- lence band can get excited and promoted to the conduc- tion band. This result in the formation of a positive charge called a hole and both the electron and the hole are free to move around the bulk material kept together by a Coulomb attraction. Together the electron and hole constitute an electron-hole pair also known as an exciton, the average distance between the electron and hole is known as the Bohr radius of the exciton. Because the excitons can be seen as particles in a box the following quantum mechanical calculations can be made. The Hamiltonian for a spherical QD of diameter R is given by [5]: 2 22 ˆ 224π eh eh e e Hmm r h r (1) The first two terms are the kinetic energy operators of the electron and the hole with masses me and mh respec- tively. The last term denotes the potential energy interac- tion of the electron and the hole that are in the positions e and h from the center of the sphere. Due to the charge on the electron (−e) and the hole (+e) the Cou- lombic attraction between the electron and the hole, where ε is the permittivity of the material and r r eh rr is the distance between the hole and the electron. Solving the Schrödinger yields the following equation for the approximation of the exciton energy [5]. 22 2 11 1.8 4π 8 ex eh h Emm R R e (2) The equation states that the exciton energy decreases with decreasing values of R and the term on the right will decrease with small R-values and therefore the exciton energy is largely of kinetic nature. Therefore the electri- cal conductivity and creation of mobile charge carriers depends on the size of the quantum dot. As a result the fluorescence emission wavelength of the QD can be tuned by size, such that the emission of smaller QDs will be blue shifted while the emission of larger QDs will be red shifted. In Figure 1 is a picture of a series of QDs emitting at wavelengths from 490 nm to 650 nm. 2.1. Optical Properties of Quantum Dots QDs distinguish themselves in several ways compared to conventional organic and protein fluorophore molecules. While the band gap of organic fluorescent dyes is defined by the π-electron system of the molecule as well as elec- tron donating and withdrawing groups, the QD band gap is determined by the size of the nanocrystal even though the material remains the same. Further, many of the most commonly applied fluorophores are characterized by relative narrow emission excitation and emission spectra. QDs on the other hand have a much wider excitation range while they exhibit very narrow and symmetrical emission spectra separated by a distinct Stokes shift. A lot of effort has been done in order to tune QD emission to cover as large a wavelength range as possible. Most commercial available QDs have emission spectra Figure 1. Image showing fluorescence emission of CdSe/ZnS with increasing size (490NC, 525NC, 565NC, 585NC, 605NC, 625NC & 650NC) and cadmium-free InGaP (700NC) Quantum dots from eBioscience. Open Access JMP 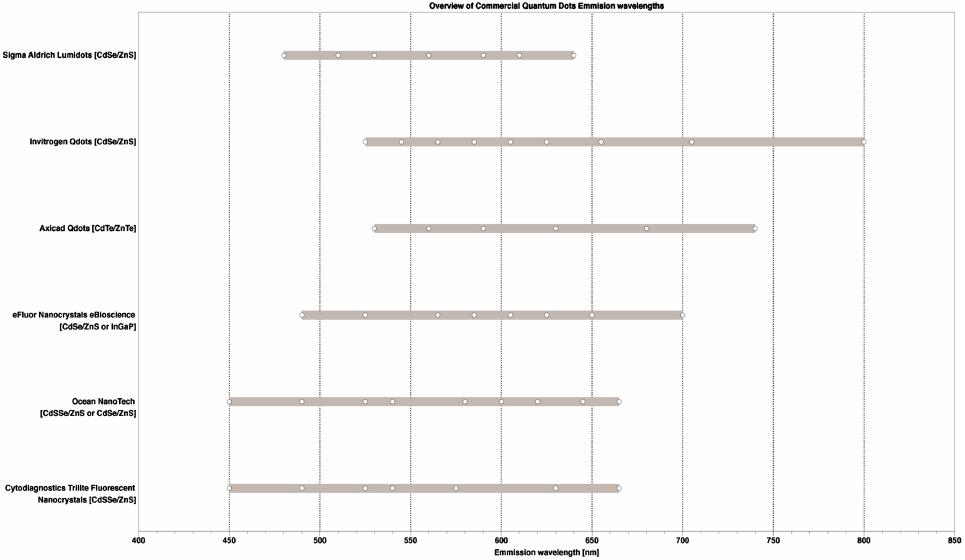 T. E. RASMUSSEN ET AL. Open Access JMP 29 situated in the visual part of the electromagnetic spec- trum and include QD materials such as (CdS, CdSe, CdTe). QDs emitting in the Ultra Violet range have mainly been made of ZnS and ZnSe while emission in the near infrared range has been accomplished with ma- terials such as CdS/HgS/CdS, InP, InAs [6]. Mainly the latter has received much attention in the development of new QD types since the penetration of NIR light into tissues is significantly higher than shorter wavelengths of light, which are susceptible to significant scattering and absorption by the tissue. Furthermore autofluorescence from biological samples is less present in this part of the spectrum resulting in a lower fluorescent background. In Figure 2 is an overview of emission wavelengths of commercially available QDs. commercially available hybrid CdSe/CdTe QDs have been shown to spend a majority of their time in a non- fluorescent dark state [7-13]. The current consensus for this observed QD intermittency is that non-charged QDs are fluorescent, while charged QDs are not [8,14]. The observed intermittency has further been shown to be par- tially inhibited by small reducing agents such as β-mer- captoethanol (BME), dithiothreitol (DTT) and mercap- toethylamine (MEA) in mM concentrations [9,15,16]. Decreased blinking has also been accomplished by growth of thick semiconductor shells around the QD cores [17,18]. Non-blinking CdZnSe/ZnSe QDs have also been reported, however, these QDs have a very broad emission spectra with three characteristic emission peaks making them non-useable for multiplexing appli- cations [14]. QDs display one more characteristic optical feature. QDs blink, periodically switching between an on and off luminescent state. The cause of this behaviour is due to trapping and untrapping of charges due to surface defects, this enables the distinction of single QDs, which is es- sential to perform single molecule measurements. When this fluorescence blinking of QDs was first observed, it was a surprise, since there was no known quantum physical mechanism which could explain this [19]. What was furthermore a surprise was that the off times of QDs were distributed according to an inverse power law rather than an exponential decay [20]. An ex- ponential dependence of the probability P that a QD is off for the time would follow the equation (= Pe , where is the off time and the slope of the linear plot on a semilog scale. A power law dependence of the probability P that a QD is off for the time would follow the equation P , where is the slope of 2.2. Blinking of Quantum Dots A major disadvantage of QDs is that the fluorescence emission is intermittent, i.e., the QDs fluctuate between a fluorescent emitting bright state and a non-emitting dark, with the dark non-fluorescence emitting states lasting for periods in the ms to second range timescale. For example, Figure 2. An overview of emission wavelengths of quantum dots from different commercial sources. 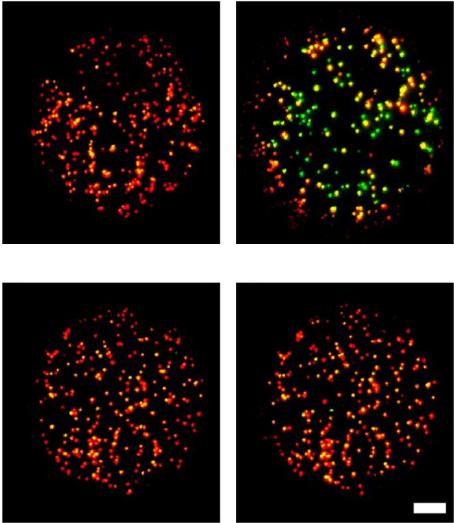 T. E. RASMUSSEN ET AL. 30 the linear plot on a log-log scale. The power law de- pendence of the blinking of QDs is unchanged for tem- peratures from 10 K to room temperature, for core radii from 15 Å to 27 Å, for different materials: CdSe, TeSe, InP, for laser intensities from 100 W/cm2 to 20 kW/cm2 and pressure from atmospheric pressure to vacuum [21-24]. Because QDs consist of two alloy compositions and the bond length in the crystal lattice of the one alloy is different from the bond length in the crystal lattice of the other alloy there are imperfections in the crystal structure. The quantum physical explanation of the blinking of the QDs proposes that the intermittency is caused by ionization of the QD. After photon excitation, two electron-hole pairs could be excited simultaneously. One pair could recombine by the Auger effect and emit one hole or one electron outside the QD. This is also called a trapped state as the emitted electron will be trapped in the surrounding medium for some time. Dur- ing this off-time the left behind electron hole in the QD will generate a very fast non-radiative Auger channel for any new excited electron-hole. The electron hole will remain inside the QD during the off-period due to dif- ferent barrier heights for electron and hole. The result is that the radiative luminescence is quenched during the off period, corresponding to the dark state of QDs [25,26]. 