Journal of Biomedical Science and Engineering
Vol.08 No.09(2015), Article ID:59498,14 pages
10.4236/jbise.2015.89056
Vitamin D3 Receptor Activation Rescued Corticostriatal Neural Activity and Improved Motor-Cognitive Function in −D2R Parkinsonian Mice Model
Azeez O. Ishola1, Babafemi J. Laoye2, Damilola E. Oyeleke1, Oluwamolakun O. Bankole2, Mujittapha U. Sirjao1, Ansa E. Cobham3, Wasiu G. Balogun4, Amin Abdulbasit5, Ibukun D. Akinrinade6,7, Olalekan M. Ogundele1*
1Department of Anatomy, College of Medicine and Health Sciences, Afe Babalola University, Ado-Ekiti, Nigeria
2Department of Biological Sciences, College of Sciences, Afe Babalola University, Ado-Ekiti, Nigeria
3Department of Anatomy, College of Medicine, University of Calabar, Calabar, Nigeria
4Department of Anatomy, College of Health Sciences, University of Ilorin, Ilorin, Nigeria
5Department of Physiology, College of Health Sciences, University of Ilorin, Ilorin, Nigeria
6Instituto Gulbenkian de Ciencia, Oerias, Portugal
7Department of Anatomy, Bingham University College of Medicine, Karu, Nigeria
Email: *ola.ogundele@abuad.edu.ng
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 23 April 2015; accepted 6 September 2015; published 9 September 2015
ABSTRACT
Background: fourth generation antipsychotics have been implicated in the blockade of calcium signalling through inhibition of dopamine receptive sites on dopaminergic D2 Receptor (D2R). As a result of the abnormal calcium signalling associated with D2R inhibition, changes occur in the motor and memory neural axis leading to the observed behavioural deficits after prolonged haloperidol. Thus, Vitamin D3 receptor (VD3R), a calcium controlling receptor in the striatum can be targeted to relief the neurological symptoms associated with haloperidol (−D2R) induced PD. Aim: This study sets to investigate the role of VD3R activation in vitro and in vivo after haloperidol- induced Dopaminergic (D2R) blockade. In addition, we examined the associated neural activity and behavioural changes in parkinsonian and VDRA intervention mice. Methods: Dopaminergic D2R inhibition was investigated in vitro using Melanocytes isolated from the scale of a Tilapia. In four separate set ups, the cells were cultured in calcium free Ringer’s solution as follows; 300 µM haloperidol, 100 µM VD3, 100 mM calcium chloride and a combination of 300 µM haloperidol and 100 µM VD3. Subsequently, dopaminergic vesicle accumulation and calcium signalling were observed in bright field microscopy using blue and green fluorescence probes. In the second phase, PD was induced in adult BALB/c mice (−D2; n = 8) after 14 days of intraperitoneal haloperidol treatment (10 mg/Kg). A set of n = 4 mice were untreated (−D2) while the other group (n = 4) received 100 mg/Kg of VD3 for 7 days (−D2/+VDR). The control groups (n = 4 each) were treated with normal saline (NS) and VD3 (+VDR) for 14 days. At the end of the treatment phase, the animals were assessed in Rotarod, parallel bar-, cylinder-, Y-Maze-, one trial place recognition- and novel object recognition-(NOR) tests. Neural activity was measured using chronic electrode implants placed in the M1 (motor cortex), CPu (striatum), CA1 (hippocampus) and PFC (prefrontal cortex). Neural activity was compared with the outcomes of behavioural tests for memory and motor functions and data was expressed as mean ± SEM (analysed using ANOVA with Tukey post-hoc test, significant level was set at 0.05). Results/Discussion: in vitro outcomes show that VDR increase calcium signalling and reverses the effect of haloperidol; specifically by reducing dopaminergic vesicle accumulation in the cell body. Similarly, in vivo neural recordings suggest an increase in calcium hyperpolarization currents in the CPu and PFC of intervention mice (−D2/+VDR) when compared with the parkinsonian mice (−D2). These animals (−D2/+VDR) also recorded an improvement in spatial working memory and motor function versus the Parkinsonian mice (−D2). These outcomes suggest the role of CPu-PFC corticostriatal outputs in the motor-cognitive decline seen in parkinsonian mice. Similarly, VDRA reduced the neural deficits through restoration of calcium currents (burst activities) in the intervention mice (−D2/+VDR). Conclusion: VDRA treatment reduced the motor-cognitive defects observed in haloperidol induced PD. Our findings suggest the role of VDRA in restoration of calcium currents associated with PFC and CPu corticostriatal outputs seen as burst frequencies in in vivo neural recording.
