Journal of Biomedical Science and Engineering
Vol.08 No.08(2015), Article ID:58840,10 pages
10.4236/jbise.2015.88047
Development of an Algorithm for Reconstructing a Comprehensive Pathway Model: Application to Saccharomyces cerevisiae
Itaru Takeda1,2, Masayuki Machida1,3, Sachiyo Aburatani2*
1Department of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, Naka-cho, Koganei, Tokyo, Japan
2Biotechnology Research Institute for Drug Discovery, National Institute of Advanced Industrial Science and Technology (AIST), Tokyo Waterfront Bio-IT Research Building, Aomi, Koto-ku, Tokyo, Japan
3Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Higashi-nijo, Tsukisamu, Sapporo, Japan
Email: *s.aburatani@aist.go.jp
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 25 June 2015; accepted 14 August 2015; published 17 August 2015
ABSTRACT
The generation of bioactive products by microbial bioprocesses is important for drug discovery, functional food development, and other beneficial purposes. Many pathways contribute to the production of these bioactive compounds, but important knowledge for improving productivity still remains in hidden pathways. Recently, an abundance of knowledge about metabolic pathways has been accumulated in metabolic pathway databases, such as BioCyc and KEGG. Many by-products are chemically transformed and actually used in other enzymatic reactions. In this work, we developed an algorithm for the reconstruction of a comprehensive genetic pathway model from a known metabolic pathway database. This model considers the interactions of the by-products, in addition to the main products. Furthermore, we developed a method for the construction of a comprehensive pathway model in a specific organism. In this study, we reconstructed a Saccharomyces cerevisiae model. From this model, the pathways among enzymes that contributed to galactose metabolism were explored. Using S. cerevisiae DNA microarray data, the activated pathways were found among the explored pathways.
Keywords:
Bioinformatics, Metabolic Pathway

1. Introduction
Many bioactive compounds, including lead compounds for the active constituents of cosmetics and novel drugs, are produced by microbial bioprocesses [1] - [3] . For example, kojic acid, a tyrosinase inhibitor, is produced by Aspergillus oryze [4] - [6] . Cyclosporine, which is produced by Tolypocladium inflatum, inhibits the signal transduction mediated by calcineurin [7] - [9] . However, the pathways that are related to the production of these useful compounds have not been clarified. To improve the productivity of useful compounds, the intracellular reactions that are related to the biosynthesis of such compounds should be identified. Furthermore, knowledge of the pathways that are activated in the biosynthesis is important for bioengineering using microorganisms.
Many studies have revealed various aspects of metabolism and other cellular processes’ in numerous organisms. This metabolic knowledge has been accumulated in databases such as KEGG [10] -[12] , BioCyc [13] , Reactome [14] and others [15] [16] . These databases are useful for the identification of biological functions from the genome sequences. A metabolic pathway database typically contains information about the chemical transformations and the enzymes catalyzing each transformation, as a metabolic network [16] . This network is represented as a chemical network and a genomic network [17] . The chemical network depicts how compounds are transformed by continuous enzymatic reactions. The genomic network represents how the enzymes are connected by the compounds that they chemically transform. In the metabolic pathway databases, these networks represent pathway maps that are classified by various metabolic categories. The metabolic network usually describes the chemical transformations of the main compounds. In actual metabolism, by-products are produced by enzymatic reactions, in addition to the main products. Since a cell is a closed space, fluctuations in the levels of the by-products are considered to affect the other reactions in the cell. It is assumed that connections of the enzymes by the by-products also exist. Consequently, the comprehensive connections among enzymes are revealed by a pathway model that considers the chemical transformations of all of the compounds, including the main compounds and the by-products.
In order to consider all of the pathways related to the production of useful bioactive compounds, we developed an algorithm for reconstructing a comprehensive pathway model (CPM), which was a comprehensive metabolism network. This network model enabled the fluctuations of compounds to be tracked comprehensively. We proposed a method for constructing a model of a specific organism. We applied our developed method to reconstruct a Saccharomyces cerevisiae model (sCPM). The pathways related to galactose metabolism were explored from sCPM. Furthermore, the activated pathways were visualized with S. cerevisiae DNA microarray data.
2. Material and Methods
2.1. KEGG Database and S. cerevisiae Gene Expression Data
In order to reconstruct a CPM, we used the KEGG database [10] -[12] (2014-04-07 release). In the KEGG database, the KEGG REACTION database contains all of the reaction formulas related to the pathway maps of metabolism, signal transduction, and other cellular phenomena [17] [18] . This database also contains additional enzymatic reactions that are not included within the pathway maps. The pathway maps are retrieved from the KEGG PATHWAY database [18] . The compounds used in these reactions are contained in the KEGG COMPOUND database [18] . The KEGG ORTHOLOGY database contains the ortholog groups, which are functional RNAs and all proteins including enzymes, transcription factors and so on [17] . In the KEGG ORTHOLOGY database, the gene IDs corresponding to each ortholog group are also provided for various organisms, including S. cerevisiae.
The DNA microarray data sets to explore the activated pathways in S. cerevisiae were retrieved from the Gene Expression Omnibus [19] (GEO: http://www.ncbi.nlm.nih.gov/geo/ accession nos. GSM990, GSM991). For the GSM990 and GSM991 data sets, the cells were grown continuously in YP media supplemented with glucose and galactose, respectively. The total RNA of each sample was labeled with Cy5-dUTP [20] , and was compared with a reference pool labeled with Cy3-dUTP [20] .
