Journal of Biomedical Science and Engineering
Vol.10 No.02(2017), Article ID:74262,17 pages
10.4236/jbise.2017.102007
A Novel Assay for Measuring Total Antioxidant Capacity in Whole Blood and Other Biological Samples
Riccardo Ciuti, Gianfranco Liguri
Post-Graduate School of Clinical Biochemistry, University of Florence, Florence, Italy

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 22, 2016; Accepted: February 17, 2017; Published: February 20, 2017
ABSTRACT
A new method for rapidly determining total antioxidant capacity (TAC) in a biological sample has been devised and evaluated. A surfactant present in the reagent causes the lysis of cells in the sample. Glutathione, ascorbic acid, protein thiols and other main antioxidant substances in the sample react with a specific redox chromogen (2,6-dichlorophenolindophenol) and produce a change in the color of the reagent, which is determined photometrically at 630 nm. This new method quantifies the overall capacity of the sample to reduce a redox chromogen and it requires the minimum volumes and time. The assay can be used on a wide variety of biological samples including whole blood, serum, plasma, tissue extracts and homogenates, food, wine, fruit juice and other beverages. Results are expressed in terms of mmol/L glutathione equivalents. TAC can be assayed manually, on automatic analyzers or micro-plate readers.
Keywords:
Total Antioxidant Capacity, Free Radicals, Total Antioxidant Status, Oxidative Stress, Intracellular TAC

1. Introduction
During the course of a lifetime, the human body is continuously exposed to potentially harmful oxidative stress caused by either an overproduction of reactive oxygen species (ROS) and reactive nitrogen species (NOS) or a depletion of endogenous protective anti-oxidative capacity [1] [2] [3] . These highly reactive moieties are generated from exogenous sources such as diet, environmental pollution, tobacco smoking, ionizing radiation, UV-light exposure, organic solvents, pesticides and medications, and from endogenous sources due to degenerative diseases, inflammation, ischemia, etc. Noticeably, valuable ROS are also generated endogenously, during normal physiological processes. These include products of mitochondrial respiration and reactions involving enzymes such as lipoxygenase and xanthine oxidase, as well as oxidants generated by phagocytic cells of the immune system [4] .
Antioxidant status reflects the balance between the antioxidant defenses and oxidants in living organisms. Oxidative stress emerges when highly oxidative species far exceed antioxidant defenses. Free radicals are unstable and so highly reactive to be able, if not properly neutralized, to damage biological structures such as DNA, proteins and lipids and to disrupt cellular functions. Oxidative stress has been implicated in the pathogenesis of major chronic diseases: neurodegenerative disorders, cardiovascular disease, diabetes, carcinogenesis and atherosclerosis [5] [6] [7] [8] [9] . For these reasons, there is strong interest by preventive healthcare and food science in avoiding oxidative stress and maintaining an antioxidant status that supports good health. In order to get protected against damage from excess of free radicals and ROS, humans and other living organism are equipped with powerful and complex antioxidant systems comprising specific enzyme systems (SOD, GPx, CAT) and non-enzymatic antioxidants such as tocopherol, retinol, reduced glutathione, ascorbic and uric acids, able to inactivate radical species. These compounds, provided with reducing power, are either produced in the body or derived from the diet [10] [11] . It should also be underlined that individual antioxidants don’t function as isolated entities, but as part of complex systems with relevant interdependence and synergistic effects.
Evaluation of the overall antioxidant status of the organism should encompass a complex and laborious process of integrating TAC values measured in all tissues of the body, yet assaying TAC in whole blood will provide a much simpler way when considering this fluid tissue continuously exchanges materials and is in metabolic equilibrium with other tissues of the body.
The role of the intake of antioxidant nutrients able to counteract oxidative stress is well established in a great number of disease including cancer, cardiovascular and neurodegenerative pathologies [12] [13] . Therefore, there is an increased interest of many research groups to evaluated TAC in foods. The consumption of fruits and vegetables, as well as of grains and nuts, containing antioxidants as polyphenols (quercetin, luteolin, anthocyanins), vitamins (A, E and C), lycopene, etc., potentially increases total antioxidant capacity of consuming population [14] [15] . The higher TAC values of foods in Mediterranean diet could in part explain why adherence to this diet reduces mortality risk of CHD [16] . Nevertheless, high antioxidant supplementation has failed to improve health and is not recommended, i.e. an excessive vitamin E intake can cause pathologies [17] [18] .
Therefore, TAC evaluation in foods and in blood represents an important tool for assessing the ability of a diet to maintain or recover an adequate redox homeostasis of the body and to avoiding ineffective or adverse supplementations.
1.1. The “Matrix” Issue
In humans, the antioxidant substances capable in various ways and capabilities of neutralizing ROS are numerous and can be found in different compartments of the organism, nonetheless they perform their most valuable action in the intracellular district.
When considering blood, antioxidants are present within cells, in cells membranes and in the extracellular environment. Antioxidant plasma/serum components are mainly represented by ascorbic acid, glutathione, tocopherols, uric acid, bilirubin, phenolic compounds of dietary origin, vitamin E, microelements, intermediate products of metabolism, etc. [19] [20] [21] .
