O. G. EL-DIEN ET AL. 669
rethrum tatsienense by the high-performance liquid chro-
ma-tographic technique coupled with photodiode array
detection [12].
Among the modern Analytical tools, HPTLC is a pow-
erful analytical method equally suitable for qualitative and
quantitative analytical tasks. HPTLC is playing an im-
portant role in today’s analytical world, not in competition
to HPLC but as a complementary method [13].
HPTLC produces visible chromatograms complex in-
formation about the entire sample available at a glance.
Multiple samples are seen simultaneously, so that refer-
ence and test samples can be compared for identification.
Similarities and differences are immediately apparent and
with the help of the image comparison. Several chroma-
tograms can be compared directly, even from different
plates. They can be evaluated either by the image based
software video-scan or by scanning densitometry with
TLC scanner, measuring the absorption and/or fluores-
cence of the substances on the plate. TLC is an offline
technique: the subsequent steps are relatively independent,
allowing parallel treatment of multiple samples during
chromatography, derivatization and detection.
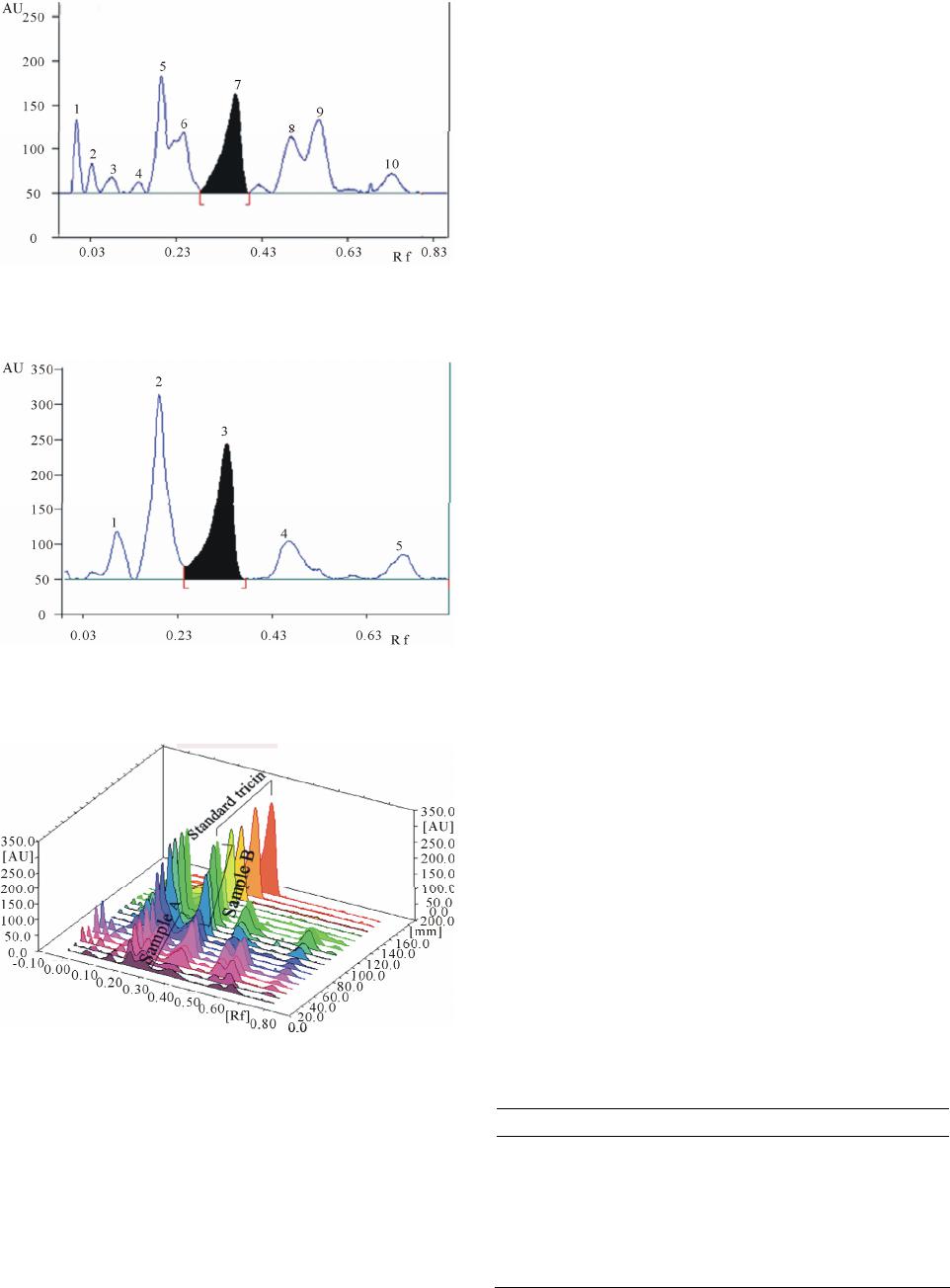
We managed to isolate tricin for the first time from
genus Spergularia. Tricin was isolated from the chloro-
form fraction of Spergularia marina; meanwhile it was
found in the chloroformic fraction of Spergularia dian-
dra in a large amount.