2.3. Bleaching of Quantum Dots A major attraction of QDs is that they are more resistant to photobleaching than is conventional fluorescent dye and protein fluorophores. But QDs have also been shown to photobleach under intense laser illumination [27,28] and more recently even with Hg arc lamp illumination [29]. However, contrary to fluorescent dyes and proteins that display single step photobleaching from a fluores- cent emitting state to a dark state, QDs have been shown to photobleach by a gradual blue-shifting of their emis- sion color eventually reaching a permanent dark state [8,27,28]. This photobleaching of QDs is indicative of a process in which the QD core is gradually shrinking as a result of photooxidation of the core [8,27,28,30,31], a hypothesis which is supported by the reported slowing of blue shifting in a nitrogen atmosphere [28]. Photooxidation at the surface of QDs has shown to re- sult in quenching of fluorescence emitted by CdSe QDs. Formation of surface quenching states will cause a de- crease in emitted fluorescence. The bleaching of QDs is accompanied with a blueshift in the emission spectrum. This bleaching process has been shown with laser powers of 20 kW/cm2 and in both air and nitrogen atmospheres. In nitrogen atmosphere the bleaching is slower but still occurs [28]. It is possible to prevent the bleaching and blueshifting of QDs by adding β-mercaptoethanol (BME) [32] or mercaptoethylamine (MAE) [29]. Figure 3 de- picts images of QD655s that were illuminated under aqueous conditions for a few minutes with and without BME added. Left and rightwimages were acquired about 7 minutes apart after continuous illumination with blue filtered light. In the absence of BME, the QD655 emis- sion color was observed to shift from an initial or- ange/red hue to a yellow-green hue. In the presence of 25 μM BME, the QD655 emission color was observed to be significantly stabilized to an orange/red hue for the dura- tion of the experiment [32]. Bleaching of QDs happens at a slower rate than other standard fluorophores. A direct comparison has been made between Alexa488 and QDs. Alexa488 fades after 60 seconds of constant illumination whereas the QDs are still emitting after 180 seconds. There are also many examples of longer full intensity periods of QDs [33]. (a) (b) (c) (d) Figure 3. Fluorescence color switching of QDs emitting at 655 nm. QDs were non-specifically adsorbed to a glass cov- erslip and imaged under aqueous conditions on a CoolS- NAP-Procf color CD camera with 10 sec integration time. Left and right images were acquired about 7 minutes apart after continuous illumination with blue filtered light from a 100 W Hg arc lamp. (a, b) QD655 in the absence of β-mercaptoethanol (BME), the QD emission color was ob- served to shift from an initial orange/red hue to a yel- low-green hue. (c, d) QD655 in the presence of 25 μM BME, the QD emission color was observed to be significantly sta- bilized to an orange/red hue for the duration of the experi- ment (scale bar is 1 m) Reprinted with permission from [32]. Open Access JMP T. E. RASMUSSEN ET AL. 31 2.4. Quantum Dot Coatings and Bioconjugation In order for QDs to be used in single molecule applica- tions certain surface modifications have to be done to the bare core/shell QDs. Both core-shell and core only QDs are coated with a thin layer of an organic ligand such as trioctylphosphine oxide (TOPO) as a result of the organic synthesis route that is used to make them [34,35]. While a hydrophobic ligand such as TOPO stabilizes the QDs in an organic solvent, these QDs need to be made more wa- ter soluble for biological relevant applications [34-38]. Keeping the original ligands in place gives the brightest QDs, and one way to do this is to use an amphiphilic co-block polymer (e.g. an octylamine-modified poly- acrylic acid) to coat the QDs [38]. The hydrophobic side chains of these polymers interdigitate with the organic ligands on the QDs, and the hydrophilic part constitute the new surface of the now water soluble QDs, adding another 1 - 2 nm to the diameter [39,40]. The hydrophilic ends of these QDs have reactive groups that are available for further bio-conjugation needed in order to direct the QD binding towards a specific target. Bio-conjugation of the QD will add to its final size and typical fully bio- functional QDs have a size of ~20 nm. Water stabilized QDs preferentially have reactive chemical groups on the surface that are available for bio- functionalization necessary in order to direct the QDs towards a specific target for biological applications [36]. Often, the initial chemical groups on the surface are car- boxylic groups, but these can be reacted with e.g. dia- mine polyethylene glycols (PEG), resulting in amino functionalized QDs. Covalent bio-functionalization of carboxylic or amine QDs is easily achieved by chemical cross-linkers, and various strategies exist depending on the conjugation partners [40,41]. For example, the cross-linker EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) can be used to conjugate carboxylic groups on QDs to amine groups on e.g. proteins or peptides [42,43]. An- other bifunctional cross-linker SMCC (succinimidyl-4- (N-maleimidomethyl)cyclohexane-1-carboxylate), which has a maleimide reactive group and an NHS ester, can be used to couple thiols on e.g. (mildly and selectively) re- duced antibodies or antibody fragments to amines on QDs [44]. QD conjugation of two amine groups has also been done using Traut’s reagent and N-succinimidyl iodoace- tate, a hetero bifunctional cross linker which couples thiols and amines (SIA) [40]. Often, a linker such as PEG is introduced between the QD and the bio-molecule to in- crease steric freedom, and to minimize unspecific bind- ing of the QDs [45,46]. Figure 4 illustrates several con- jugation strategies between QDs and organic molecules. 3. Single QD Applications in Biology The enhanced optical properties of QDs, in particular the significant brightness and photostability, make these ma- terials highly suitable for use in biological applica- tions requiring even single molecule sensitivity. 3.1. Single Particle Tracking (SPT) The foremost single molecule application where QDs have been used is single particle tracking (SPT) [48]. In SPT, single molecules of interests (MOIs) are sparsely labeled with a luminescent or scattering probe and the movement of the MOI:probe complexes is imaged by time-lapse microscopy at repetition rates ranging from a few to 50,000 Hz depending on the process investigated and the signal of the probe [48]. In this way, trajectories describing the motion of single MOI:probe conjugates can be constructed with nanometer precision and milli- second time resolution to provide details of the molecular dynamic that ensures cellular structure and function. More detail on technical aspects and data analysis in SPT can be found in recent reviews (e.g. [48]). The most common MOIs in SPT for biological appli- cations are amphiphilic molecules such as lipids, lipid anchored proteins, and transmembrane proteins. In this case, the investigated motion of the MOIs is most fre- quently restricted to lateral diffusion in two dimensions within the plane of e.g. the plasma membrane or in a model membrane. In this particular application more leeway in the choice of probe is given because the limit- ing factor in the lateral motion of the MOI:probe com- plex is the viscosity of the membrane within which the hydrophobic part of the MOI is residing. This is because the viscosity of the membrane is 100 times that of vis- cosity in the surrounding aqueous solution. For this rea- son it is generally accepted that even probes that are much larger than a particular MOI will only have a minor impact of the motion of the MOI. In contrast, in the case of an aqueous soluble MOI the motion will be severely affected by large probes. In fact even fluorescent proteins, e.g. green fluorescent protein which has a molecular weight of about 28 kDa and a hydrodynamic radii of about 3.4 nm will have a dramatic effect on the motion of most biological MOIs. Initially SPT experiments were done using interfere- ence contrast video microscopy measuring the scatter from micrometer-sized latex beads or 40 - 100 nm gold nanoparticles [49]. Subsequently the technique has been extended to track single fluorescent dyes and proteins (sometimes called single molecule fluorescent tracking, SMFT) [50]. By studying the motion of single molecules, different modes of motion can be distinguished, and of- ten the motion turns out to be very heterogeneous in a way that cannot be described by ensemble measurements which involve averaging over a large pool of indistin- guishable molecules [51]. However, neither the latex Open Access JMP 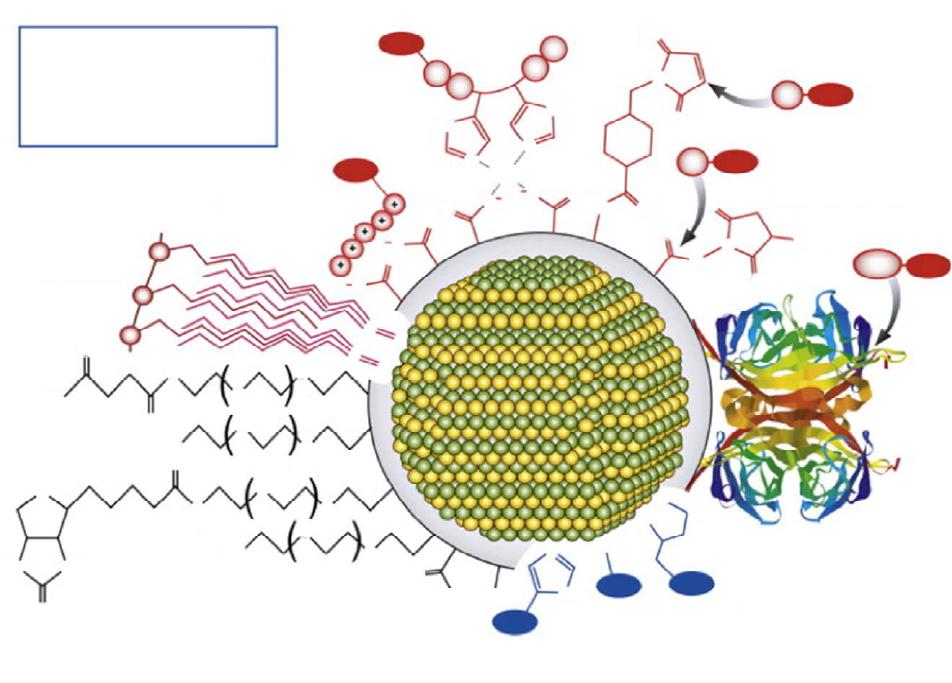 T. E. RASMUSSEN ET AL. Open Access JMP 32 R = small molecule, peptide, protein, antibody, enzyme, oligonucleotide, aptamer, lipid, surface, etc. (g) Histidine-Nickel-Carboxyl Coordination (h) Maleimide Activation (i) Active Esters (f) Electrostatic Assembly (e) Hydrophobic interactions (j) Streptavidin (a) Thiol Coordination (a) Imidazole/Histidine Coordination (d) Functionalized PEG Coatings (c) Amine