Relevant membrane-bound antioxidants comprise α-tocopherol, polyunsaturated fatty acids β-carotene and ubiquinone [22] . Glutathione is far the most abundant antioxidant defense inside cells, together with ascorbic acid, low molecular weight thiols and sulfhydryl groups of proteins and other reducing species. Remarkably, intracellular TAC is higher than one order of magnitude with respect of extracellular TAC and this datum is coherent with the physiological role of these antioxidants in cellular metabolism.
Furthermore, as the match between free radicals vs. antioxidants takes place primarily in intracellular environment, it would make sense to study/quantify the players of the game in this compartment. However, almost the whole literature dealing with oxidative stress issue in humans use serum or plasma as the sample matrix. Additionally, no consistent reviews or meta-analysis can be retrieved assessing a highly significant correlation between serum/plasma TAC and the “actual” total capacity of cells to counteract free radicals.
On the other hand, a high level of antioxidants in plasma or serum, corresponding to elevated TAC value, may not be necessarily a desirable physiological condition. Indeed, in some cases such as renal failure (hyperuricemia), icteric status (hyperbilirubinemia), hepatic damage (hypoalbuminemia) and other dishomeostatic diseases, abnormal levels of such metabolites falsely modifies TAC measured in plasma [23] [24] . Consequently, plasma or serum measurements are biased and may provide conflicting and inconsistent measurements.
Measuring the redox status inside cells thus represents a highly desirable tool, and blood cells are definitely the most easily available cells for diagnostic purposes. In past, a few methods have been suggested to measure TAC in erythrocytes, by 1) assaying individual markers (GSH, Vitamin E, etc.) [25] [26] ; 2) determining the sensitivity of erythrocytes to hemolysis, by measuring the time re- quired to hemolyzing 50% of the RBC exposed to a controlled free radical attack [27] ; 3) utilizing nitroblue tetrazolium (NBT-test) [28] , that is reduced to forma- zan by incubating with blood-sample at 37˚C and read at 570 nm. Although such approaches are more reliable and accurate than measuring TAC in plasma or se- rum they are laborious, time consuming and not suitable for screening purposes.
1.2. Methods Overview
In recent years, research on redox balance in cells, tissues, organisms, foods, and biological samples grew exponentially due to the relevant interest of oxidative stress in many degenerative pathologies. Unbalanced redox status of the body could be assessed by directly evaluating the level of oxygen-reactive molecules, by measuring the biomarkers of oxidative damage to cellular molecules, or measuring the antioxidant capacity in the organism. In the first case the difficulty of accurately quantifying ROS and nitrogen reactive species is not affordable, given their ephemeral life expectancy, unless sophisticated and expensive methodologies, such as electron spin resonance (ESR) are used [29] . Alternatively, markers of specific oxidized lipids, proteins and nucleotides can be determined, but these biomarkers should be individually measured and represent only a part of the whole oxidative stress. Furthermore biomarkers, such as oxidized LDL, 4-Hydroxynonenal, 8-hydroxyguanosine, 8-isoprostane, malondialdehyde, advanced glycation end products, show poor time correlation to the oxidative events they should witness.
Differently from approaches focused on the oxidative insult, the determination of total antioxidant capacity allows the evaluation of the effective defense power of the organism to counteract damages from excess of free radicals and reflects the actual redox balance of the organism.
Lastly, the redox balance evaluation could be carried out using electrochemical methods, for example by means of electrodes for measuring the redox potential (oxidation reduction potential, ORP) [30] . These methods, however, are expensive and are characterized by instability of the signal, long measurement times and poor accuracy, such that their use is not compatible with the common laboratory practice.
As regards TAC assay, the chemical and biochemical methods currently available for the determination of antioxidant capacity provide inaccurate or partial values and are unfit for accurately measuring total antioxidants contained within cells and/or because they cannot detect the complex of all antioxidant species present in the samples. In principle, one possible approach to overcome these drawbacks could rely on the integration of levels of all single antioxidants assayed separately. However, the approach of analyzing various individual antioxidants is expensive, time consuming and essentially unreliable since they contribute synergistically to the antioxidant systems and the bare integration of single contributions simply is not representative of the whole antioxidant status, not to consider the complex role played by enzymes involved in the redox intracellular equilibrium [31] . Therefore, assaying total antioxidant activity may give more biologically relevant information than merely measuring concentrations of individual antioxidants [32] [33] .
A plurality of analytical methods has been devised out during last decades for assaying total antioxidant capacity in biological samples [34] [35] . Results obtained from these various methods are hardly comparable because of the different mechanisms, redox potentials, pH and differing nature of specific antioxidant detected and other analytical parameters of various assays [36] .
Most of the methods currently available for TAC assay are performed in serum or plasma based on two different approaches: 1) spectrophotometric assays, which measure the capacity of plasma antioxidants to reduce a specific oxidant chromogen; 2) assays based on the measure of the capability of antioxidants to quench free radicals generated by the reagent.