In view of the fore-mentioned points, the aim of this
work is to develop a simple, non expensive, accurate,
specific and repeatable HPTLC method for the determi-
nation of tricin in Genus Spergularia. Literature review
revealed that only one HPLTC method has been reported
for the analysis of tricin among other five flavonoids, but
the method was not thoroughly validated [14].
2. Experimental
2.1. Chemicals and Reagents
HPTLC analyses were performed on Merck 20 cm × 10
cm (0.25 mm) plates. Tricin used as standard material
was obtained from the Department of Pharmacognosy,
Faculty of Pharmacy, University of Alexandria and
tested for its purity by TLC and spectroscopic method.
All the reagents used in the experiment were of analytical
grade and were supplied by Merck, Darmstadt, Germany.
2.2. Preparation of Standard Solution
A weight of 4.0 mg of standard tricin was accurately
weighed, quantitatively transferred into a 10 ml volumet-
ric flask, dissolved in methanol and the volume was ad-
justed with the same solvent.
2.3. Preparation of Sample Solutions
Fresh aerial parts, 500 g of each of Spergularia marina
and Spergularia diandra were accurately weighed and
exhaustively extracted using (2 L) 70% ethanol at room
temperature. Each alcoholic extract was concentrated to
200 ml volume, fractionated successively by (600 ml)
petroleum ether, then by (600 ml) chlorofrorm. The
chloroform fraction was evaporated under reduced pres-
sure to give 440 mg of extract for S. marina and 770 mg
for S. diandra. A weight of 225 mg of each of the
chloroformic fraction of Spergularia marina and Sper-
gularia diandra were accurately weighed, dissolved in a
mixture of methanol and chloroform (8:2), transferred
quantitatively to 10 ml volumetric flask, adjusted to
volume with the same mixture and shaken to mix thor-
oughly to give samples A and B, respectively.
2.4. HPTLC—Densitometric Procedure
2.4.1. Instrumentation
Sample solutions for HPTLC analyses were applied by
means of a CAMAG (Wilmington, NC) Linomat IV
automated spray-on band applicator. Zones were quanti-
fied by linear scanning at 270 nm with a CAMAG TLC
Scanner 3 with a deuterium source in the reflection mode,
slit dimension settings of length 6 and width 0.1, mono-
chromator bandwidth 20 nm, and a scanning rate of 15
mm·s−1. The peak areas of chromatograms were deter-
mined using CATSTLC software (version 4.X).
2.4.2. Chromatographic Procedure
Sample and standard solutions were applied in the form
of bands on pre-coated HPTLC silica gel plates 60 F-254
(20 cm × 10 cm with 250 µm thickness) by means of
Linomat IV automated spray-on band applicator operated
with the following settings: band length 6 mm, applica-
tion rate 15 s/µl, distance between each two bands 4 mm,
distance from the plate side edge 1 cm, distance from the
bottom of the plate 1 cm.
The volumes applied for routine analysis were dupli-
cate 1.0 μl, 3.0 μl and 5.0 μl of the TLC tricin standard
(0.4, 1.2 and 2.0 μg) and triplicate 6 μl and 4 μl aliquots
of samples solutions A and B respectively. A volume of
20 ml of mobile phase (system IV) were used for devel-
opment.
Linear ascending development of the plates was car-
ried out in 20 cm × 20 cm Camag HPTLC twin trough
chamber saturated with the mobile phase. The optimized
chamber saturation time for the mobile phase was 30 min
at room temperature. Plates were developed to a distance
of 8 cm beyond the origin. The development time was 17
min. After development, the plates were air-dried for 5
min. Densitometric scanning was performed on Camag
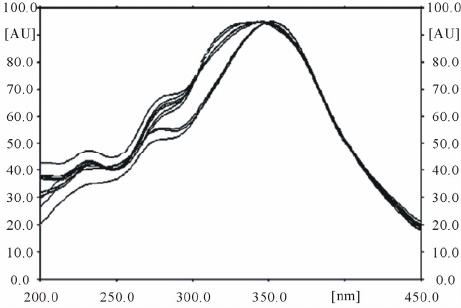
TLC scanner III in the reflectance-absorbance mode at λ
270 nm and operated by WINCATS software (V. 3.1).
The source of radiation utilized was deuterium lamp
Open Access AJAC