or Carboxyl Coatings HN NH O S H 2 N H 3 N + O O O O O O O O O O O O O - HO H N HO N H n n n n O O p p O O OO O OOOO O O O O N HN O N O NH N HN NNi 2+ SO 3 - NH NS - S - S - R RR R R R R R HisHis His His HS Biotin NH 2 Figure 4. An illustration of some selected surface chemistries and conjugation strategies that are applied to QDs. The grey periphery around the QD represents a general coating. This coating can be associated with the surface of the QD via (e) hy- drophobic interactions, or ligand coordination. Examples of the latter include: (a) monodentate or bidentatethiols, (b) imida- zole, polyimidazole (e.g. polyhistidine), or dithiocarbamate (not shown) groups. The exterior of the coating mediates aqueous solubility by the display of (c) amine or carboxyl groups, or (d) functionalized PEG. Common strategies for bioconjugation include: (a) thiol modifications or (b) polyhistidine or metallothionein (not shown) tags that penetrate the coating and interact with the surface of the QD; (f) electrostatic association with the coating; (g) nickel mediated assembly of polyhistidine to car- boxyl coatings; (h) maleimide activation and coupling; (i) active ester formation and coupling; (j) biotin-labeling and strepta- vidin-QD conjugates (not to scale) Reprinted with permission from [47]. beads or gold particles nor the fluorescent dyes or pro- teins are ideal probes for SPT [52]. The former suffer from being bulky, and the latter have limiting optical properties, and none of them are ideal for multiplexing studies. QDs on the other hand, are a great compromise between those two categories of probes. They have a moderate size, an extreme brightness, and an excellent resistance to photobleaching, and are ideal for multi- plexing studies even with simple setups [53]. The ease, by which they are bio-functionalized, further makes it possible to direct their binding towards almost any mo- lecular target of interest, and makes them a preferred choice for SPT (or single quantum dot tracking, SQT) studies [40]. In a typical SPT experiment, the motion of sparsely labeled single molecules is monitored by recording a time-lapse image series [54,55]. Subsequently, the indi- vidual main intensity peaks of the diffraction limited point spread functions from the well separated probes are detected and fitted computationally to a 2D Gaussian distribution in order to localize the centroid positions with a sub-diffraction limited spatial resolution of 10 - 40 nm. These centroid positions are then linked between successive frames using advances linking algorithms to build up single molecule trajectories [56-58]. In the case of tracking QDs these algorithms take QD blinking into account and are able to track molecules even if the QD is “off” for some frames. The resulting trajectories are typically analyzed by calculating the mean squared dis- placement (MSD), or alternatively by calculating the probability distributions of the squared displacements [54,55]. For this analysis, QDs has the advantage that the T. E. RASMUSSEN ET AL. 33 trajectories are long enough to allow for analysis of the single trajectories. This is in contrast to tracking with fluorescent dyes or proteins where the limited photosta- bility of these probes requires that all trajectories are pooled to give an analysis averaged in space and time hindering the observation of transient and rare events. By the shape of a MSD-vs-time plot it is possible to classify the mode of motion. A freely moving molecule undergo- ing Brownian motion will be a straight line in an MSD- vs-time plot, according to MSD = 4 D t, with D being the diffusion coefficient. Normally, however, plasma mem- brane molecules in live cells experience hindrance in their motion. This results in a MSD curve with a steep slope at small times and a more moderately increasing slope or a flat slope at longer times, indicating a time dependent diffusion. The reasons for the confinement are many, including interactions with membrane domains, cytoskeleton barriers, molecular crowding, membrane topology, and specific interactions with other membrane molecules [59-62]. Since the first paper on tracking single QDs appeared 10 years ago [63], many have followed, and contributed to the present understanding of structural, dynamical, and functional aspects of the plasma membrane. Studies have shown heterogeneous motion of individually labeled plasma membrane proteins and lipids, and that e.g. actin [64], the extracellular matrix [65], lipid microdomains [66,67], and cholesterol all affect the movement of these molecules. In neurobiology, single QD studies have been used extensively to study the motion of specific receptors in synapses under various cellular conditions and stimuli (for review see [68]). For instance, it was shown how GABAA receptors distribute asymmetrically across the axon growth cone in a microtubule and calcium depend- ent manner in response to a GABA gradient [69], and how AMPA receptor mobility is functional in recovery of synaptic activity [70]. Details of non-neural signal transduction pathways have also been revealed. Lidke and co-workers tracked EGF-conjugated QDs targeting the EGF receptor (erbB1) that is often found dysregu- lated in many cancers. Upon QD-ligand binding homo/ hetero dimerization and endosomal uptake was followed, and further, a previously unknown mechanism of retro- grade transport of the QD-EGF-EGFR complex from the filopodia to the cell body was found [71,72]. Tracking of membrane species using orthogonal multicolor QD la- beling strategies have been conducted tracking the same membrane species [64,73-76]. Recently, Clausen et al. has extended this to the orthogonal and simultaneous tracking of three different species, a lipid, a lipid-an- chored protein, and a transmembrane protein (In press PLOS ONE). Most QD tracking experiments are re- corded at video rate (25 or 30 Hz), however, using a camera with fast read-out, the extreme brightness of QDs allows for imaging at up to 1750 Hz [77]. 3.2. Single Particle Tracking of Hybrid Lipid-DNA Analogues Using Quantum Dots Labelling specificity is a major concern when performing SPT experiments. Preferably the conjugation system used to couple QDs to the target biomolecule should exhibit high specificity and strong avidity towards the target and as high degree of monovalency as possible. Another con- sideration when designing a conjugation system for SPT could be to make it as interchangeable as possible so the same system could be designed to bind different sized QDs to different targets enabling easy labelling for multi- color SPT experiments. As a result Vogel et al. have synthesized lipid-DNA analogues based on a polyaza crown ether depicted in Figure 5 [78]. The membrane anchors are linked to the nitrogens of the polyaza crown ether building block and can be interchanged to encom- pass either acyl chains or sterols such as cholesterol, which in turn can be used to probe different environ- ments in the plasma membrane or an artificial membrane system. The lipid-DNA analogue is then coupled to an oligomer of 17 bases at the 3’ end, which in turn can bind to a complimentary strand that has a 5’-biotinylated oli- gomer. Finally, streptavidin coated QDs can be bound to the biotin end of the complimentary strand with high specificity and avidity forming the SPT complex. The inherent advantage of this conjugation system is the specificity of the DNA complexation, since several dif- ferent lipid-DNA analogues with different membrane- anchor moieties could be labeled individually by using unique DNA sequences that bind only to certain colored QDs. 3.3. Single Particle Tracking Studies in Supported Lipid Bilayers SPT studies were conducted to investigate the application of previously mentioned lipid-DNA analogues in sup- ported lipid bilayers made from a binary lipid mixture of the phospholipids 1,2-Dipalmitoyl-sn-Glycero-3-phospho- choline (DPPC) and 1,2-Dioleoyl-sn-Glycero-3-phospho- choline (DOPC) in the ratio 1:1 supported on glass cov- erslips. The supported lipid bilayers were made as de- scribed in [79], briefly 24 mm in diameter glass cover- slips were cleaned in a basic piranha solution composed of Milli-Q water, ammonia and hydrogen peroxide in the ratio 6.2:1:1 respectively and heated to 100˚C for two hours. The processed cover slips were