The assay proposed by Benzie belongs to the first group [37] . It is based on the “ferric reducing?antioxidant power” (FRAP), the reduction of ferric ions to ferrous ions by plasma antioxidants being measured spectrophotometrically at 593 nm. Similarly, the CUPRAC (cupric reducing antioxidant capacity) assay utilizes the antioxidant capacity of the sample to reduce the chromogen from Cu(II)-neocuproin (λmax = 453 nm) to Cu(I)-neocuproin [38] . Another widely used TAC method is ORAC (oxygen radical absorbance capacity), developed by Cao’s et al. [39] . The ORAC method exploits a competitive reaction in which plasma constituents and substrate (β-phycoerythrin) kinetically compete for peroxyl radicals formed from thermal decomposition of the ABAP (2,2’-azobis,2- aminopropane). Fluorescence decay of β-phycoerythrin (excitation wavelength of 540 nm and emission wavelength of 565nm) is then measured. Miller et al. described the “Trolox-equivalent antioxidant capacity” (TEAC) method, based on the formation of the ABTS•+ cation [2,2’-azinobis (3-ethylben- zothiazoline-6-sulfonic acid)] and its scavenging by sample’s antioxidants measured spectrophotometry [40] . In this assay the absorbance decay of the green/ blue chromophore is inversely associated with antioxidants content in the sample and Trolox (a hydrophilic vitamin E analog) is used as the calibrator. Likewise, the method of Chrzczanowicz is based on the scavenging ability of the sample toward 2,2-diphenyl-picrylhydrazyl (DPPH) radical [41] . Kampa et al. developed a method based on the measurement of the inhibition caused by sample’s antioxidants on the bleaching of crocin by ABAP [42] . Finally, Whitehead et al. reported the use of a luminol enhanced chemiluminescent reaction to detect total antioxidants of biological samples [43] .
In summary, none of the existing methods fulfill all desirable features for a reliable and accurate TAC assay, namely: 1) direct measurement of all physiological, reducing substances contributing to TAC; 2) easiness to be automated; 3) possibility to use whole blood or tissue homogenates as samples, due to hemoglobin or to other interferences; 4) aptness to measure TAC in food and beverages.
A new original, reliable and fast method based on the two electrons reduction of the chromogen 2,6-dichlorophenolindophenol (DCPIP) has been devised out in order to assay a comprehensive antioxidant capacity of a biological sample that could encompass both extra- and intra-cellular compartments. This chromogen is a well known reagent for ascorbic acid quantitation [44] . This method is able to quantify total antioxidant capacity in a variety of biological samples. In particular, it can be used to quantify the reduced metabolites such as glutathione, the most concentrated intracellular species that plays a pivotal role in the protection from free radicals, in whole blood in order to assess the effective redox status of the organism.
2. Materials & Methods
2.1. Chemicals
All reagents were analytical grade. 2,6-Dichlorophenol indophenol was from Sigma-Aldrich (Milwaukee, WI). TAC reagents and calibrators were kindly supplied by Solosale Srl, Florence, Italy. All other reagents were from Sigma (St Louis, MO), Merck (Darmstadt, DE) or other commercially available sources.
2.2. Samples
Whole blood was collected by venipuncture in the presence of an anticoagulant agent (Na2-EDTA, preferably, or heparin). Blood samples can be stored at room temperature for 30 min, at 2˚C - 6˚C for 2 hours or below −18˚C for two days prior to assay. When frozen, samples may be used within 30 min after thawing.
TAC of tissues or cells from cell cultures can be measured by the present method following their 1:5 (sample to buffer volumes) homogenization. TAC was measured in the supernatant following ultracentrifugation at 10,000 g for 10 min at 4˚C or filtering the homogenate through 0.45 μm filter to remove particulate.
Clear beverages, such as wine or beer, can be used as samples without pre- processing. Beverages with suspended particulate, such as fruit juices and milk, should be properly centrifuged, extracted or filtered in order to get a clear solution before assaying.
Solid foods, such as fruit, grain, flesh, etc., should be grinded, pressed or finely minced, extracted or homogenized in the presence of a suitable buffer and then filtered (0.45 μm) or centrifuged (10,000 g for 10 min at 4˚C) in order to obtain a clear juice.
Fatty and oily samples can be assayed for their TAC providing a previous extraction with a water-soluble, low-polarity solvent is carried out.
Samples other than blood can be stored in the same conditions described above for blood samples.
2.3. Procedure
Total antioxidant capacity was measured by mixing sample and reagent to a volume ratio interval ranging from 1:10 to 1:200, depending on the TAC level in the samples.
Reagent was prepared by dissolving 0.08 mmol 2,6-dichlorophenolindophenol, 177 mL ethanol, 75 mmol phosphate buffer pH 8.8, 3 mL THESIT and 5 mmol Na2EDTA in 500 mL bidistilled water. Upon complete solubilization, water was added to 1000 mL final volume. Reduced glutathione was used as the calibrator.