2.2. Algorithm for Reconstructing the CPM
We developed an algorithm to reconstruct a comprehensive pathway model from KEGG data base, by the following three steps. This algorithm uses the data sets of the ortholog group and the enzymatic reactions in the KEGG database.
Step 1 Construction of Compound Combinations
Compound combinations were constructed from substrate-product pairs in an enzymatic reaction. The information about the ortholog groups and the reactions was obtained from the KEGG ORTHOLOGY database and the KEGG REACTION database, respectively. In Figure 1(a), Reaction1 stands for the enzymatic reaction catalyzed by the enzyme of Ortholog1. Cpd.1, Cpd.2, Cpd.3 and Cpd.4 are compounds in Reaction 1. The interactions of Cpd.1- > Cpd.3, Cpd.1- > Cpd.4, Cpd.2- > Cpd.3 and Cpd.2- > Cpd.4 with an index of Ortholog1 were constructed from Reaction1 (Figure 1(b)). The combinations of compounds were constructed from reverse reactions as well as forward reactions. The ortholog groups, which are related to some enzymatic reactions, were indexed to all compound combination pairs. In the entire KEGG REACTION database, we considered all compound combinations for each enzymatic reaction with both the forward and reverse directions.
Step 2 Reconstruction of the Whole Compound Network
The whole compound network model, Dcpd, was compiled from all compound combinations (Figure 1(c)). The graph of Dcpd is expressed as follows:
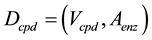 (1)
(1)
where Vcpd and Aenz denote nodes and arcs, respectively. In Dcpd, the ortholog groups are the connections between the compounds. Dcpd contains all compounds related to all enzymatic reactions in the KEGG REACTION database, as nodes, and indicates the comprehensive chemical transformation by enzymes.
Step 3 Reconstruction of the CPM
The comprehensive pathway model, Denz, was reconstructed from Dcpd. In Dcpd, the nodes were replaced with arcs. The nodes and the arcs of Denz were the ortholog groups and the compounds, respectively (Figure 1(d)). In Dcpd, each compound was non-redundant. In contrast, some ortholog groups were present as multiple arcs. In Denz, each ortholog group was non-redundant and some compounds were present as multiple arcs. The graph of Denz is expressed as follows:
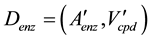 (2)
(2)
where
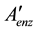 is a non-redundant subset of
is a non-redundant subset of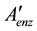 .
.
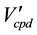 is the set that is constructed by multiplexing elements of
is the set that is constructed by multiplexing elements of
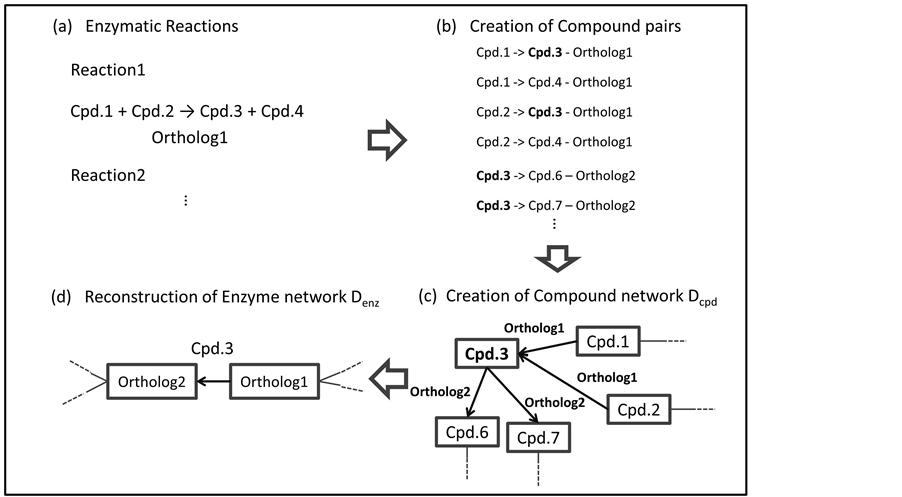
Figure 1. Overview of the algorithm for reconstructing CPM. (a) The abbreviation Cpd. represents compound. In Reaction1, Cpd.3 and Cpd.4 are produced from Cpd.1 and Cpd.2. This enzymatic reaction is catalyzed by Ortholog1; (b) The interactions of Cpd.1- > Cpd.3, Cpd.1- > Cpd.4, Cpd.2- > Cpd.3 and Cpd.2- > Cpd.4 were indexed by Ortholog1; (c) The nodes of Cpd.1 and Cpd.3 are connected by an arc labeled Ortholog1. (d) The nodes are replaced by arcs. The nodes of Ortholog1 and Ortholog2 are connected by an arc labeled Cpd.3.
Vcpd, depending on the number of connections. The constructed Denz was a comprehensive pathway model that is connected by compounds. This model represents all of the enzymatic reactions within a cell. Consequently, a product of one reaction is used as a substrate of another reaction, even if these reactions are far apart in the known metabolic pathways.