rinsed 3 times in Milli-Q water and dried in the oven prior to use. The phospholipids DPPC and DOPC (Avanti Polar Lipids) were dissolved in a mixture of 90% chloroform and 10% acetonitrile at a total lipid concentration of 10 mM. In order to visualize the membn the microscope 0.5 ranes i Open Access JMP  T. E. RASMUSSEN ET AL. Open Access JMP 34 Lipid/DNA analogue (SNV090) Biotinylated olygomer Stre p t av idi n 605-QD 3′ - T T T T G T G G A A G A A G T T G G T G - 5′ A C A C C T T C T T C A A C C A C - 3′ HO O O O O O O O N N N N H H H H H H Figure 5. Schematic representation of the Streptavidin-coated 605 QD coupled to the lipid-DNA analogue by a biotinylated complementary oligomer (not drawn to scale). mol% of the fluorescent lipid dye NBD-PC (Avanti Polar Lipids) was added to the lipid mixture. The supported lipid bilayers were prepared on the glass cover slips by spincoating using a Chemat technology spincoater KW- 4A. Spincoating is performed by adding 50 μl of the lipid solution is placed in the middle of the coverslip and was done using the following settings: Spincoater settings Steps Duration Speed 1 3 sec. 500 rpm 2 40 sec. 3000 rpm Afterwards the spincoated coverslips are stored in a vacuum desiccator for at least 24 hours to ensure the solvent has evaporated. Depending on the lipid concen- tration and of the applied volume of stock solution it is possible to form several bilayer on top of each other. Hydration of the dry spincoated bilayers is accomplished by placing the spin-coated coverslip into a special heat- ing stage composed of an aluminum holder and is fixed in place by a plastic ring. The hydration of the multi- laminar bilayer system enables one to wash of the top layers by gentle pipetting of the revealing the bottom bilayers on the coverslip. Heating of the setup is essential since the phase transition of the lipid mixture is above RT and it also aides in removing the top bilayers due to the increased mobility of the lipids in the bilayers. The hydration of the DOPC:DPPC 1:1 supported lipid bilay- ers was performed in Dulbecco’s phosphate buffered saline (PBS) (Sigma Aldrich) and the sample was heated to 60˚C well above the phase transition of the mixture and left to incubate for 30 minutes to ensure the mem- brane is its fluid state. Gentle pipetting was used to re- move excess lipid and to reveal the bottom bilayers on the coverslip. Below the phase transition of the lipid mixture DOPC should phase segregate to form ridged flower-shaped domains in which the fluorescent dye NBD-PC will not be present. Our microscopy studies have shown that these flower-shaped domains do not occur in the bottom lipid bilayer, possibly due to interact- tions between the glass and the lipids as well as due to the roughness of the glass surface. Therefore SPT studies were only performed in bilayers that exhibited the flower-like domains upon cooling below 40˚C - 36˚C. Once a planar patch of membrane has been established and characterized by fluorescent microscopy 100 μl of a 100 pM lipid-DNA analogue was added locally and in- cubated for 30 minutes. The PBS was exchanged in order to remove any non-incorporated membrane anchors. Af- terwards 100 μl of a 100 pM biotin coupled complimen- tary strand DNA solution was added and left to incubate for 15 minutes. The PBS was exchanged in order to re- move any unbound excess complimentary DNA, fol- lowed by the addition of 10 μl of a 1 nM streptavidin coated QD605 solution (Invitrogen), which was left to incubate for 10 min. The unbound QDs were removed by a final washing step. SPT studies were done by first bleaching away the signal from the NBD-PC in order to visualize the QDs bound to the membrane surface, see Figure 6. Thereafter, varying lengths (300 & 900 frames) were recorded and from these the individual trajectories of the QDs were extracted using ImageJ and a plugin called Spot tracker. The mean squared displacements of these trajectories were calculated using Wolfram Mathematica. From the preliminary results obtained from the SPT 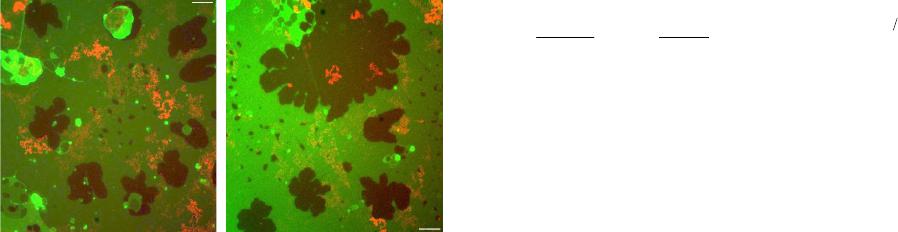 T. E. RASMUSSEN ET AL. 35 Figure 6. Epi-fluorescence images showing the membrane (shown in green) labelled with 1 mol% NBD-PC and su- perimposed tracks of QD605 coupled to the lipid-DNA ana- logue imbedded in the membrane (shown in red) (scalebar is 8 μm).The diffusion coefficient outside domains was 1.84 ± 1.08 μm2/sec whereas the diffusion coefficient of mole- cules in contact with domains was 1.22 ± 0.00 μm2/sec. The diffusion coefficient inside domains was 0.42 ± 0.20 μm2/sec. experiments three distinct diffusion patterns have been observed: unhindered diffusion outside domains, hin- dered diffusion at the border of the liquid and gel phase domains and finally unhindered diffusion inside the gel domains. It was unexpected to observe diffusion in the supposedly immobile DPPC gel phase present in the flower shaped domains—most likely this is due to the phase not being completely equilibrated. As to be ex- pected, the calculated 2D diffusion coefficients decrease in relation to the amount of contact the SPT complex has to the immobile phase. The most predominant diffusion events were observed to be the unhindered diffusion in the liquid phase. There were several observations of dif- fusion along the edge of the flower domains. Finally, some occasional events showed the SPT complex leaving the rim of a domain at continuing the diffusion into the fluid phase, whereas diffusion from inside a domain into the fluid phase was not observed. For biological membranes it has been reported that a typical lateral diffusion values range between 1 - 4 μm2 sec−1 and relating the values from this preliminary SPT study the values seem comparable. As a control Fluores- cence Recovery after Photobleaching (FRAP) was per- formed on the NBD-PC labeled fluid phase of the sup- ported bilayers. The measurements were performed on both membrane systems with a confocal laser-scanning microscope (CLSM) of the type (Zeiss LSM 510). Usu- ally when doing FRAP only a spot is bleached in order to determine the diffusion. However based on the work of Braeckmans et al. line FRAP is now possible [80]. A region of interest (ROI) was chosen to be 100*1 pixels, which would be used for all measurements. Compared to ordinary FRAP this improved technique is faster due to the scanning motion and is able to determine the diffusion constant and mobile fraction in more localized areas [80]. 12 022 00 0 0 0 0, ! n ec ne n K Ft rnra nr Fn (3) ,,0 ,,FytFykFyt Fy 0 (4) From the FRAP measurements the lateral diffusion coefficient of the fluid phase was determined to be 2.9 ± 1.8 μm2·sec−1, which is significantly higher than the 1.8 ± 1.1 μm2·sec−1 calculated from SPT measurement. This is not surprising since the acquisition rate was slower in the FRAP studies. When also keeping in mind that only the mobile fraction is included in the FRAP studies, no direct comparison between these numbers. Furthermore, it cannot be excluded that the QDs studied by SPT are bound to the membrane by more than only one lipid DNA analogue. The quantum dots used for these ex- periments from Invitrogen have approximately 37 strep- tavidin binding sites on one quantum dot. Therefore cross-linking of one quantum dot to several membrane anchors can be a possibility. Although only preliminary SPT data has been collected, the application of the lipid-DNA analogue seems to be promising. Additional data needs to be collected in order to evaluate the diffu- sion coefficient values reported so far as well as if the lipid-DNA analogue affects the physiochemical proper- ties of the supported bilayer. One of the most interesting applications of the anchor molecules is multi-color im- aging, which can be performed with different sizes (and hence colors) of quantum dots. If the anchors could be designed to have a particular preference for a particular lipid environment such as lipid domains these could then be specifically labeled. If such a labeling probe could be developed in vitro it would be possible to label domains in cells and thus one would be able study the dynamics of lipid rafts below the diffraction limit. 