The following solution was used as a calibrator for the assay: 2 mM reduced glutathione, 50 mM Phosphate buffer, pH 6.5, 1 mM Na2EDTA in the presence of a biocide stabilizer. Gallic acid can be used in place of glutathione when foods or beverages are analyzed. Calibrator solutions are stable for several months when stored at −20˚C.
Absorbance readings at a wavelength between 610 and 700 nm were taken at T0, T1 and T2 after mixing the sample or calibrator with the reagent.
Total antioxidant capacity expressed in terms of mmol/L of reduced glutathione was computed as follows:
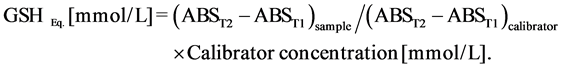
When blood was used as the sample, results were normalized with respect to the sample’s hemoglobin, to take into account the effect of hematocrit on TAC values. Actually, due to the much higher levels of glutathione and other antioxidants inside cells than in extracellular compartments, ratio between total cell volume and intercellular matrix greatly affects TAC. When cells or tissues are assayed, results were normalized dividing TAC values by protein concentration of the sample.
2.4. Spectrophotometric Assay
Automatic assay was carried out using the “fixed-time” method on Viva-E Siemens Healthcare Italia autoanalyzer at 630 nm.
Samples were previously hemolyzed by adding 80 μL 0.3% THESIT in 50 mM Tris-HCl, pH 8.8 to 20 μL sample and mixing. Lysates were kept at room T and processed within 60’. Fixed-time was selected as analytical algorithm. T1 = 20” and T2 = 80”, 1:20 lysed sample-to-reagent volume ratio and 37˚C were setup as analytical parameters.
2.5. Assay Using a Microplate Reader
TAC assay was performed on a Bio-Rad Model 3550 microplate reader at 655 nm, using the above-described protocol. Briefly, 10 μL lysed samples (see previous paragraph) were added by a multi-pipette to a 96 wells microplate prefilled with 200 μL reagent/well. Analytical parameters were the same used for the autoanalyzer.
2.6. Decentralized, Fast Assay
Small portable photometer working at fixed wavelength of 630 nm and capillary blood drawn by finger-pricking can be used to carried out this analytical procedure for rapidly assaying TAC in decentralized points-of-care. For this purpose, 10 μL capillary blood is added to a cuvette prefilled with 1000 μL reagent using a glass or plastic capillary or a fixed-volume pipette, and rapidly mixed by gently inverting the cuvette twice. Readings are taken at 20” and 80” after mixing. Results are computed as described above.
2.7. Statistics
All analyses were carried out with SPSS V.18.0 (SPSS Statistics v. 18.0, SPSS Inc., Chicago, IL, USA). t-student test was performed to assess significance of differences between averages; a value of p < 0.05 was considered statistically significant.
3. Results
Firstly, the chromogen recipe was optimized for the quantitative determination of total antioxidant capacity of whole blood and other biological samples with respect to reactivity, sensitivity, stability and specificity.
A screening of most commonly used redox indicators was therefore carried out. DCPIP was chosen as the assay chromogen thanks to its peculiar features. In particular this reagent is water soluble, elicits a mild oxidizing activity towards almost all physiological TAC-related metabolites due to its intermediate redox potential (about 108 mV at pH 8.8), shows acceptable thermal stability in the reagent mixture and, last but not least, its absorbance spectrum allows to avoid optical interference by hemoglobin.
Interference by hemoglobin was avoided by choosing 610 to 700 nm as the wavelengths interval for carrying out the absorbance readings.
Chromogen solution was buffered at pH 8.8 for optimal stability and maximum extinction coefficient. DCPIP reagent, checked during a 12 months period at 4˚C, elicited a color fading at 630 nm of about 4% per month. The reagent proved therefore usable for at least 1 year when properly stored (0˚C - 4˚C away from light).
The other key constituent of the reagent was the lysing agent, which is necessary to cause the rapid disintegration of the cells of the biological sample and make the intracellular substances react with the chromogen. The use of hydroxypolyethoxydodecane (Thesit®) has proved surprisingly effective. For the purpose of complexing bivalent metal ions, potential catalyzers of the oxidation reactions of the chromogen, and therefore to increase its stability, sodium ethylenediaminetetraacetate (Na2EDTA) was added as a further component of the reagent. Finally, in order to ensure an adequate thermal stability and to avoid any bacterial contamination of the reagent, a biocide as preservative was added.
Figure 1 shows the reaction mechanism of 2,6-DCPIP. The absorbance decrease at 630 nm can be measured at end-point, at fixed time or in kinetic mode. The analytical procedure performed on a fully automated analyzer requires a delay time T1 (20 seconds), reading the absorbance A1 and subsequent reading of absorbance A2 after 60 seconds (T2) at a temperature of 37˚C. As shown in Figure 2, the kinetic of the reaction is quite linear within this time interval. Quantification is performed using 2 mM GSH as a calibrator.