2.3. Elimination of Compounds from the CPM
Some compounds in Denz should be eliminated from the reconstructed model. Since some compounds connect many pairs of ortholog groups in the Denz model, the amount of fluctuation is considered to be buffered. Furthermore, since these compounds are usually essential for growth, sufficient amounts are assumed to exist in the cell. In order to simplify the CPM, the compounds corresponding to many arcs were eliminated in this process. The compounds were arranged in the descending order of the number of arcs corresponding to them. The high ranking compounds and the nodes with no arcs were eliminated from Denz. The remaining model was the graph Denz_celi, defined as follows.
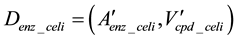 (3)
(3)
where
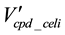 is the arcs remaining after the compound elimination against
is the arcs remaining after the compound elimination against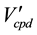 .
.
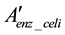 is the nodes remaining after the elimination of the nodes that lack connections by arcs.
is the nodes remaining after the elimination of the nodes that lack connections by arcs.
2.4. Method for Constructing an Organism-Specific CPM
To construct a specific CPM for an individual species, we extracted the substructure from the CPM. The CPM contains all ortholog groups, regardless of the organisms. To identify the ortholog groups corresponding to the enzymes encoded in the genome of a specific organism, we utilized the information from the KEGG ORTHOLOGY database. The nodes that did not correspond to a specific organism were eliminated from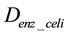 . The remaining model was expressed as a graph for a specific organism, DSO.
. The remaining model was expressed as a graph for a specific organism, DSO.
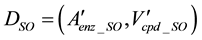 (4)
(4)
where
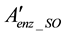 is the nodes remaining after the elimination of the nodes without annotated genes.
is the nodes remaining after the elimination of the nodes without annotated genes.
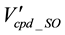 is the arcs connecting the remaining ortholog groups.
is the arcs connecting the remaining ortholog groups.
2.5. Path Finding Algorithm
To find the comprehensive pathways from the CPM, we developed an algorithm for path finding. To resolve some problems, some nodes were distinguished in this algorithm. In Denz, all arcs coming into and going out from a node must be a substrate-product pair in an enzymatic reaction. However, some ortholog groups indexed multiple reactions. This means that two compounds, which were coming into and going out from one ortholog group in our model, were not transformed from one to the other in a real reaction. This problem occurred because the substrate and the product were generated from different reactions. A similar problem occurred when considering the forward and reverse reactions for each enzymatic reaction. The forward and reverse reactions can be defined as two reactions that are one-way reactions with different directions. Consequently, two compounds that are coming into and going out from one ortholog group may not be a substrate-product pair. For example, when the forward and reverse reactions were considered as different reactions, the substrates of the forward reaction were identical to the products of the reverse reaction. The products of the forward reaction were also identical to the substrates of the reverse reaction. If two compounds that were coming into and going out from one ortholog group were generated from different reactions, then they were a substrate-substrate pair or a product-product pair. In order to resolve these problems, we distinguished an index for each reaction. The forward and reverse reactions were also distinguished. For example, if one ortholog group indexes three enzymatic reactions with two directions (forward and reverse), then this ortholog group is distinguished to 6 (3*2) indexes.
The possible interactions between the ortholog groups located within the constrained paths were explored comprehensively, by a path finding algorithm. The main function of the algorithm is as follows. First, a variable for the counting depth of exploration was initialized (Figure 2(a)). Next, a beginning node was obtained (Figure 2(a)). In this algorithm, the counter and the beginning node were defined as Cpath and v0, respectively. Finally, a defined function of PATH FINDING was employed (Figure 2(a)). This function explores nodes that are connected by one path from a provided node, and occurs recursively until the depth of exploration achieves fixed paths (Figure 2(b)). In this algorithm, Cmax is fixed as the maximum depth of path finding. The terms v0,1, v0,2, v0,3, … and v0,n stand for the nodes that are connected by one path from v0 (Figure 2(b)). The subscript number denotes the depth of exploration. For example, v0,1,2 is a node that is connected to v0 for two paths. The parents of v0,1,2 are v0 and v0,1. Spath is a stack that contains the nodes from v0 to the current node (Figure 2(b)). The v0 is accumulated on Spath as an initial value. The Scpd is a stack with the compounds corresponding to the arcs (Figure 2(b)). The compounds contained in Scpd connect the nodes from v0 to the current node. The initial value of Scpd is null. In Figure 2(b), Process1~Process10 are described as follows.
PROCESS 1 Increment of the path counter Cpath
Cpath is increased by 1.
PROCESS 2 Acquisition of 1 path connected nodes
All nodes that are directly connected to v0 are obtained. In Figure 2(b), the number of obtained nodes is n.
PROCESS 3 Ortholog group check
In order to simplify the explored paths, the ortholog group of a node was checked. The nodes of v0,1, v0,2, v0,3, … and v0,n, which were obtained in PROCESS2, are compared to the nodes in Spath one by one. If the current node is not equal to the node in Spath, then PROCESS4 is performed. Otherwise, this process is finished. After this process is finished, the next node of v0,1, v0,2, v0,3, … or v0,n is compared to the nodes in Spath. If the last node v0,n exists in Spath, then PROCESS8 is performed. The nodes with the same ortholog group were defined as being equal, even if they were different nodes.