4. Quantum Dots as Probes for Two-Photon Fluorescence Correlation Spectroscopy Fluorescence correlation spectroscopy [FCS] is a very useful technique to study the movement and interactions of fluorescently labelled biomolecules and fluorophores. Elson, Magde et al. [81] introduced FCS in 1972 as an analogous technique to Dynamic Light Scattering and Relaxation Kinetics, in an attempt to overcome inherent limitations in both techniques [82]. In comparison with optical scattering, fluorescence is much more sensitive and selective enabling measurements with a low back- ground intensity and detection sensitivity that spans from nanomolar concentrations to the single molecule regime. FCS can be performed on either a confocal microscope or a two-photon fluorescence microscope [83], neverthe- less in both experimental setups a laser is focused down to a small focal volume in the order of a few femtoliters. Open Access JMP 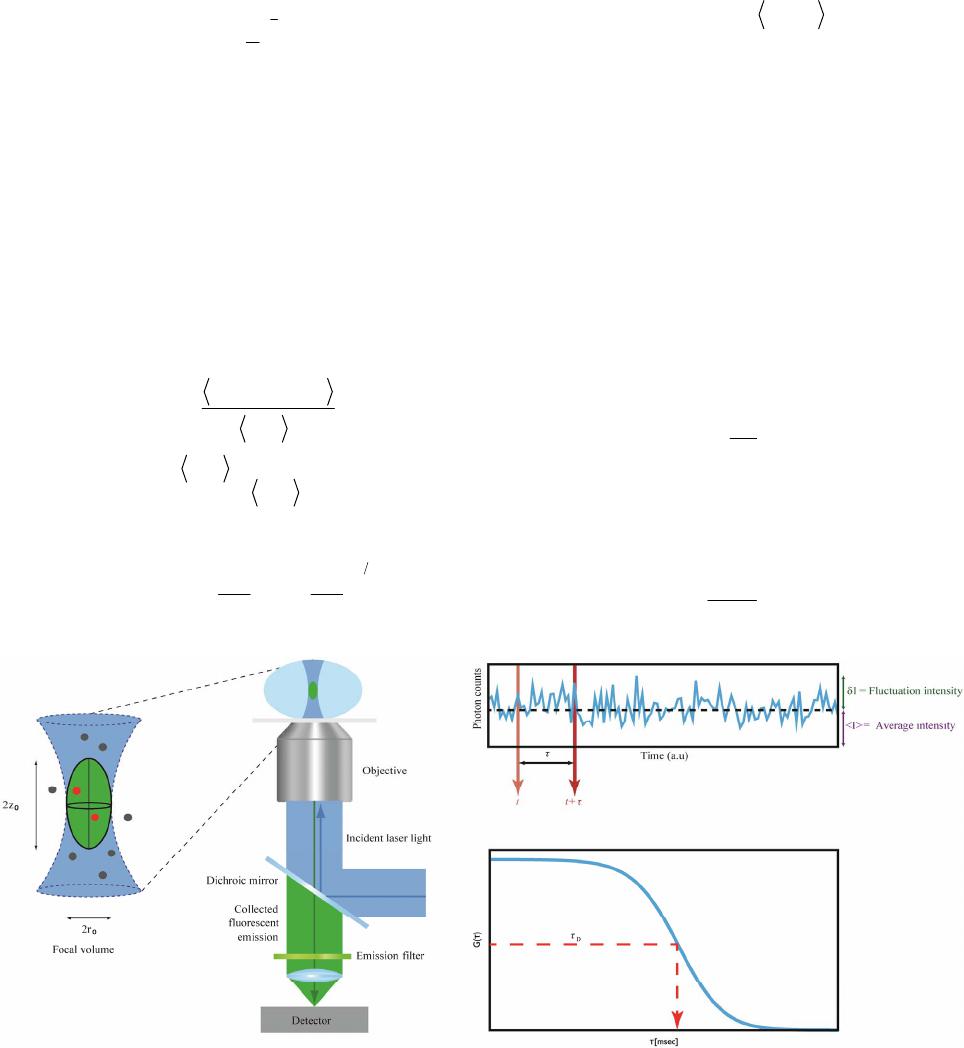 T. E. RASMUSSEN ET AL. Open Access JMP 36 In the confocal setup this is achieved by the confocal pinhole whereas the focal volume is inherent with a two- photon setup since only a small volume has high enough photon densities for two-photon excitation process to occur [84]. In Figure 7 is a schematic representation of a two-photon fluorescence correlation spectroscopy mi- croscope setup. The amplitude of the autocorrelation function is de- noted 0 G and is inversely proportional to the aver- age number of fluorescent species present in the focal volume, and therefore can be used to determine the con- centration of the fluorescent species. Consequently, the amplitude will increase with low concentrations and de- crease with high concentrations of the fluorescent species respectively. The focal volume for a two-photon setup is defined as 1 1 0Neff GNCV (8) 3 22 00 π 2 eff V rz (5) The decay rate of the autocorrelation function yields information about the occupancy of the fluorescent spe- cies in the focal volume. The function shows the prob- ability of the fluorescent species being in the focal vol- ume at 0t and still remaining in the focal volume at a later time point t . Since large molecules diffuse slower than smaller molecules the probability of finding the same molecule in the focal volume at higher values of t will also increase—in turn autocorrelation curves of large and small molecules will be shifted to the right and left respectively. where r0 is the beam waist in the radial direction and z0 is the beam waist in the axial direction. In order to deter- mine the size of the focal volume and the values of r0 and z0 the setup is calibrated by measuring the diffusion of a well-known fluorescent size standard such as a fluoro- phore of a fluorescent polymeric bead. When fluorescent or fluorescently labelled species diffuse randomly through the focal volume due to Brownian motion the resulting fluctuation fluorescence signal is recorded as a function of time. These fluctuations can be analyzed by the use of autocorrelation function G from which a diffusion value can be attained. The diffusion coefficient can be determined by relating the diffusion time τD, which is the τ value corresponding to the half value of the autocorrelation amplitude, to the following relation [86]. 2 GIt It It (6) 2 0 8 D r D (9) where tIt It is the deviation from the mean fluorescence intensity, t. The auto-correla- tion function for translational diffusion using two-photon excitation can be calculated as [85]. Furthermore, assuming that the diffusion molecules are spherical, the Stokes Einstein equation can be used to determine the hydrodynamic radius, R, from the deter- mined diffusion coefficient, D 11 022 00 88 11 DD GG rz 2 (7) 6π B kT DR (10) Figure 7. A schematic representation of a two-photon fluorescence correlation setup (a). Fluctuation measurements and the esulting autocorrelation analysis (b). r  T. E. RASMUSSEN ET AL. 37 where kB is the Boltzmann constant, T is the absolute temperature (kelvin), η is the viscosity of the medium quantum dots from Invitrogen. The FCS m and R is the hydrodynamic radius of the diffusion mole- cule [87]. In Figure 8 we present auto correlation curves and fits of 605sAv easurements were performed in 50 mM sodium borate pH 8.2 with 1% (w:v) BSA at RT (293 K). The experi- ments were performed on a custom built system which is described in [88] and the data was fitted using the Globalssoftware package developed at the Laboratory for Fluorescence Dynamics at the University of California Irvine. The excitation wavelength was 820 nm. It can be seen from the figure that fitting the data to the simple diffusion model (equation 8) does not fit the data satis- factorily. Adding a term to equation 8 which takes the power law nature [89] of the blinking of the QDs into account is seen to greatly improve the fit to the data. The auto-correlation function for translational diffusion and blinking two-photon excitation takes the form 112 G 022 00 11 88 11 DD Grz B B (11) where B is the blinking amplitude and is the exponent. The measured diffusion coefficient, of the QDs, found from a global analysis of the data using the model in- cluding the power law term was 19.7 ± 2 m2/sec. Using Stokes Einstein’s equation, as in [76] we find the hydro- dynamic radius to be 10.5 ± 1 nm. 1e-4 1e-3 1e-2 1e-1 Time [sec.] Data 0.5 nM Data 1 nM Fit+ex 0.5 nM Fit 1 nM Fit+ex 1nM Fit 0.5 nM 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 G(0) Figure 8. Auto correlation curves from FCS measurements carried out on 605 sAv QDs from Invitrogen. The solid line 5. Optical Trapping of Quantum Dots and mi- s are for 0.5 nM concentrations and the dotted lines are for 1 nM concentrations of QDs. The green lines represent the data, the red lines are the best fit of a model for a single freely diffusing species. The black lines are the best fit of a model for a single freely diffusing molecule together with a power law term to fit the blinking. It is clear from the figure that a simple diffusion model does not completely describe the data. Adding a term which takes the blinking of the QDs into account, greatly improves the fit. Optical tweezers are formed by tightly focusing a laser beam. Most often, optical tweezers are implemented in a microscope, equipped also with a quadrant photo-diode or a CCD for position detection with nanometer cro-second resolution. Using an optical trap, particles in the nanometer to micrometer range can be manipulated in 3D and corresponding values of forces (typically in the pico-Newton range) and distances can be measured. An inducible dipole in an optical field experiences a force in the direction of the field gradient. This 3D restoring force is harmonic in all three dimension and directed towards the focus of the laser beam: F = κx, where κx is denoted the trap stiffness in the x-direction and x is the position of the particle with respect to the equilibrium position within the trap, the center of the potential sketched in Figure 9(a). From tracking the Brownian motion and knowledge of the size of the trapped object and the vis- cosity and temperature of the surrounding fluid, one can deduce the force, F, acting on the trapped particle. QD optical tweezers (a) 200 10 3 Laser power (mW) 10 4 10 5 10 6 Intensity (S/N) (b) Figure 9. (a) Schematic of an optically trapped QD. The particle performs thermal motions in the harmonic poten- tial while the QD is simultaneously two-photon excited and emits polarized red light. b) Emitted intensity (signal/noise) versus laser power for QD agates of varying initial size. ggre The dotted line has a slope of 2. The different aggregates have different initial intensities; probably because of their different sizes. Regardless of initial aggregate size, all traces scales with a slope of 2; the characteristic of two-photon absorption. Reprinted with permission from. Reprinted with permission from [90]. Copyright (2012) American Che- mical Society. Open Access JMP  T. E. RASMUSSEN ET AL. 38 QDs, with inducible dipole moments, can be optically trapped by an infrared CW laser beam [91]. They serve as excellent combined force handles and visualization markers for investigations of individual cells, proteins, and biological filaments. In general, a fluorescent mole- cule can be two-photon excited if it simultaneously ab- sorbs two photons, and if the energy difference between the ground and excited state of the molecule corresponds to the added value of the energies of the two photons. QDs can be