3.1. Imprecision
Glutathione standard at the indicated concentrations (Table 1) were run 20 times according to the above Assay Procedure.

Figure 1. Mechanism of reduction reaction of 2,6-DCPIP.
Inter assay imprecision was determined by running the same glutathione standards used for intra-assay imprecision (Table 2). Each standard was run 8 times in each session.
3.2. Accuracy
Table 3 and Figure 3 shows results from the recovery assay. Increasing amounts of GSH were spiked to aliquots of pooled venous whole blood samples. TAC was measured by this method within 30’. Values are averages from 10 tests. An overall good recovery performance was assessed for the assay.
3.3. Linearity
A standard curve of 0 - 15 mmol/L of glutathione was assayed on VIVA mod. SIEMENS auto-analyzer. The obtained plot was acceptably linear (r2 > 0.99) over

Figure 2. Reaction kinetics. Capillary whole blood was used as a sample. Kinetics was carried out on a Viva autoanalyzer.

Table 1. Within-run imprecision.
Table 2. Between-run imprecision.
Table 3. Relative reactivity between common reducing substances with respect to GSH.

Figure 3. Plotted data from Table 3.
the range 0 - 15 mmol/L (Figure 4), a concentration range including the majority of the values elicited by blood samples (see later results). Values are means from 5 measurements. A limit of quantitation of 0.2 mM was computed on the basis of standard deviation of lowest detectable amount and the slope of the calibration curve.
3.4. Differential Reactivity
Different reagents were challenged with 2,6-DCPIP in order to select the most suitable one to be used as the calibrator for the assay. Five commonly used reducing chemicals were chosen to this aim. Ascorbic acid far resulted as the most reactive one, Trolox being the less reactive. Noticeably, glutathione and gallic acid exhibited almost the same reactivity (Table 4). The reason for this differential reactivity could be ascribed to the specific reaction mechanisms of each reductant with DCPIP, more than to their different redox potential, as this method is based on kinetic measurements. Glutathione was chosen as the reference because of its physiological role and its relatively high abundance within cells.

Figure 4. TAC assay linearity. Points are mean of five measurements.
Table 4. Relative reactivity between common reducing substances with respect to GSH.
3.5. Human Blood Samples
Venous whole blood samples were collected from hospitalized patients (N = 122, 70 males and 52 females) suffering from various age-related degenerative diseases and from outdoor healthy subjects (N = 24, 14 males and 10 females) of equivalent age (ranging between 46 and 72 y) as controls. Sodium EDTA or Lithium heparin were used as anticoagulants. Samples were kept at room temperature and processes within 60 min, or frozen at −20˚C until used. Total hemoglobin concentration was assayed on a Hitachi 704 chemistry analyzer using Drabkin’s reagent. Figure 5(a) shows results from hospitalized patients and Figure 5(b) from outdoor subjects, expressed in terms of glutathione equivalents normalized for hemoglobin concentration. As expected, TAC values were lower in hospitalized patients (mean ± SD: 21.9 ± 4.3 μEqGSH/g Hb) as compared to controls (24.0 ± 2.9 μEqGSH/g Hb). Computed p value (0.0235) indicates the difference is statistically significant.
3.6. Wines
White (N = 4) and red (N = 13) Italian wines differing with regard to the land of origin and year of production have been analyzed, using 2 mM gallic acid as the calibrator. Figure 6 shows results, white wines (N = 4) being at the far left in the graph (clear columns). It may be noticed the great difference in antioxidants content between white and red wines. Even red wines considerably differ each other for TAC values, the one with the lowest antioxidant content eliciting less than half antioxidant capacity with respect to the highest one.
The remarkable difference between white and red wines may be ascribed to the relevant amount of substances characterized by antioxidant activity, such as polyphenols, released by grape pomace during the red wine manufacturing process. White wine, on the other hand, is typically produced by white grapes cultivars or, more frequently, by removing grape pomace during the fermentation stage.
 (a)
(a) (b)
(b)
Figure 5. (a) (upper graph) TAC distribution among hospitalized patients. Values are expressed as GSH equivalents (mmol/L) divided by hemoglobin concentration (g/L); (b) (lower graph) TAC distribution among outdoor subjects. Values are expressed as GSH equivalents (mmol/L) divided byhemoglobin concentration (g/L).

Figure 6. TAC levels in a selected set of white and red Italian wines.
4. Discussion
Free radical activity and oxidative damage have been implicated in the pathogenesis of a number of diseases. In particular, their role in the development of vascular disorders and on the progression of neurodegenerative syndromes has received more and more attention. For this reason an accurate, rapid and simple automatable method of measuring antioxidant capacity in the blood and other biological samples would elicit great potential benefits.