PROCESS 4 Compound check
The Compound check is performed for the same reason as PROCESS 3. The current compound that connects the current node with the top node of Spath is compared to the compounds in Scpd. If Scpd is a null set, then PROCESS 5 is performed. If the current compound is not contained in Scpd, then PROCESS 5 is also performed. Otherwise, this process is finished and the next node of v0,1, v0,2, v0,3, … or v0,n is checked in PROCESS 3. If the compound that connects the last node v0,n with the top node of Spath exists in Scpd, then PROCESS 8 is performed.
PROCESS 5 Pushing a node and arc onto stacks
The current node, which is v0,1, v0,2, v0,3, … or v0,n, is pushed to the top of Spath. The current compound is also pushed to the top of Scpd.
PROCESS 6 Terminus check
This process assesses whether the pathway of Spath achieves the target ortholog group. If the top node of Spath is equal to the node corresponding to the target ortholog group, then PROCESS 7 is performed. Otherwise, PROCESS8 is performed.
PROCESS 7 Acquisition pathway
The set of nodes in Spath is defined as one of the pathways from v0 to the target ortholog group.
PROCESS 8 Depth check
In this process, the depth of exploration is checked. If Cpath is smaller than Cmax, then the function of PATH FINDING is performed recursively, using the top node of Spath as the beginning node.
PROCESS 9 Eliminating a node and an arc of stacks
The top values of Spath and Scpd are eliminated.
PROCESS 10 Decrement of the path counter Cpath
Cpath is decreased by 1. If Cpath is equal to zero, then the control returns to the main function. The exploration of the pathway is finished. Otherwise, the control returns to the point where the PATH FINDING function occurs.
This exploration revealed the connected pathways from one ortholog group to the specific ortholog group. The pathways consisted of continuous enzymatic reactions. The number of paths on the pathway means the number of enzymatic reactions occurring from a substrate to a product.
3. Results and Discussion
3.1. Overview of the Whole CPM
In this model, each ortholog group was arranged as asingle node. The arcs were also arranged with the nodes. Since one node indicates some different enzymes in this graph, some node pairs were connected by multiple arcs
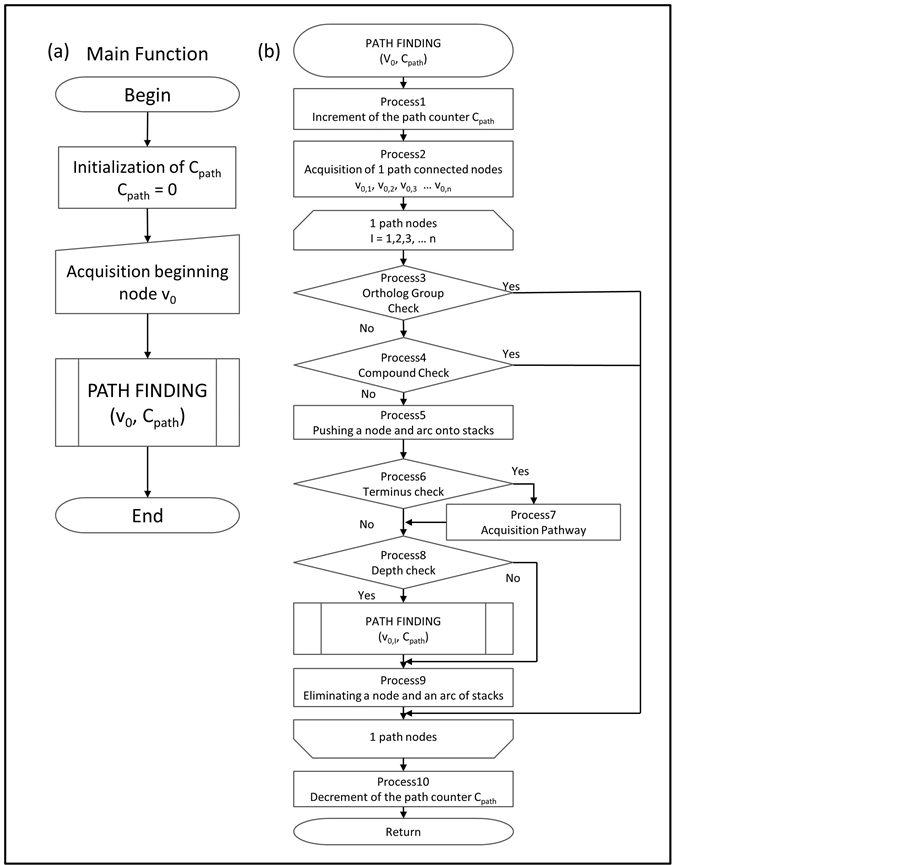
Figure 2. Flowchart of the Path Finding Algorithm. (a) This flow chart is the main function. The variable Cpath records the depth, and is initialized to 0. The beginning node v0 is obtained. The function of PATH FINDING is called in the main function; (b) This flow chart is a sub function called in the main function. The processes from 1 to 10 are performed according to this flow chart. The v0,1, v0,2, v0,3, … and v0,n, are nodes. This function calls itself recursively.
indexed with different compounds. We defined the arcs indexed with the same compound as a single connection. The forward and reverse reactions were considered for all of the enzymatic reactions. Node pairs were connected by two arcs with conflicting directions. These two arcs were regarded as a single, bidirectional arc. In the graph Denz, 3872 nodes were connected by 2,634,193 arcs. Among the 17,685 KEGG registered ortholog groups, approximately 21.9% of the ortholog groups were contained in Denz. In addition, 2801 kinds of compounds were contained in Denz (Table 1).