Keywords:
Dopamine, Calcium Signalling, D2R, VD3R, Corticostriatal, Neural System

1. Introduction
Fourth generation antipsychotics are often characterized by their ability to centrally block dopaminergic D2 receptors in the striatum (CPu), thus inhibiting the excitatory motor neural pathway (Motor cortex: M1, basal ganglia and substantianigra) which leads to rigidity and akinesia in limbs [1] -[3] . After prolonged use of dopaminergic D2 blockers in the treatment of depression or schizophrenia, movement disorders and tardive dyskinesia have been reported in humans [4] [5] . As a result of effect of antipsychotics on Dopaminergic D2 transmission, they are capable of generating extrapyramidal symptoms-jerky movements of the eyes, oral regions and limbs [6] -[8] . Although dopamine depletion and loss of dopaminergic neurons have been described as the main cause of Parkinson disease (PD), recent studies have shown that D2 receptor sensitivity can also induce PD in the presence of normal striatal dopamine concentration [9] [10] .
Haloperidol, a typical antipsychotic medication and Dopaminergic D2 receptor blocker, has been known to induce toxicity in the motor neural system. Furthermore, its major mechanism of toxic induction has been identified to involve weak binding of haloperidol to the dopamine receptive site on D2 receptor domain; this often results to receptor sensitivity after a prolonged duration [11] [12] . This in effect prevents the heteromeric combination of dopaminergic D2 with D1 receptors; required for calcium signalling in dopaminergic neurotransmission [11] [13] . Furthermore, D2-D1 calcium signalling has been shown to be involved in the activation of BDNF, vesicle clearance (autophagy) and CamKII α activation: all of which are required for synaptic plasticity and motor neural function [14] .
Vitamin D3 Receptor (VD3R), a candidate calcium controlling receptor, is relatively abundant in the brain; thus giving an advantage of selective targeting of pigmented dopaminergic neurons in mammalian models of degenerative diseases [15] [16] . Similarly, VD3RA (VD3R-Agonist), a steroid, has been reported to be involved in the activation calcium for BDNF signalling required for neuronal survival under various pathological conditions [17] [18] . Other studies have reported the role of VD3RA in transcription of genes involved in neuronal repair, protein synthesis, dopamine metabolism and glia-dependent neurotropin release in the brain [18] [19] .
Previously, we have shown that VD3R activation holds promising therapeutic effects in relieving motor-cog- nitive dysfunctions observed in mice models of haloperidol-induced PD. Electrophysiological (extracellular) neural recordings from the motor cortex (M1; Layer2/3) showed a decline in extracellular neural summation (epoch-time activity; Hz/secs) after an acute treatment with intraperitoneally administered haloperidol. Interestingly, normal neural activity was restored after VD3RA treatment in vivo. In addition, immunohistochemical demonstration of neuron and astrocyte populations revealed an increase in cell proliferation in the M1 post VD3RA treatment and corroborated the improved motor-cognitive function observed in behavioural studies [20] . The current study examines the role of prolonged haloperidol use in D2 sensitivity in the motor-cognitive neural axis. Subsequently, we examined the role of VD3R activation on D2-dependent neural activity (calcium signalling) in the motor-cognitive neural axis and how it moderates motor-cognitive behavioural changes in parkinsonian mice.
2. Materials and Methods
Materials: All chemical reagents were sourced from Sigma-Aldrich, Germany. The Melanocytes were isolated from Tilapia obtained from a river around the University. The Tilapia scale was dissected and perfused with Ringer’s solution in a petridish at room temperature for 2 hours before treatment. Haloperidol Injection was procured from Kanada Pharmacy, Nigeria and re-suspended in dextrose saline. VD3 was procured from Standard Pharma, Nigeria and dissolved in normal saline. Haloperidol and VD3 solutions were prepared weekly as needed and stored at 4˚C.
Animal Source: Live Tilapia fishes (n = 5) was obtained from a nearby river with the help of professional fishermen and were kept in a tank filled with flowing freshwater before use. Adult BALB/c mice were procured form the animal holding facility of Afe Babalola University and were maintained in the same facility throughout the duration of the study.