In the present work, we propose a spectrophotometric assay for the assessment of total antioxidant capacity in human whole blood or other liquid and/or biological tissues, based on the use of a reagent containing 2,6-dichlorophenolin- dophenol (DCPIP) with the function of redox chromogen. This chromogen behaves as an electron acceptor that is blue-colored in its oxidized status and colorless when it is reduced by antioxidants present in the sample. The rate of discoloration, measured at 630 nm, correlates to the overall level of antioxidants in the sample. The choice of DCPIP was made on the basis of its oxidation-reduc- tion potential (ORP) at alkaline pH, which makes it capable to react with most of antioxidants that occur in blood and other biological matrices. Furthermore, this chromogen allows the sensitivity far enough for accurately and precisely detecting glutathione, the main TAC contributor, in human tissues (see “linearity” section). Thanks to the presence of a cytolytic agent, the present TAC assay is able to completely lyse cells of most tissues, and then to measure the total antioxidant capacity of the whole sample, i.e. the overall contribution to the redox equilibrium of the antioxidants present in both intracellular and extracellular compartments. The use of this chromogen offers the additional advantage to overcome the interference by hemoglobin and other hemeproteins, as the absorbance spectrum of the heme group does not overlap to the chromogen spectrum. These features, which are pivotal for determining TAC on whole blood, are advantageous also for analyzing biological samples of different kinds, regardless of their animal or vegetal nature. The test can indeed be used for analyzing any biological material, such as tears, saliva, semen, vaginal secretions, amniotic liquid, maternal milk, cord blood, etc. The present assay can also be advantageously used to analyze foods and beverages, such as juices or extracts of fruits or vegetables, dairy products, grains, meats and fishes, etc.
As an overall evaluation, the present TAC assay procedure elicits the following advantages over the existing methods for TAC measurement: direct detection of analytes; assay of antioxidants present in foods and beverages; assay of very different biological matrices, such as extracellular and intracellular fluids; dependence of analytical output on the weighted contributions of all antioxidants of the samples, each one eliciting different specific reactivity; results expressed as glutathione (the most abundant physiological antioxidant) equivalents per unit volume; easiness and rapidity of implementation that makes it profitably exploitable in decentralized settings.
The present method, based on the use of an appropriate redox chromogen, provides analytical results which are similar to, but more accurate than, those obtained by electrochemical or by other analytical methods based on the use of chemical reagents, while being not affected by their drawbacks. The described procedure actually, thanks to the specific redox potential of the chromogen and to the presence of an agent able to rapidly cause the complete lysis of cells, enables the measurement of the overall contribution of all antioxidants present in the sample to its redox balance.
Cite this paper
Ciuti, R. and Liguri, G. (2017) A Novel Assay for Measuring Total Antioxidant Capacity in Whole Blood and Other Biological Samples. J. Biomedical Science and Engineering, 10, 60-76. https://doi.org/10.4236/jbise.2017.102007
References
- 1. Halliwell, B. (1994) Free Radicals, Antioxidants and Human Disease: Curiosity, Cause or Consequence. The Lancet, 344, 721-724.
https://doi.org/10.1016/S0140-6736(94)92211-X - 2. Cross, C.E., Halliwell, B., Borish, E.T., Pryor, W.A., Ames, B.N., Saul, R.L., McCord, J.M. and Harman, D. (1987) Oxygen Radicals and Human Disease. Annals of Internal Medicine, 107, 526-545.
https://doi.org/10.7326/0003-4819-107-4-526 - 3. Schieber, M. and Chandel, N.S. (2014) ROS Function in Redox Signaling and Oxidative Stress. Current Biology, 24, R453-R462.
https://doi.org/10.1016/j.cub.2014.03.034 - 4. Valko, M., Leibfritz, D., Moncol, J., Cronin, M.T., Mazur, M. and Telser, J. (2007) Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. The International Journal of Biochemistry & Cell Biology, 39, 44-84.
https://doi.org/10.1016/j.biocel.2006.07.001 - 5. Ames, B.N., Shigenaga, M.K. and Hagen, T.M. (1993) Oxidants, Antioxidants, and the Degenerative Diseases of Aging. Proceedings of the National Academy of Sciences of the United States of America, 90, 7915-7922.
https://doi.org/10.1073/pnas.90.17.7915 - 6. Leinonen, J.S., Ahonen, J.-P., Lönnrot, K., Jehkonen, M., Dastidar, P., Molnár, G. and Alho, H. (2000) Low Plasma Antioxidant Activity Is associated with High Lesion Volume and Neurological Impairment in Stroke. Stroke, 31, 1889-1892.
https://doi.org/10.1161/01.STR.31.1.33 - 7. Jenner, P. (1994) Oxidative Damage in Neurodegenerative Disease. The Lancet, 344, 798-799.
https://doi.org/10.1016/S0140-6736(94)92347-7 - 8. Ferrari, C.K.B. (2001) Oxidative Stress Pathophysiology Searching for an Effective Antioxidant Protection. Internal Medicine Journal, 8, 175-184.
- 9. Giacco, F. and Brownlee, M. (2010) Oxidative Stress and Diabetic Complications. Circulation Research, 107, 1058-1070.
https://doi.org/10.1161/CIRCRESAHA.110.223545 - 10. Stes, H. (2007) Total Antioxidant Capacity: Appraisal of a Concept. Journal of Nutrition, 137, 1493-1495.