3.2. Graph Simplification
We eliminated some arcs from the graph, to simplify the CPM. The details of elimination are denoted as follows. In Denz, some compounds connected many pairs of ortholog groups. The fluctuations of these compounds are likely to be buffered in the cell, and a compound with many arcs is considered to hardly vary. In this study, we eliminated 17 compounds with arc numbers greater than 10,000: H2O, H+, oxygen, NADP+, NADPH, ATP, NAD+, NADH, diphosphate, CO2, orthophosphate, ADP, CoA, ammonia, S-adenosyl-L-methionine, AMP and S-adenosyl-L-homocysteine. The resultant graph is expressed as Denz_celi. Approximately 94.0% of the arcs were eliminated from Denz. Furthermore, the 43 nodes with no connections were eliminated. Since the eliminated nodes were connected by compounds that are present in sufficient quantities in the cell, the eliminated nodes were considered not to affect the fluctuation of compounds. Thus, the expression of those enzymatic genes didnot contribute to the fluctuation of metabolites. Although the initial Denz is too complicated to find new pathways, the elimination of too many compounds will cause important interactions to be missed. We assumed that the optimum number of eliminated compounds depends on the focused functions of the enzymes.
3.3. Construction of the S. cerevisiae Specific CPM
To infer the by-pass of a known metabolic pathway, we constructed the S. cerevisiae-specific CPM (sCPM). An overview of sCPM is provided in Figure 3. The multiplexing arcs between the nodes are shown as an arc. In the KEGG database, there are 2693 ortholog groups corresponding to 3570 genes. The reconstructed graph was defined as DSC. In DSC, 547 nodes and 8085 arcs remained from Denz_celi (Table 1). The number of nodes in DSC was approximately 14.3%, in comparison with that in Denz_celi. These eliminated nodes suggested two possibilities: 1) these enzymes were not preserved in S. cerevisiae, although they were preserved in other fungal species or other kingdoms or 2) the genes encoding these enzymes were not found in S. cerevisiae, although these genes were encoded in the genome. Depending on the elimination of nodes, about 94.9% of the arcs were deleted from Denz_celi. In DSC, the average number of arcs per node was 15, while it was 41 in Denz_celi. This means that the nodes that function as hubs connected by many arcs were eliminated. Furthermore, the kinds of compounds that corresponded to the arcs decreased from 2784 to 550 in DSC (Table 1). Recently, the entire genomes of various organisms have been determined [21] [22] . If the specific CPM to these organisms is reconstructed, then the ortholog ID in the KEGG database should be assigned to the annotated genes of these organisms by KAAS [23] .

Table 1. Overview of the reconstructed models.
1The number of nodes in each graph; 2Percentage of the nodes among the total nodes of the entire CPM; 3The number of arcs in each graph; 4The average number of arcs per node; 5The kinds of compounds in each graph.
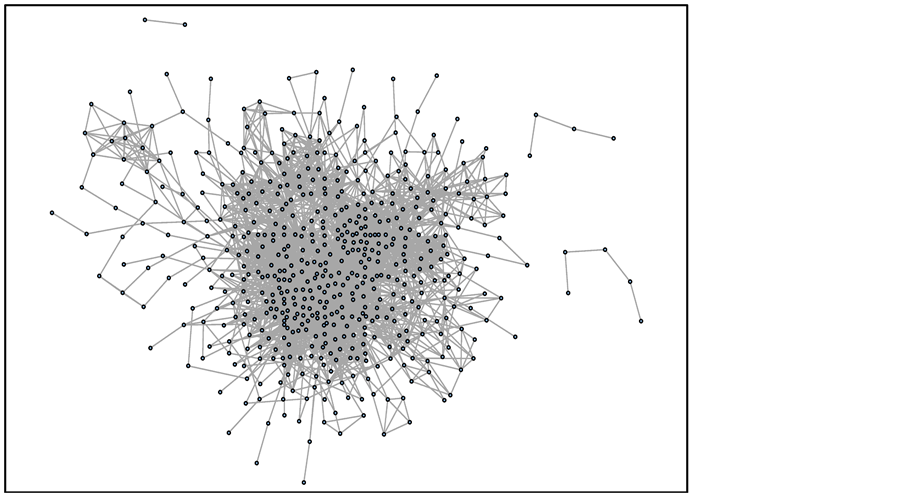
Figure 3. Whole image of sCPM. Representation of the entire network image of sCPM.