excited by a two-photon absorption process [92,93], with a two-photon absorption cross section which is large in comparison to other fluorophores used in multiphoton microscopy [94-97]. Jauffred and Odder- shede showed that QDs are readily excited through two- photon excitation by a relatively weak CW infrared laser beam, which can also be used to trap the QDs [98]. For a two-photon absorption the emitted intensity scales as P2, where P is the laser power, as shown in Figure 9(b). One hallmark of optical trapping is that there exists a linear relationship between κ and P. As shown in Figure 10(a), this is also true for optical trapping of two-photon excited QDs, thus supporting that the nonlinear absor- tion is only a weak perturbation that does not alter trap- p ping properties considerably. It is important to correctly choose the QD such that it matches the given experi- mental goals and conditions, e.g., available excitation 0.3 0.4 0.5 0.6 0.7 0.8 0 0.2 0.4 0.6 0.8 1 1.2 x 10-4 κ(pN/nm) P(W) ) (a) b) 10 15 20 25 30 1 1.5 2 x 10-4 κ(pN/nm) d (nm) (b) Figure 10. (a) Trapping strength versus laser power for individual QDs. The error bars denote one SEM. The dot- ted line is a linear fit to the data and it has a slope of 1.5 × 20−3 pN/nm/W. Reprinted with permission from [90]. Copy right (2012) American Chemical Society. (b) Trapping strength dependence on diameters, d. All error bars denote erred to the trapped parti- cl n limit, will align with the laser’s po- la rod would rotate in an oscillatory man- ne - one SEM and the dotted line is the mean value of κ: κ = 1.6 × 10−4 pN/nm. Reprinted with permission from [99]. lasers and filters. The trapping strength for QDs with different emission wavelengths λ (from 525 nm to 800 nm) and different physical sizes have similar trapping capabilities, as seen in Figure 10(b). The typical trap- ping strength measured for QDs is comparable to that obtained from optical trapping of silver and gold nano- particles of similar sizes [100-104]. See Reference [105] for a review on optical tweezing and nanoparticles. In summary, the QD can serve both as a handle for ma- nipulation and controlled force transduction and at the same time for visualization through two-photon absorp- tion of the trapping laser light. Optical trapping relies on light scattering, a linear property. Within the recent years efforts were done also to take advantage of the angular momentum of the laser beam [106,107]. Angular momentum is transf e by changing the direction of the laser polarization vector. Most often optical tweezers are based on linearly polarized light and an elongated trapped object, smaller than the diffractio rization vector, as shown in Figure 11(a). Objects lar- ger than the diffraction limit, as for instance bacteria, will align along the propagation direction of the trapping laser. By changing the direction of the laser’s polarization vec- tor, e.g., by using circularly polarized light, a trapped elongated nano-particle can be forced to rotate in a con- trolled fashion. This was successfully done by Head et al. [108], who used a focused laser bam to trap and two-photon excite elongated quantum rods (with an aspect ratio of 10). They used circularly polarized light and hence, the trapped quantum r. The quantum rod emitted light polarized along its long axis and by analyzing emitted light using a polariza- tion filter (as sketched in Figure 11(b)), they were able to QD optical tweezers 150 180 0 30 90 120 x PMT y x 210 240 270 300 330 PMT (a) (b) Figure 11.Schematic of an optically trapped quantum rod that aligns along the laser polarization but still perform significant 3D Brownian; (b) Schematic of the polarized emission from an optically trapped semiconductor quantum rod. The rod is polarized ot shows (a) light emitted from the trapped quantum along its long axis. The polar intensity pl the light ideally measured by two orthogonally polarized photomultipliers (x-PMT and y-PMT) as a function of the polarization angle measured from the x-axis, this is indeed consistent with experimental measurements [108]. Open Access JMP  T. E. RASMUSSEN ET AL. 39 detect that the trapped quantum rod was indeed rotating with the same frequency as the applied polarization vec- tor was changing with. The trapped particle could follow field rotations of, at least, 320 Hz [108]. These results pave the way for a system where the trapping and visu- alization is further combined with the ability to measure and transduce torque. Altogether, with a single infrared tightly focused laser beam one can optically trap, rotate, and perform two- photon excitation of individual colloidal QDs. Hence, individual quantum dots serve both as force and torque tranducers as well as visualization markers. antage article tracking, where their low ake them favorable in comparison t and for designed nano-scopic molecular electronics. 6. Conclusion In this review we demonstrated the versatility of using individual quantum dots as markers for individual mole- cules and as force probes. QDs can be advously applied for single p bleaching rates mo conventional fluorophores. Also, they give clear results in FCS experiments, and can be individually optically trapped, thus serving as force and torque transducing handles. The great spectral properties of QDs include high photostability, narrow emission spectra, blinking and low bleaching rate. The photo-physical properties of QDs make them ideal for multicolour experimental set- ups, since several differently coloured QDs can be dis- tinguished at the same time, while being illuminated by a single light source. When performing single particle tracking experiments, the blinking of QDs is a disadvan- tage as certain visited positions remain undetected. How- ever, blinking is also an advantage in localization mi- croscopy, in particular in the novel super-resolution techniques, as a subset of the QDs will emit in each im- age frame. For super-resolution localization microscopy QDs have a high potential in experiments involving posi- tion localization microscopy of lipids and proteins in the plasma membrane in live mammalian cells [7,109]. In live-cell experiments as well as in vivo experiments in animals, QDs have a strong potential as fluorescent probe as they can be easily inserted into the cytoplasm and then individually detected, conjugated and optically manipu- lated [37,110-118]. In FCS experiments, the great advan- tages of QDs are their well defined size and high fluores- cence emission, which make QDs a trust-worthy refer- ence or a good label of membrane proteins or lipids. The possibility to excite QDs with two-photon excitation also makes QDs applicable in skin permeability or drug up- take receptor studies in which deep tissue microscopy is necessary. Finally, as QDs can be individually manipu- lated and visualized using a single laser beam, they serve as the optimal handle and marker for the expanding ef- forts in uncovering the action of individual molecules 7. Acknowledgements We thank The Danish Council for Independent Research for grant to EAC and funding from The Danish Research Infrastructure to the Danish Molecular Biomedical Im- aging Center and Dr. Mathias Clausen for reading the manuscript before submission. REFERENCES [1] M. Bruchez, et al., Science, Vol. 281, 1998, pp. 2013- 2016. http://dx.doi.org/10.1126/science.281.5385.2013 [2] X. Gao, et al., Current Opinion in Biotechnology, Vol. 16, 2005, pp. 63-72. http://dx.doi.org/10.1016/j.copbio.2004.11.003 [3] B. O. DabbouPhysical Chemistry B, Vol. 101, 1 si, et al., The Journal of 997, pp. 9463-9475. http://dx.doi.org/10.1021/jp971091y [4] M. A. Hines and P. Guyot-Sionnest, The Journal of Physical Chemistry, Vol. 100, 1996, pp. 468-471. http://dx.doi.org/10.1021/jp9530562 [5] T. Kippeny, L. A. Swafford and S. J. Rosenthal, Journal of Chemical Education, Vol. 79, 2002, pp. 1094-1100. http://dx.doi.org/10.1021/ed079p1094 [6] Z. Deng, et al., Journal of the American Chemical Society, Vol. 134, 2012, pp. 17424-17427. http://dx.doi.org/10.1021/ja3081023 [7] B. C. Lagerholm, et al., Biophysical Journal, Vol. 91, 2006, pp. 3050-3060. http://dx.doi.org/10.1529/biophysj.105.079178 [8] S. F. Lee and M. A. Osborne, ChemPhyschem, Vol. 10, 2009, pp. 2174-2191. http://dx.doi.org/10.1002/cphc.200900200 [9] S. Hohng and T. Ha, Journal of the