Animal Handling and Care: The marine specie (tilapia) was kept in a holding tank with adequate water current for respiration. Adult BALB/c mice were kept in well aerated rooms with alternating 12 hours light and 12 hours darkness. The animals were adequately fed (normal rat chow) and had constant supply of drinking water. All animal handling protocols were in accordance with the IACUC animal use guidelines and were approved by the Animal Use Ethical Committee of the Afe Babalola University, Nigeria [Protocol Number: ABUAD/015/ 0075].
In vitro Melanocyte Preparations: Melanocytes were obtained from the scale of live Tilapia fish. The scales were washed and softened in 10 mM Na2HCO3 prepared in calcium free Ringer’s solution [D-Glucose: 1.8 g/L, MgCl2: 0.0468 g/L, KCl: 0.34 g/L, NaCl: 7.0 g/L, Na2HPO4 (dibasic): 0.1 g/L, Na2HPO4: 0.18 g/L and 1.26 gm of sodium bicarbonate (pH 7.4)] to allow percolation of fluid into the cavity of the scale where the melanocytes are located.
D2 Inhibition and Ca2+ Signalling In vitro: A light microscope (Roachoscope; Backyard Brains, MI, USA) equipped with green and blue fluorescent light sources were used for this study. The light sources were made incident on the cells in culture while the fluorescence images were acquired (Cameroscope 5.1 MP). Subsequent analysis involved measurement of the fluorescence intensity on the cell body and cellular processes using Image J (NIH, USA) to determine the maximum wavelength of several single fluorescence spots on the cell body and cellular processes.
Animal Treatment (In vivo): BALB/c mice (N = 16) were separated into 4 groups based on body weight distribution. PD was induced after 14 days of treatment with 10 mg/Kg BW of dopaminergic D2 blocker (n = 8). Subsequently, n = 4 animals were treated with 100 mg/Kg BW (i.p. 7 days) VD3 (−D2/+VDRA) while the remaining set of n = 4 animals were untreated (−D2). A separate group of n = 4 animals received VD3 for 14 days (100 mg/Kg BW intraperitoneal; +VDR) while the control (n = 4) received normal saline for 14 days (NS; intraperitoneal).
2.1. Motor and Memory Function
At the end of the treatment phase, (Day 14 for control, −D2, +VDR; Day 21 for −D2/+VDR) the animals were examined in various tests for motor and memory function. All animals were familiarized with the behavioural tools during the treatment phase and were moved to the testing area 72 hours before the commencement of the tests.
2.2. Motor Function and Coordination
Parallel Bar Test: Motor coordination was accessed on two raised 1 m long (1 mm) parallel bars (3 cm apart) mounted on the 60 cm high wooden frame. The mice was placed at the 0.5 m mark (centre of the raised bars) and allowed to roam freely on the bar. The duration taken for the mice to make a 90˚ turn was recorded as the latency of turn (LOT) for a 3 minutes trial.
Rotarod: The test involved three trials of 3 minutes each (T1, T2 and T3) separated by an inter trial time of 60 minutes. The time spent on the Rotarod in T1, T2 and T3 were determined and averaged to determine the latency of fall (LOF) for each group.
Cylinder Test: Each animal was placed in a 500 ml beaker (cylinder) and was allowed to explore the walls of the cylinder with the forelimbs while standing on the two hind limbs. Subsequently, we determined the count of exploratory forelimb-to-wall contacts made by the mice during a 3 minutes test to calculate the score of climbing attempts.
2.3. Memory Function
Novel Object Recognition (NOR): Non-spatial working was accessed in NOR test for object recognition. In this study, the test was modified as the objects were placed in close proximity (10cm apart) to eliminate the effect of motor dysfunction on object exploration by the animals. In the first trial (T1), the animal was allowed to explore two identical objects (green) for 5 minutes following which an intertrial (I.T.) time of 60 minutes was observed. For the second trial (T2), one of the familiar objects was replaced with a novel object (red) for the test duration (5 minutes). Subsequently, the time spent on exploration of familiar and novel objects were measured in T2 to determine the memory index (MI).