- 11. Shapiro, H.M. (1972) Redox Balance in the Body: An Approach to Quantitation. Journal of Surgical Research, 13, 138-152.
https://doi.org/10.1016/0022-4804(72)90057-1 - 12. Ferrari, C.K.B. (2004) Functional Foods, Herbs and Nutraceuticals: Towards Biochemical Mechanisms of Healthy aging. Biogerontology, 5, 275-289.
https://doi.org/10.1007/s10522-004-2566-z - 13. Kijoon, K., Vance, T.M. and Chun, O.K. (2016) Greater Total Antioxidant Capacity from Diet and Supplements Is Associated with a less Atherogenic Blood Profile in US Adults. Nutrients, 8, 15.
https://doi.org/10.3390/nu8010015 - 14. Korotkova, E.I., Voronova, O.A. and Dorozhko, E.V. (2012) Study of Antioxidant Properties of Flavonoids by Voltammetry. Journal of Solid State Electrochemistry, 16, 2435-2440.
https://doi.org/10.1007/s10008-012-1707-6 - 15. Cao, G., Booth, S., Sadowsky, J.A. and Prior, R.L. (1998) Increases in Human Plasma Antioxidant Capacity after Consumption of Control Diets High in Fruit and Vegetables. American Journal of Clinical Nutrition, 68, 1081-1087.
- 16. Pitsavos, C., Panagiotakos, D.B., Tzima, N., Chrysohoou, C., Economou, M., Zampelas, A. and Stefanadis, C. (2005) Adherence to the Mediterranean Diet Is Associated with Total Antioxidant Capacity in Healthyadults: The ATTICA Study. American Journal of Clinical Nutrition, 82, 694-699.
- 17. McCall, M.R. and Frei, B. (1999) Can Antioxidant Vitamins Materially Reduce Oxidative Damage in Humans? Free Radical Biology and Medicine, 26, 1034-1053.
https://doi.org/10.1016/S0891-5849(98)00302-5 - 18. Miller, E.R., Pastor-Barriuso, R., Dalal, D., Riemersma, R.A., Appel, L.J. and Guallar, E. (2005) Meta-Analysis: High-Dosage of Vitamin E Supplementation May Increase All-Cause of Mortality. Annals of Internal Medicine, 142, 37-46.
https://doi.org/10.7326/0003-4819-142-1-200501040-00110 - 19. Evans, P. and Halliwell, B. (2001) Micronutrients: Oxidant/Antioxidant Status. British Journal of Nutrition, 85, S67-S74.
https://doi.org/10.1079/bjn2000296 - 20. Young, I.S. (2001) Measurement of Total Antioxidant Capacity. Journal of Clinical Pathology, 5, 339.
https://doi.org/10.1136/jcp.54.5.339 - 21. Bartosz, G. (2003) Total Antioxidant Capacity. Advances in Clinical Chemistry, 37, 219-292.
https://doi.org/10.1016/S0065-2423(03)37010-6 - 22. Halliwell, B. (1989) Free Radicals, Reactive Oxygen Species and Human Disease: A Critical Evaluation with Special Reference to Atherosclerosis. British Journal of Experimental Pathology, 70, 737-757.
- 23. Ghiselli, A., Serafini, M., Natella, F. and Scaccini, C. (2000) Total Antioxidant Capacity as a Tool to Assess Redox Status: Critical View and Experimental Data. Free Radical Biology and Medicine, 29, 1106-1114.
https://doi.org/10.1016/S0891-5849(00)00394-4 - 24. Bergesio, F., Monzani, G., Ciuti, R., Pinzani, P., Fiaschi, N., Priami, F., Cirami, C., Ciccarelli, C. and Salvadori, M. (1998) Total Antioxidant Capacity (TAC): Is It an Effective Method to Evaluate the Oxidative Stress in Uremia? Journal of Bioluminescence and Chemiluminescence, 13, 315-319.
https://doi.org/10.1002/(SICI)1099-1271(1998090)13:5<315::AID-BIO491>3.0.CO;2-6 - 25. Beutler, E., Duron, O. and Kelly, B.M. (1963) Improved Method for the Determination of Blood Glutathione. Journal of Laboratory and Clinical Medicine, 61, 882-888.
- 26. Kayden, H.J., Chow, C.K. and Bjornson, L.K. (1973) Spectrophotometric Method for Determination of Tocopherol in Red Blood Cells. Journal of Lipid Research, 14, 197-205.