3.4. Path Finding from sCPM
We applied a path finding algorithm to sCPM. The maximum depth of the path finding was determined to be 5; in other words, the maximum length of a pathway was 5 paths. In sCPM, we explored the pathways that are related to the transformation from α-D-galactose to α-D-glucose-6-phosphate [24] . To explore the pathways, two restrictions were set for the path finding algorithm: 1) the node that the arcs corresponding to α-D-galactose were coming into and 2) the node that the arcs corresponding to α-D-glucose-6-phosphate were goingout from. The arc corresponding to α-D-galactose was coming into the node of “catabolic enzyme of α-D-galactose”. The arc corresponding to α-D-glucose-6-phosphate was going out from the node of “biosynthetic enzyme of α-D- glucose-6-phosphate”. Two beginning nodes and seven end nodes were selected from the KEGG ORTHO- LOGY database. Within the 5 path length, our algorithm found pathways from K00849 to K00844, K01810, K01792, K01835, K16055 and K00697. All of the pathways from the beginning node to the end nodes are presented in Figure 4(a). The pathways were merged for visualization. In order to simplify the depiction of the pathways, the multiplexed arcs between nodes are represented as single arcs (Figure 4). The information about all of the ortholog groups is provided in Table 2. Since multiple genes corresponded to an ortholog group, some ortholog groups are described several times in Table 2.
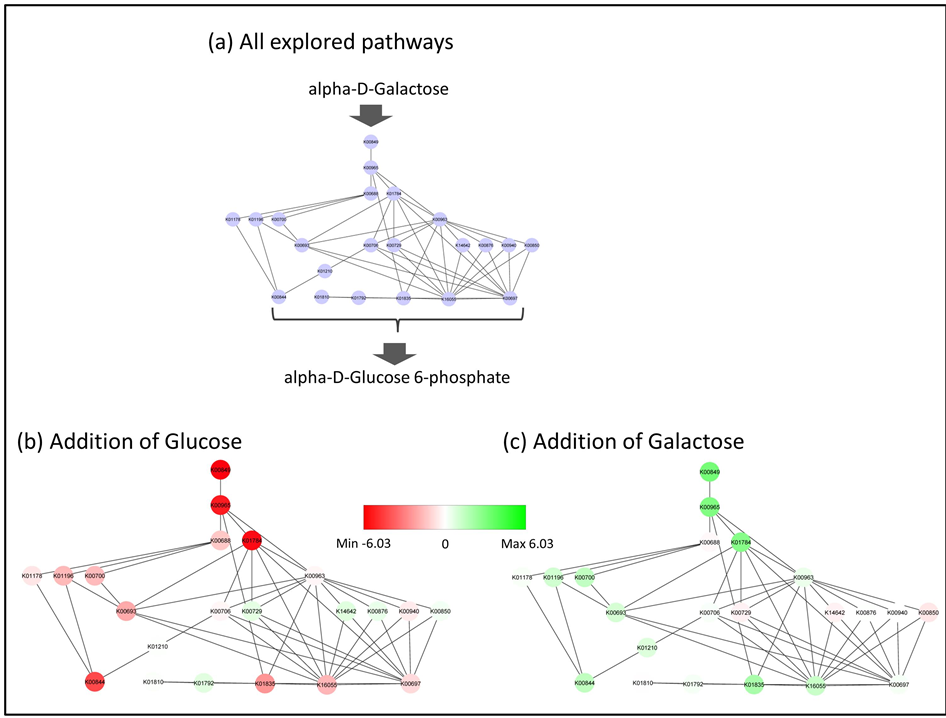
Figure 4. All pathways from α-D-galactose to α-D-glucose-6-phosphate. (a) All pathways explored from sCPM are shown. The ID labels in the nodes represent the ortholog group IDs in the KEGG database. The beginning node is K00849. The end nodes are K00844, K01810, K01792, K01835, K16055 and K00697. The merged pathways are shown. If nodes are connected by multiple arcs, then the arcs are shown as one arc; (b) Mapping of the gene expression data to all pathways. The data from the addition of glucose were used. The induction and repression of the genes are represented by the colors of the nodes. The deeper green represents larger induction, while the deeper red represents larger repression. White is used for the genes without any changes in their expression; (c) The gene expression data are mapped to all pathways. The data from the addition of galactose were used. The color code is the same as in Figure 4(b).
Table 2. Compound ranking according to the number of arcs.
*If the same ortholog groups were represented, then the gene expression data in this row were mapped in Figure 4(b) and Figure 4(c); aGene expression under the conditions of glucose addition; bGene expression under the conditions of galactose addition; cIf the ortholog group ID was included in the pathway map00052 in the KEGG PATHWAY database, then T was assigned. Otherwise, F was assigned.
To identify the different activated pathways for nutrient conditions, the DNA microarray data sets were mapped onto these pathways. In Figure 4(b), we mapped the value of log2(Cy5/Cy3), in which the ratio is the addition of glucose and a reference pool [20] . In Figure 4(c), we also mapped the value of log2(Cy5/Cy3), in which the ratio is the addition of galactose and a reference pool [20] . If some of the spots corresponded to the same gene on the DNA microarray, then the mean value of these spots was used as the value of the gene. If multiple genes corresponded to an ortholog group, then the value of a gene marked with “*” in Table 2 was mapped. In Figure 4(b) and Figure 4(c), the induction and repression of gene expression are represented by colors. The deeper green represents strong induction and the deeper red represents strong repression. If the gene expression minimally varied, then the color of the node is close to white. In these pathways, the expression of many genes was repressed by the addition of glucose (Figure 4(b); Table 2). In contrast, the expression of many genes was induced by the addition of galactose (Figure 4(c); Table 2). It is well known that some GAL genes are inducted and repressed by galactose and glucose, respectively [25] -[27] . Those GAL genes are represented as some nodes in Figure 4: GAL1 as K00849, GAL7 as K00965 and PGM2 as K01835. Furthermore, many other genes were identified as galactose metabolism related genes in our explored pathways. Interestingly, those other genes were also activated by galactose. The “Galactose Metabolism” column in Table 2 indicates whether the ortholog group is included in the KEGG pathway map of Galactose Metabolism (KEGG ID map00052). In Table 2, the genes such as YEL011W and YFR015C were not included in the pathway map of Galactose Metabolism. However, some genes were induced by galactose and repressed by glucose. These genes could have influenced galactose metabolism with other general genes in the pathway map of Galactose Metabolism.