Y-Maze: The Y-Maze set up was placed in an isolated sound-proof room with adequate illumination. The animal was gently placed into the central triangle of the Y-Maze and allowed to explore the three arms of the maze. Subsequently, we determined the frequency of alternation between the three arms of a Y-Maze (a, b and c) for the duration of the test (10 minutes) to determine the memory index (percentage alternation between successive arms of the maze)
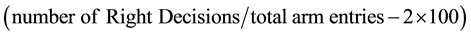
One Trial Place Recognition Test: The Y-Maze apparatus was set up in an isolated room with a digital camera to cover three arms of the maze. One arm (a) of was blocked while the animal was placed in the centre of the maze facing the two open arms (b and c). For the first trial (T1) the animal was allowed to explore the two open arms (b and c) for 10 minutes following which an intertrial time of 60 minutes was observed; animal was returned to the cage during the intertrial period. In the second trial (T2; 10 minutes), the animal was returned to the Y-Maze with the three arms a, b and c open. The exploration time for the arms was recorded to determine the duration spent in the novel arm (a).
2.4. Electrophysiology
We obtained electrophysiological recording from the basal nucleus (CPu) Motor cortex (M1; L4-L6), Hippocampus (CA1) and the Prefrontal cortex (PFC) using chronically implanted wire electrodes. Thirty minutes before the implant, animals received 2 mg/Kg i.p meloxicam and were deeply anesthetised using 100 mg/Kg Ketamine combined with 5 mg/Kg Diazepam. Using a stereotaxic frame, the scalp was dissected above the bregma to expose the cranium. Periosteal tissue was removed using hydrogen peroxide solution and a cotton bud. The positions of the M1, CPu, PFC and Hippocampus were located using a stereotaxic grid while the depth was an approximate equivalent of the electrode length [coordinates relative to the bregma: M1 (AP: +3.34 mm ML: +3 mm DV: +2.5 mm), CPu coordinates (AP: +2.28 mm ML: +3 mm DV: +6 mm), Hipp (AP: −4.4 mm ML: +2 mm DV: +3 mm), PFC (AP: +2.2 mm ML: +1 mm DV: +2.5 mm)]. A dental drill was used to make holes in the cranium following which insulated wire electrodes were implanted to the appropriate depth. The ground electrode was placed on the cranium of the contralateral side. Subsequently, the implant was covered by orthodontic resin to hold the electrodes in position during the recording procedure. The terminal wires of the electrodes were connected to the amplifier through small head-sockets in preparation for immobile awake recordings. The data from the amplifier (Spiker Box; Backyard Brains, Michigan, USA) was captured on the Audacity software v4.2 and analysed in SigView v2.1 (Signal Labs, USA) to determine the extracellular summation epoch neural activity (calcium signals) expressed as Frequency (Hz) per unit time.
Statistical Analysis: Data were presented as mean ± SEM; analysed using ANOVA and Tukeys Post-Hoc test. Statistical Significance was set as P < 0.05*, P < 0.01**, P < 0.001***.
3. Results
Cells Autofluorescence in vitro: We have shown that Tilapia melanocytes emitted white and green fluorescence when exposed to blue light at 500 nm and green light at 580 nm (Figure 1). White fluorescence was seen when melanocytes were cultured in calcium-free Ringer’s solution (pH 7.4) containing 300 µM Dopaminergic D2 blocker (melanophores accumulation; Figure 1(A) and Figure 1(B). Subsequent analysis show that inhibition of Dopaminergic D2 receptors (WT/-D2), activation of VDR (WT/+VDRA) and a combination of both (WT/-D2/ +VDR) caused no significant change in light intensity measured on the cell body (Figure 1(C) and Figure 1(D)). By contrast, green fluorescence was emitted by the melanocytes when 100 mM calcium chloride (CaCl2) was added to calcium free Ringer’s solution (calcium signal; Figure 1(F)) However, Dopaminergic D2 inhibition increased fluorescence intensity in the cellular processes when compared with the control (untreated) and 100 mM CaCl2 treatment. In addition, VD3RA treatment (300 µM) increased the number of cells exhibiting fluorescence in calcium free Ringer’s solution and caused an increase in intrinsic calcium signalling seen as green fluorescent bands (Figure 1(D)). This was also associated with a reduction of white fluorescence (increased vesicle clearance; Figure 1(D)) when compared with the−D2 (Figure 1(C)). In a separate set up, Tilapia melanocytes were incubated with VD3R agonist and D2 antagonist (300 µM respectively) in calcium free Ringer’s solution for 60 minutes. As a result of this treatment, a moderate intensity of white and green fluorescence was observed on the cell body and cellular processes (Figure 1(E)); similar to the control (Figure 1(A)). These findings suggest that VD3RA activation can restore calcium signalling (green band) and improve vesicle clearance (reduced white fluorescence intensity) following haloperidol induced D2 inhibition.