American Chemical Society, Vol. 126, 2004, pp. 1324-1325. http://dx.doi.org/10.1021/ja039686w [10] W. E. Moerner and M. Orrit, Science, Vol. 283, 1999, pp. 1670-1676. http://dx.doi.org/10.1126/science.283.5408.1670 [11] M. Nirmal, et al., Nature, Vol. 383, 1996, pp. 802-804. http://dx.doi.org/10.1038/383802a0 [12] N. Durisic, et al., ACS Nano, Vol. 3, 2009, pp. 1167-1175. http://dx.doi.org/10.1021/nn800684z [13] N. Durisic, et al., Biophysical Journal, Vol. 93, 2007, pp. 1338-1346. http://dx.doi.org/10.1529/biophysj.107.106864 [14] X. Wang, et al., Nature, Vol. 459, 2009, pp. 686-689. http://dx.doi.org/10.1038/nature08072 , pp. 5174-5178. [15] A. Biebricher, M. Sauer and P. Tinnefeld, The Journal of Physical Chemistry B, Vol. 110, 2006 http://dx.doi.org/10.1021/jp060660b [16] V. Fomenko a pp. 287-293. nd D. J. Nesbitt, Nano Letters, Vol. 8, 2008, 9http://dx.doi.org/10.1021/nl072660 ty, http://dx.doi.org/10.1021/ja711379k [17] Y. Chen, et al., Journal of the American Chemical Socie Vol. 130, 2008, pp. 5026-5027. Open Access JMP  T. E. RASMUSSEN ET AL. 40 [18] B. Mahler, et al., Nature Materials, Vol. 7, 2008, pp. 659-664. http://dx.doi.org/10.1038/nmat2222 [19] M. Nirmal, et al., Physical Review Letters, Vol. 75, 1995, pp. 3728-3731. http://dx.doi.org/10.1103/PhysRevLett.75.3728 [20] K. T. Shimizu, et al., Physical Review B, Vol. 6320, 2001, Article ID: 205316. [21] P. A. Frantsuzov and R. A. Marcus, Physical Review B, Vol. 72, 2005, Article ID: 155321. http://dx.doi.org/10.1103/PhysRevB.72.155321 120601. 1 [22] X. Brokmann, et al., Physical Review Letters, Vol. 90, 2003, Article ID: http://dx.doi.org/10.1103/PhysRevLett.90.12060 . [23] M. Kuno, et al., Journal of Chemical Physics, Vol. 115, 2001, pp. 1028-1040 http://dx.doi.org/10.1063/1.1377883 [24] M. Kuno, et al., The Journal of Ch 112, 2000, pp. 3117-3120. emical Physics, Vol. http://dx.doi.org/10.1063/1.480896 [25] L. Wang, The Journal of Ph 2001, pp. 2360-2364. ysical Chemistry B, Vol. 105, http://dx.doi.org/10.1021/jp0032053 [26] A. L. Efros and M. Ros 78, 1997, pp. 1110-1113. en, Physical Review Letters, Vol. http://dx.doi.org/10.1103/PhysRevLett.78.1110 [27] S. F. Lee and M. A. Osbor Chemical Society, Vol. 129, 2007, p ne, Journal of the American p. 8936-8937. http://dx.doi.org/10.1021/ja071876+ [28] W. G. J. H. M. van Sar Chemistry B, Vol. 105, 2001, pp. 8281 k, et al., The Journal of Physica -8284. l http://dx.doi.org/10.1021/jp012018h [29] H. Chen, H. Gai and E. S. Y tions, No. 13, 2009, pp. 1676-1678. eung, Chemical Communica- http://dx.doi.org/10.1039/b819356h [30] J. E. B. Katari, V. L. Colvin and A. P. Alivisatos Journal of Physical Chemistry, Vol. , The 98, 1994, pp. 4109- 4117. http://dx.doi.org/10.1021/j100066a034 [31] P. Hoyer, et al., Nano Letters, Vol. 11, 2011, pp. 245-250. http://dx.doi.org/10.1021/nl103639f [32] E. A. Christensen, P. Kulatunga and B. C. Lagerholm, PLoS One, Vol. 7, 2012, Article ID: e44355. e.0044355http://dx.doi.org/10.1371/journal.pon [33] Y. Xing, Z. Xia and J. Rao, IEEE Transactions on Nano- Bioscience, Vol. 8, 2009, pp. 4-12. http://dx.doi.org/10.1109/TNB.2009.2017321 [34] X. Michalet, et al., Science, Vol. 307, 2005, pp. 538-544. http://dx.doi.org/10.1126/science.1104274 [35] T. Pons and H. Mattoussi, Annals of Biomedical Engi- neering, Vol. 37, 2009, pp. 1934-1959. http://dx.doi.org/10.1007/s10439-009-9715-0 [36] M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, Vol. 281, 1998, pp. 2013-2016. 3 http://dx.doi.org/10.1126/science.281.5385.201 ol. 298, 2002 pp. 1759-1762. [37] B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, V, http://dx.doi.org/10.1126/science.1077194 [38] X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J way, J P. Larson, N. F. Ge, F. Peale and M. . A. Tread- P. Bruchez, Nature Biotechnology, Vol. 21, 2002, pp. 41-46. http://dx.doi.org/10.1038/nbt764 [39] T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, /10.1021/nl035172j A. L. Rogach, S. Keller, J. Rädler, G. Natile and W. J. Parak, Nano Letters, Vol. 4, 2004, pp. 703-707. http://dx.doi.org J. R. McBride [40] S. J. Rosenthal, J. C. Chang, O. Kovtun, and I. D. Tomlinson, Chemistry & Biology, Vol. 18, 2011, pp. 10-24. http://dx.doi.org/10.1016/j.chembiol.2010.11.013 [41] G. T. Hermanson, “Bioconjugate Techniques,” Academic 46. Press, San Diego, 1996. [42] I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mat- toussi, Nature Materials, Vol. 4, 2005, pp. 435-4 http://dx.doi.org/10.1038/nmat1390 [43] S. K. Chakraborty, J. A. J. Fitzpatrick, J. A. Phillippi, S. Andreko, A. S. Waggoner, M. P. Bruchez and B. Ballou, Nano Letters, Vol. 7, 2007, pp. 2618-2626. http://dx.doi.org/10.1021/nl0709930 [44] S. Pathak, M. C. Davidson and G. A. Silva, Nano Letters, Vol. 7, 2007, pp. 1839-1345. http://dx.doi.org/10.1021/nl062706i [45] T. Jamieson, R. Bakhshia, D. Petrovaa, R. Pococka Iman and A. M. Seifalian, Biomate , M. rials, Vol. 28, 2007, pp. 4717-4732. http://dx.doi.org/10.1016/j.biomaterials.2007.07.014 [46] E. L. Bentzen, I. D. Tomlinson, J. Mason, P R. Warnement, D. Wright, E. Sande . Gresch, M. rs-Bush, R. Blakely and S. J. Rosenthal, Bioconjugate Chemistry, Vol. 16, 2005, pp. 1488-1494. http://dx.doi.org/10.1021/bc0502006 [47] W. R. Algar, A. J. Tavares and U. J. Krull, Analytica Chimica Acta, Vol. 673, 2010, pp. 1-25. http://dx.doi.org/10.1016/j.aca.2010.05.026 l of [48] D. Alcor, G. Gouzer and A. Triller, European Journa Neuroscience, Vol. 30, 2009, pp. 987-997. http://dx.doi.org/10.1111/j.1460-9568.2009.06927.x [49] M. J. Saxton and K. Jacobson, Annual Review of Bio- physics and Biomolecular Structure, Vol. 26 373-399. , 1997, pp. http://dx.doi.org/10.1146/annurev.biophys.26.1.373 [50] C. Joo, H. Balci, Y. Ishitsuka, C. Buranach Annual Review of Biochemistry, Vol. 77, 200 ai and T. Ha, 8, pp. 51-76. http://dx.doi.org/10.1146/annurev.biochem.77.070606.10 1543 [51] M. Brameshuber and G. J. Schutz, Nature Methods, Vol. 5, 2008, pp. 133-134. http://dx.doi.org/10.1038/nmeth0208-133 [52] M. P. Clausen and B. C. Lagerholm, Current Protein Peptide Science, Vol. 12, 2011, pp. 699-713. & [53] F. Pinaud, S. Clarke, A. Sittner and M. Dahan, Nature Methods, Vol. 7, 2010, pp. 275-285. http://dx.doi.org/10.1038/nmeth.1444 [54] S. Wieser and G. J. Schutz, Methods, Vol. 46, 2008, pp. Open Access JMP  T. E. RASMUSSEN ET AL. 41 131-140. http://dx.doi.org/10.1016/j.ymeth.2008.06.010 [55] H. Bannai, S. Lévi, C. Schweizer, M. Triller, Nature Protocols, Vol. 1, 2006, pp. 2628- Dahan and A. 2634. http://dx.doi.org/10.1038/nprot.2006.429 [56] A. Serge, N. Bertaux, H. Rigneault and D. Marguet, Na- ture Methods, Vol. 5, 2008, pp. 687-694. http://dx.doi.org/10.1038/nmeth.1233 . Kuwata, S. Grin- [57] K. Jaqaman, D. Loerke, M. Mettlen, H stein, S. L. Schmid and G. Danuser, Nature Methods, Vol. 5, 2008, pp. 695-702. http://dx.doi.org/10.1038/nmeth.1237 [58] I. F. Sbalzarini and P. Koumoutsakos, Journal of Stru tural Biology, Vol. 151, 2005, pp. 182-195 c- . http://dx.doi.org/10.1016/j.jsb.2005.06.002 [59] J. Adler, A. I. Shevchuk, P. Novak, Y. E. Parmryd, Nature Methods, Vol. 7, 2010 Korchev , pp. 170-171. and I. http://dx.doi.org/10.1038/nmeth0310-170 [60] A. Kusumi, Y. M. Shirai, I. Koyama-Honda, K. G. N. Suzuki and T. K. Fujiwara pp. 1814-1823. , FEBS Letters, Vol. 584, 2010, http://dx.doi.org/10.1016/j.febslet.2010.02.047 [61] D. Lingwood and K. Simons, Science, Vol. 3 46-50. 27, 2010, pp. 4621http://dx.doi.org/10.1126/science.117 l Society, V [62] P. S. Niemela, M. S. Miettinen, L. Monticelli, H. Ham- maren, P. Bjelkmar, T. Murtola, E. Lindahl and I. Va lainen, Journal of the American Chemica ttu- ol. 132, 2010, pp. 7574-7575. http://dx.doi.org/10.1021/ja101481b [63] M. Dahan, S. Lé and A. Triller, Science, Vol. 302, 2003, pp. 442- vi, C. Luccardini, P. Rostaing, B. R 445. iveau http://dx.doi.org/10.1126/science.1088525 [64] N. L. Andrews, K. A. Lidke, J. R. Pfeiffer, A. R. Burns, B. S. Wilson, J. M. Oliver and D. S. Lidke, Nature Cell Bi- ology, Vol. 10, 2008, pp. 955-963. http://dx.doi.org/10.1038/ncb1755 [65] R. Frischknecht, M. Heine, D D. Choquet and E. D. Gundelfinger, Natur . Perrais, C. I. Seidenbecher e Neuroscience , , Vol. 12, 2009, pp. 897-904. http://dx.doi.org/10.1038/nn.2338 [66] F. Pinaud, X. Michalet, G. Iyer, E. Margeat, H.