We confined and identified the hidden pathways from CPM. Even though the numerous nodes and arcs make the graph complicated, our path finding algorithm can find the sequential compound reactions that are influenced strongly by the fluctuation. In order to explore the pathways related to the production of a useful compound, it is important that the candidate enzymes are chosen with consideration of the distance on the pathway map.
4. Conclusion
In this work, we developed an algorithm for the reconstruction of a comprehensive pathway model. This algorithm was applied to S. cerevisiae. The path finding from sCPM revealed the hidden pathways. Some pathways included enzymatic genes that were induced or repressed, as with other known genes in the known pathway maps. The pathways were explored effectively by the combination with the known pathway maps.
Acknowledgements
This work was supported by a grant from the commission for the Development of Artificial Gene Synthesis Technology for Creating Innovative Biomaterial, from the Ministry of Economy, Trade and Industry (METI), Japan.
Cite this paper
ItaruTakeda,MasayukiMachida,SachiyoAburatani, (2015) Development of an Algorithm for Reconstructing a Comprehensive Pathway Model: Application to Saccharomyces cerevisiae. Journal of Biomedical Science and Engineering,08,500-510. doi: 10.4236/jbise.2015.88047
References
- 1. Wiemann, P. and Keller, N.P. (2014) Strategies for Mining Fungal Natural Products. Journal of Industrial Microbiology & Biotechnology, 41, 301-313.
http://dx.doi.org/10.1007/s10295-013-1366-3 - 2. Hwang, K.S., Kim, H.U., Charusanti, P., Palsson, B.O. and Lee, S.Y. (2014) Systems Biology and Biotechnology of Streptomyces Species for the Production of Secondary Metabolites. Biotechnology Advances, 32, 255-268.
http://dx.doi.org/10.1016/j.biotechadv.2013.10.008 - 3. Bentley, R. (2006) From miso, saké and shoyu to Cosmetics: A Century of Science for Kojic Acid. Natural Product Reports, 23, 1046-1062.
http://dx.doi.org/10.1039/b603758p - 4. Saruno, R., Kato, F. and Ikeno, T. (1979) Kojic Acid, a Tyrosinase Inhibitor from Aspergillus albus. Agricultural and Biological Chemistry, 43, 1337-1338.
- 5. Cabanes, J., Charzarra, S. and Garcia-Carmona, F. (1994) Kojic Acid, a Cosmetic Skin Whitening Agent, Is a Slow-Binding Inhibitor of Catecholase Activity of Tyrosinase. Journal of Pharmacy and Pharmacology, 46, 982-985.
http://dx.doi.org/10.1111/j.2042-7158.1994.tb03253.x - 6. Terabayashi, Y., Sano, M., Yamane, N., Marui, J., Tamano, K., Sagara, J., Dohmoto, M., Oda, K., Ohshima, E., Tachibana, K., Higa, Y., Ohashi, S., Koike, H. and Machida, M. (2010) Identification and Characterization of Genes Responsible for Biosynthesis of Kojic Acid, an Industrially Important Compound from Aspergillus oryzae. Fungal Genetics and Biology, 47, 953-961.
http://dx.doi.org/10.1016/j.fgb.2010.08.014 - 7. Liu, J., Farmer, J.D. Jr., Lane, W.S., Friedman, J., Weissman, I. and Schreiber, S.L. (1991) Calcineurin Is a Common Target of Cyclophilin-Cyclosporin A and FKBP-FK506 Complexes. Cell, 66, 807-815.
http://dx.doi.org/10.1016/0092-8674(91)90124-H - 8. Kunz, J. and Hall, M.N. (1993) Cyclosporin A, FK506 and Rapamycin: More than Just Immunosuppression. Trends in Biochemical Sciences, 18, 334-338.
http://dx.doi.org/10.1016/0968-0004(93)90069-Y - 9. Survase, S.A., Kagliwal, L.D., Annapure, U.S. and Singhal, R.S. (2011) Cyclosporin A—A Review on Fermentative Production, Downstream Processing and Pharmacological Applications. Biotechnology Advances, 29, 418-435.
http://dx.doi.org/10.1016/j.biotechadv.2011.03.004 - 10. Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H. and Kanehisa, M. (1999) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research, 27, 29-34.
http://dx.doi.org/10.1093/nar/27.1.29 - 11. Kanehisa, M. and Goto, S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research, 28, 27-30.