3.1. In Vivo
Motor Function Tests
Rotarod Test: Haloperidol induced PD (−D2) caused a decline in motor function seen as a reduction in LOF when the −D2 treatment was compared with the control (P < 0.01). Subsequent treatment with VD3RA (−D2/ +VDR) improved motor function in this group as the mice recorded an increase in LOF when compared with the control and the parkinsonian group (−D2). Although VDRA intervention after haloperidol induced PD caused a significant improvement in motor function, however, VD3RA treatment without prior induced PD caused a decline in motor function when +VDR was compared with the control (P < 0.05) (Figure 2(A)).
Parallel Bar Test: In this test a significant increase or decrease in LOT scores were considered as abnormal motor coordination when the treatment groups were compared against the control. Haloperidol treatment (−D2) increased the LOT significantly versus the control (P < 0.05) as the mice showed characteristic decline in motor coordination on the raised bars. After VD3RA intervention (−D2/+VDR), a decline in LOT was observed when the −D2/+VDR was compared with the −D2 (P < 0.01). In addition, the LOT values were similar to the control scores and represent an increase in motor coordination (no significant increase or decrease versus the control). Similar to our observations in Rotarod test, VD3RA treatment without prior haloperidol induced PD caused a significant decrease in LOT when compared with the control (P < 0.01); thus, we inferred that VD3R treatment induced motor dysfunction when administered without prior haloperidol treatment (Figure 2(B)).
Cylinder Test: We determined the climbing attempts in a glass cylinder as a measure of motor function after haloperidol induced PD and subsequent VD3RA intervention. Decreased climbing attempts score represents a decline in motor function when the treatment groups were compared with the control. Haloperidol induced PD (−D2) caused a significant decrease in climbing attempts (impaired) when compared with the control (P < 0.05). In the intervention group (−D2/+VDR), motor coordination (number of climbing attempts) increased significantly after 7 days of VD3RA treatment when compared with the −D2 (P < 0.05). No significant change in motor function was observed in the +VDR group when compared with the control (Figure 2(C)).
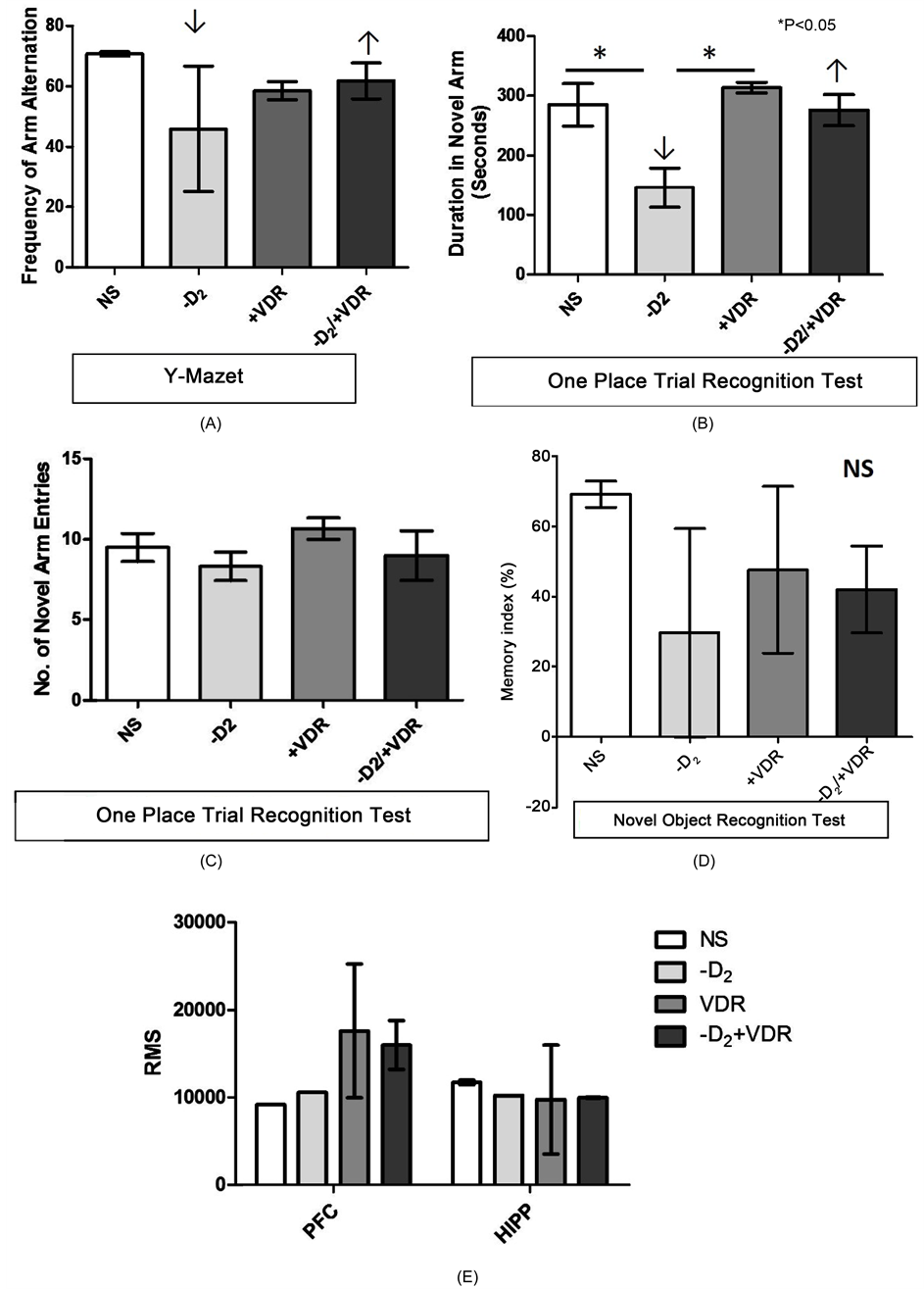
Motor Epoch Neural Activity: Haloperidol induced PD caused an increase in summation activity in the M1 and CPu (Figure 2(D) and Figure 2(E)) and was associated with a decline in motor function (Figures 2(A)-(C)). After VD3RA intervention (−D2/+VDR) a reduction in epoch neural activity was observed in the M1 while a