-P. Moore and S. Weiss, Traffic, Vol. 10, 2009, pp. 691-712. http://dx.doi.org/10.1111/j.1600-0854.2009.00902.x [67] I. R. Bates, B. Hébert, Y. S. Luo, J. L. Kolin, P. W. Wiseman and J. W Liao, A. I. Bachir, D . Hanrahan, Biophysi- . cal Journal, Vol. 91, 2006, pp. 1046-1058. http://dx.doi.org/10.1529/biophysj.106.084830 [68] A. Triller and D. Choquet, 359-374. Neuron, Vol. 59, 2008, pp. j.neuron.2008.06.022http://dx.doi.org/10.1016/ [69] C. Bouzigues, M. Morel, A. Triller and M. Dahan, Pro- ceedings of the National Academy of Sciences of the States of America, Vol. 104, 2007, pp. 11251-11256. United http://dx.doi.org/10.1073/pnas.0702536104 [70] M. Heine, L. Groc, R. Frischknecht, J.-C. Béïque, B. Lou- nis, G. Rumbaugh, R. L. Huganir, L. Cogne quet, Science, Vol. 320, 2008, pp. 201-205. t and D. Cho- http://dx.doi.org/10.1126/science.1152089 [71] D. S. Lidke, K. A. Lidke, B. Rieger, T. M. Jovin and D. J. Arndt-Jovin, Journal of Cell Biology, Vol. 170, 2005, pp. 619-626. http://dx.doi.org/10.1083/jcb.200503140 [72] D. S. Lidke, P. Nagy, R. Heintzmann, D. J. Arndt-Jo N. Post, H. E. Grecco, E. A. Jares-Erijma vin, J. n and T. M. Jovin, Nature Biotechnology, Vol. 22, 2004, pp. 198-203. http://dx.doi.org/10.1038/nbt929 [73] S. T. Low-Nam, K. A. Lidke, P. J. Cutler, R. C. P. M. van Bergen en Henegouwen, B. S. W Roovers, ilson and D. S. Lidke, Nature Structural and Molecular Biology, Vol. 18, 2011, pp. 1244-1249. http://dx.doi.org/10.1038/nsmb.2135 [74] C. J. You, S. Wilmes, O. Beutel, S. Löchte, Y. Podople- lowa, F. Roder, C. Richter, T. Seine, D. Schaible, G. Uzé, S. Clarke, F. Pinaud, M. Dahan and J. Piehler, Ange- wandte Chemie International Edition, Vol. 49, 2010, pp. 4108-4112. http://dx.doi.org/10.1002/anie.200907032 [75] N. L. Andrews, J. R. Pfeiffer, A. M. Martinez, D. M. Haaland, R. W. Davis, T. Kawakami, J. M. Oliver, B. S. Wilson and D. S. Lidk 469-479. e, Immunity, Vol. 31, 2009, pp. muni.2009.06.026http://dx.doi.org/10.1016/j.im [76] E. C. Arnspang, J. R. Brewer and B. C. Lagerholm, PLoS ONE, Vol. 7, 2012, Article ID: e48521. http://dx.doi.org/10.1371/journal.pone.0048521 [77] M. P. Clausen and B. C. Lagerholm, Nano Letters, Vol. 13, 2013, pp. 2332-2337. http://dx.doi.org/10.1021/nl303151f [78] K. Rohr and S. Vogel, Chembiochem: A European Jour- nal of Chemical Biology, Vol. 7, 2006, pp. 463-470. http://dx.doi.org/10.1002/cbic.200500392 [79] A. C. Simonsen and L. A. Bagatolli, Langmuir: The ACS Journal of Surfaces and Colloids, Vol. 20, 9720-9728. 2004, pp. http://dx.doi.org/10.1021/la048683+ [80] K. Braeckmans, K. Remaut, R. E. Vandenbroucke, B. Lucas, S. C. De Smedt and J. Dem Journal, Vol. 92, 2007, pp. 2172-218 eester, Biophysical 3. http://dx.doi.org/10.1529/biophysj.106.099838 [81] D. Magde, W. W. Webb and E. Elson, Physical Rev Letters, Vol. 29, 1972, pp. 705-708. iew http://dx.doi.org/10.1103/PhysRevLett.29.705 [82] E. L. Elson, Journal of Biomedical Optics, Vol. 9, 2004, pp. 857-864. http://dx.doi.org/10.1117/1.1779234 -4 [83] K. M. Berland, P. T. C. So and E. Gratton, Biophysical Journal, Vol. 68, 1995, pp. 694-701. http://dx.doi.org/10.1016/S0006-3495(95)80230 306.132 [84] E. Haustein and P. Schwille, Annual Review of Biophysics and Biomolecular Structure, Vol. 36, 2007, pp. 151-169. http://dx.doi.org/10.1146/annurev.biophys.36.040 612 [85] P. Schwille, U. Haupts, S. Maiti and W. W. Webb, Bio- physical Journal, Vol. 77, 1999, pp. 2251-2265. http://dx.doi.org/10.1016/S0006-3495(99)77065-7 [86] E. Haustein and P. Schwille, Current Opinion in Struc tural Biology, Vol. 14, 2004, pp. 531-540. - http://dx.doi.org/10.1016/j.sbi.2004.09.004 [87] A. Einstein, Annalen der Physik, Vol. 322, 1905, pp. 549- 560. http://dx.doi.org/10.1002/andp.19053220806 [88] J. Brewer, J. B. de la Serna, K. Wagner and L. A. Baga- Open Access JMP  T. E. RASMUSSEN ET AL. Open Access JMP 42 tolli, Biochimica et Biophysica Acta, Vol. 1798, 2010, pp. 1301-1308. http://dx.doi.org/10.1016/j.bbamem.2010.02.024 [89] A. I. Bachir, N. Durisic, B. Hebert, P. Grütter and P. W. Wiseman, Journal of Applied Physics, Vol. ticle ID: 064503. 99, 2006, Ar- .2175470 http://dx.doi.org/10.1063/1 [90] L. Jauffred and L. B. Oddershede, Nano Letters, Vol. 10, 2010, pp. 1927-1930. http://dx.doi.org/10.1021/nl100924z [91] L. Jauffred, A. C. Richardson and L. B. Oddershede, Nano letters, Vol. 8, 2008, pp. 3376-3380. http://dx.doi.org/10.1021/nl801962f [92] M. Schmidt, S. A. Blanton, M. A. Hines and P. Guyot- Sionnest,. Physical Review B, Vol. 53, 1996, pp. 12629- 12632. http://dx.doi.org/10.1103/PhysRevB.53.12629 [93] S. A. Blanton, A. Dehestani, P. C. Lin and P. Guyot-Sion- nest, Chemical Physics 322. Letters, Vol. 229, 1994, pp. 317- 614(94)01057-9 http://dx.doi.org/10.1016/0009-2 f Fluorescence, Vol [94] T. Wang, J. Y. Chen, S. Zhen, P. N. Wang, C. C. Wang, W. L. Yang and Q. Peng, Journal o 19, 2009, pp. 615-621. . http://dx.doi.org/10.1007/s10895-008-0452-9 [95] L. Pan, A. Ishikawa and N. Tamai, Physical Review B, Vol. 75, 2007, p. 161305. http://dx.doi.org/10.1103/PhysRevB.75.161305 [96] S. C. Pu, M. J. Yang, C.-C. Hsu, C.-W. Lai, C.-C. Hsieh, S. H. Lin, Y. M. Cheng and P. T. Chou, Small, Vol. 2, 2006, pp. 1308-1313. http://dx.doi.org/10.1002/smll.200600157 [97] D. R. Larson, W. R. Zipf M. P. Bruchez, F. W. Wise and W. W. Webb, el, R. M. Williams, S. W. Clark Science , , Vol. 300, 2003, pp. 1434-1436. http://dx.doi.org/10.1126/science.1083780 [98] L. Jauffred and L. B. Oddershede, Nano Letters, Vol. 10, 2010, pp. 1927-1930. http://dx.doi.org/10.1021/nl100924z [99] L. Jauffred, M. Sletm shede, Quantum dots as handles for optica oen, F. Czerwinski and L. Odder- l manipulation. arXiv.org, 2010. physics.optics. [100] F. Hajizadeh and S. N. S. Reihani, Optics Express, Vol. 18, 2010, pp. 551-559. http://dx.doi.org/10.1364/OE.18.000551 [101] C. Selhuber-Unkel, I. Zins, O. Schubert and C. Sönnich- sen, Nano letters, Vol. 8, 2008, pp. 2998-3003. http://dx.doi.org/10.1021/nl802053h [102] L. Bosanac, T. Aabo, P. M. Bendix and L. B. Oddershede, Nano letters, Vol. 8, 2008, pp. 1486-1491. http://dx.doi.org/10.1021/nl08049 0+ 5, pp. 1937-1942. [103] P. M. Hansen, V. K. Bhatia, N. Harrit and L. Oddershede, Nano letters, Vol. 5, 200 http://dx.doi.org/10.1021/nl051289r [104] K. Svoboda and S. M. Block, Optics Let 1994, pp. 930-932. ters, Vol. 19, http://dx.doi.org/10.1364/OL.19.000930 [105] M. Dienerowitz, M. Mazilu and K. Dholak Nanophotonics, Vol. 2, 2008, Article ia, Journal of ID: 021875. http://dx.doi.org/10.1117/1.2992045 [106] M. Padgett and R. Bowman, Nature Pho 2011, pp. 343-348. tonics, Vol. 5, http://dx.doi.org/10.1038/nphoton.2011.81 [107] K. Dholakia and T. Čižmár, Nature Photonics, 2011, pp. 335-342. Vol. 5, http://dx.doi.org/10.1038/nphoton.2011.80 [108] C. R. Head, E. Kammann, M. Zanella, L. Mannabc and P. G. Lagoudakis, Nanoscale , Vol. 4, 2012, p p. 3693-3697. http://dx.doi.org/10.1039/c2nr30515a [109] K. Lidke, B. Rieger, T. Jovin and R. Heintzman Express, Vol. 13, 2005, pp. 7052-7062. n, Optics http://dx.doi.org/10.1364/OPEX.13.007052 [110] P. Pierobon, S. Achouri, S. Courty, A. R. Dunn, J. A. Spudich, M. Dahan and G. Cappello, Biophysical Journal, 45 Vol. 96, 2009, pp. 4268-4275. http://dx.doi.org/10.1016/j.bpj.2009.02.0 . Gelfand and [111] C. Kural, H. Kim, S. Syed, G. Goshima, V. I P. R. Selvin, Science, Vol. 308, 2005, pp. 1469-1472. http://dx.doi.org/10.1126/science.1108408 [112] L. B. Oddershede, Nature Chemical Biology pp. 879-886. , Vol. 8, 2012, http://dx.doi.org/10.1038/nchembio.1082 50- .035 [113] A. Biebricher, W. Wende, C. Escudé, A. Pingoud and P. Desbiolles, Biophysical Journal, Vol. 96, 2009, pp. L L52. http://dx.doi.org/10.1016/j.bpj.2009.01 et- [114] Y. Ebenstein, N. Gassman, S. Kim, J. Antelman, Y. Kim, S. Ho, R. Samuel, X. Michalet and S. Weiss, Nano L ters, Vol. 9, 2009, pp. 1598-1603. http://dx.doi.org/10.1021/nl803820b [115] I. J. Finkelstein, M. L. Visnapuu and E. C. Greene, Na- ture, Vol. 468, 2010, pp. 983-987. http://dx.doi.org/10.1038/nature09561 [116] A. Seitz and T. Surrey, The EMBO 2006, pp. 267-277. Journal, Vol. 25, http://dx.doi.org/10.1038/sj.emboj.7600937 [117] D. M. Warshaw, G. G. Kennedy, S. S. mentsova, S. Beck and K. M. Trybus, Work, E. B. Kre- Biophysical Jour- 1529/biophysj.105.061903 nal, Vol. 88, 2005, pp. L30-L32. http://dx.doi.org/10. emsey, L. B. 8105 [118] M. Eriksen, P. Horvath, M. A. Sørensen, S. S Oddershede and L. Jauffred, Journal of Nanomaterials, Vol. 2013, 2013, Article ID: 468105. http://dx.doi.org/10.1155/2013/46
|