- 27. Rossi, R., Pastorelli, G. and Corino, C. (2013) Application of KRL Test to Assess Total Antioxidant Activity in Pigs: Sensitivity to Dietary Antioxidants. Research in Veterinary Science, 94, 372-377.
https://doi.org/10.1016/j.rvsc.2012.08.005 - 28. Konuganti, K., Seshan, H., Zope, S. and Delphine, W. (2012) A Comparative Evaluation of Whole Blood Total Antioxidant Capacity Using a Novel Nitrobluetetrazolium Reduction Test in Patients with Periodontitis and Healthy Subjects: A Randomized, Controlled Trial. Journal of Indian Society of Periodontology, 16, 620-622.
https://doi.org/10.4103/0972-124X.106937 - 29. Kohno, M.J. (2010) Applications of Electron Spin Resonance Spectrometry for Reactive Oxygen Species and Reactive Nitrogen Species Research. Journal of Clinical Biochemistry and Nutrition, 47, 1-11.
https://doi.org/10.3164/jcbn.10-13R - 30. Rael, L.T., Bar-Or, R., Salottolo, K., Mains, C.W., Slone, D.S., Offner, P.J. and Bar-Or, D. (2009) Injury Severity and Serum Amyloid A Correlate with Plasma Oxidation-Reduction Potential (ORP) in Multi-Trauma Patients: A Retrospective Analysis. Scandinavian Journal of Trauma, Resuscitation and Emergency Medicine, 17, 57-64.
https://doi.org/10.1186/1757-7241-17-57 - 31. Duthie, G.G. (1999) Determination of Activity of Antioxidants in Human Subjects. Proceedings of the Nutrition Society, 58, 1015-1024.
https://doi.org/10.1017/S0029665199001330 - 32. Apak, R., Gorinstein, S., Böhm, V., SchaichÖzyürek, M. and Güçlü, K. (2013) Methods of Measurement and Evaluation of Natural Antioxidant Capacity/Activity. Pure and Applied Chemistry, 85, 957-998.
https://doi.org/10.1351/PAC-REP-12-07-15 - 33. Somogyi, A., Rosta, K., Pusztai, P., Tulassay, Z. and Nagy, G. (2007) Antioxidant Measurements. Physiological Measurement, 28, R41-R55.
https://doi.org/10.1088/0967-3334/28/4/R01 - 34. Stes, H. (2007) Total Antioxidant Capacity: Appraisal of a Concept. Journal of Nutrition, 137, 1493-1495.
- 35. Shapiro, H.M. (1972) Redox Balance in the Body: An Approach to Quantitation. Journal of Surgical Research, 13, 138-152.
https://doi.org/10.1016/0022-4804(72)90057-1 - 36. Cao, G. and Prior, R.L. (1998) Comparison of Different Analytical Methods for Assessing Total Antioxidant Capacity of Human Serum. Clinical Chemistry, 44, 1309-1315.
- 37. Benzie, I.F.F. and Strain, J.J. (1996) The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Analytical Biochemistry, 239, 70-76.
https://doi.org/10.1006/abio.1996.0292 - 38. Apak, R., Güçlü, K., Ozyurek, M., Karademir, S.E. and Altun, M. (2005) Total Antioxidant Capacity Assay of Human Serum Using Copper(II)-Neocuproine as Chromogenic Oxidant: The CUPRAC Method. Free Radical Research, 39, 949-961.
https://doi.org/10.1080/10715760500210145 - 39. Cao, G., Alessio, H.M. and Cutler, R.G. (1993) Oxygen-Radical Absorbance Capacity Assay for Antioxidants. Free Radical Biology and Medicine, 14, 303-311.
https://doi.org/10.1016/0891-5849(93)90027-R - 40. Miller, N.J., Rice-Evans, C., Davies, M.J., Gopinathan, V. and Milner, A. (1993) A Novel Method for Measuring Antioxidant Capacity and Its Application to Monitoring The antioxidant Status in Premature Neonates. Clinical Science, 84, 407-412.
https://doi.org/10.1042/cs0840407 - 41. Chrzczanowicz, J., Gawron, A., Zwolinska, A., de Graft-Johnson, J., Krajewski, W., Krol, M., Markowski, J., Kostka, T. and Nowak, D. (2008) Simple Method for Determining Human Serum 2,2-Diphenyl-1-Picryl-Hydrazyl (DPPh) Radical Scavenging Activity—Possible Application in Clinical Studies on Dietary Antioxidants. Clinical Chemistry and Laboratory Medicine, 46, 342-334.
https://doi.org/10.1515/CCLM.2008.062 - 42. Kampa, M., Nistikaki, A., Tsaousis, V., Maliaraki, N., Notas, G. and Castanas, E. (2002) A New Automated Method for the Determination of the Total Antioxidant Capacity (TAC) of Human Plasma, Based on the Crocin Bleaching Assay. BMC Clinical Pathology, 2, 3.
https://doi.org/10.1186/1472-6890-2-3 - 43. Whitehead, T.P., Thorpe, G.H.G. and Maxwell, S.R.J. (1992) Enhanced Chemiluminescent Assay for Antioxidant Capacity in Biological Fluids. Analytica Chimica Acta, 266, 265-277.
https://doi.org/10.1016/0003-2670(92)85052-8 - 44. VanderJagt, D.J., Garry, P.J. and Hunt, W.C. (1986) Ascorbate in Plasma as Measured by Liquid Chromatography and by dichlorophenolindophenol Colo-rimetry. Clinical Chemistry, 32, 1004-1006.