- 12. Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. and Tanabe, M. (2012) KEGG for Integration and Interpretation of Large-Scale Molecular Data Sets. Nucleic Acids Research, 40, 109-114.
http://dx.doi.org/10.1093/nar/gkr988 - 13. Karp, P.D., Ouzounis, C.A., Moore-Kochlacs, C., Goldovsky, L., Kaipa, P., Ahrén, D., Tsoka, S., Darzentas, N., Kunin, V. and López-Bigas, N. (2005) Expansion of the BioCyc Collection of Pathway/Genome Databases to 160 Genomes. Nucleic Acids Research, 33, 6083-6089.
http://dx.doi.org/10.1093/nar/gki892 - 14. Croft, D., O’Kelly, G., Wu, G., Haw, R., Gillespie, M., Matthews, L., Caudy, M., Garapati, P., Gopinath, G., Jassal, B., Jupe, S., Kalatskaya, I., Mahajan, S., May, B., Ndegwa, N., Schmidt, E., Shamovsky, V., Yung, C., Birney, E., Hermjakob, H., D’Eustachio, P. and Stein, L. (2011) Reactome: A Database of Reactions, Pathways and Biological Processes. Nucleic Acids Research, 39, 691-697.
http://dx.doi.org/10.1093/nar/gkq1018 - 15. Karp, P.D. and Caspi, R. (2011) A Survey of Metabolic Databases Emphasizing the MetaCyc Family. Archives of Toxicology, 85, 1015-1033.
http://dx.doi.org/10.1007/s00204-011-0705-2 - 16. Stobbe, M.D., Jansen, G.A., Moerland, P.D. and van Kampen, A.H. (2014) Knowledge Representation in Metabolic Pathway Databases. Briefings in Bioinformatics, 15, 455-470.
http://dx.doi.org/10.1093/bib/bbs060 - 17. Kanehisa, M. (2013) Chemical and Genomic Evolution of Enzyme-Catalyzed Reaction Networks. Federation of European Biochemical Societies, 587, 2731-2737.
http://dx.doi.org/10.1016/j.febslet.2013.06.026 - 18. Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. and Hattori, M. (2004) The KEGG Resource for Deciphering the Genome. Nucleic Acids Research, 32, 277-280.
http://dx.doi.org/10.1093/nar/gkh063 - 19. Barrett, T. and Edgar, R. (2006) Gene Expression Omnibus: Microarray Data Storage, Submission, Retrieval, and Analysis. Methods in Enzymology, 411, 352-369.
http://dx.doi.org/10.1016/S0076-6879(06)11019-8 - 20. Gasch, A.P., Spellman, P.T., Kao, C.M., Carmel-Harel, O., Eisen, M.B., Storz, G., Botstein, D. and Brown, P.O. (2000) Genomic Expression Programs in the Response of Yeast Cells to Environmental Changes. Molecular Biology of the Cell, 11, 4241-4257.
http://dx.doi.org/10.1091/mbc.11.12.4241 - 21. Umemura, M., Koyama, Y., Takeda, I., Hagiwara, H., Ikegami, T., Koike, H. and Machida, M. (2013) Fine de Novo Sequencing of a Fungal Genome Using Only SOLiD Short Read Data: Verification on Aspergillus oryzae RIB40. PLoS ONE, 8, e63673.
http://dx.doi.org/10.1371/journal.pone.0063673 - 22. Takeda, I., Tamano, K., Yamane, N., Ishii, T., Miura, A., Umemura, M., Terai, G., Baker, S.E., Koike, H. and Machida, M. (2014) Genome Sequence of the Mucoromycotina Fungus Umbelopsis isabellina, an Effective Producer of Lipids. Genome Announcements, 2, e00071-14.
http://dx.doi.org/10.1128/genomea.00071-14 - 23. Moriya, Y, Itoh, M., Okuda, S., Yoshizawa, A.C. and Kanehisa, M. (2007) KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Research, 35, 182-185.
http://dx.doi.org/10.1093/nar/gkm321 - 24. Sellick, C.A., Campbell, R.N. and Reece, R.J. (2008) Galactose Metabolism in Yeast-Structure and Regulation of the Leloir Pathway Enzymes and the Genes Encoding Them. International Review of Cell and Molecular Biology, 269, 111-150.
http://dx.doi.org/10.1016/S1937-6448(08)01003-4 - 25. Conrad, M., Schothorst, J., Kankipati, H.N., Van Zeebroeck, G., Rubio-Texeira, M. and Thevelein, J.M. (2014) Nutrient Sensing and Signaling in the Yeast Saccharomyces cerevisiae. FEMS Microbiology Reviews, 38, 254-299.
http://dx.doi.org/10.1111/1574-6976.12065 - 26. Johnston, M. (1987) A Model Fungal Gene Regulatory Mechanism: The GAL Genes of Saccharomyces cerevisiae. Microbiological Reviews, 51, 458-476.
- 27. Bro, C., Knudsen, S., Regenberg, B., Olsson, L. and Nielsen J. (2005) Improvement of Galactose Uptake in Saccharomyces cerevisiae through Overexpression of Phosphoglucomutase: Example of Transcript Analysis as a Tool in Inverse Metabolic Engineering. Applied and Environmental Microbiology, 71, 6465-6472.
http://dx.doi.org/10.1128/AEM.71.11.6465-6472.2005
NOTES

*Corresponding author.