Figure 1. (A)-(F). Bright field imaging of Tilapia melanocytes with blue and green fluorescent light sources. (A)-(B) Wild type (WT) Tilapia melanocytes cultured in calcium free Ringer’s solution. Red circles denotes cells emitting fluorescence when a blue light was made incident on the cells in vitro. Subsequently, the light source was removed and the melanophores retained the fluorescence in bright field imaging (Figure 1(B)). (C) Extensive cellular processes were observed 60 minutes after the cells were incubated in a Calcium free Ringer’s solution containing 300 µM haloperidol (D2-Antagonist). The extent of dopaminergic inhibition was seen as an increase in melanophore fluorescence intensity when the WT:−D2 was compared with the control. This was presumably due to the inhibition of Ca2+ signalling as no green fluorescence was observed. (D) Tilapia melanocytes were incubated with 300 µM of VD3RA in calcium free Ringer’s solution for 60 minutes. Green and white fluorescence were seen after this treatment. The green fluorescence (calcium signal) was observed in the centre of the cell (outlined with dotted black lines) while the white fluorescence (melanophore) was restricted to the cell body and showed a decrease in fluorescence when compared with the haloperidol treated cells (Figure 1(C)). (E) A combined treatment with 300 µM VD3RA and 300 µM D2-Antagonist showed the presence of green (calcium) and white fluorescence with reduced intensity in both cases (similar to the control; Figure 1(A)). This show that VD3R activation facilitates intrinsic calcium signalling, thus reducing the extent of vesicle accumulation when compared with haloperidol treatment (Figure 1(C)). (F) WT melanocytes in Ringer’s solution containing 100 mM CaCl2. Prominent green fluorescence was seen in the cell body and cellular processes; especially the terminal part of melanocytes (Magnification ×140). Figures 1(G)-(H). Measurement of fluorescence intensity (nm) on the cell body and cellular processes of melanocytes. (G) No significant change was seen in fluorescence intensity for the cell bodies when the WT/−D2, WT/+VDRA and WT/+VDRA/−D2 were compared with the control (WT). However, all groups were significant versus the 100 mM CaCl2 treated group (P < 0.001***). (H) Dopaminergic D2 inhibition caused the most significant increase in fluorescence observed on the cellular processes and this was reduced when VDRA was co-incubated with D2 antagonist (WT/+VDRA/−D2) in vitro (See Figure 1(E)). Extensive cellular processes green fluorescence was observed in cells co-incubated with 100 mM CaCl2. All groups recorded a significant increase in fluorescence when compared against the control (P < 0.001***); similarly D2 inhibition was significant versus CaCl2 treatment (P < 0.01**).

 (D)
(D)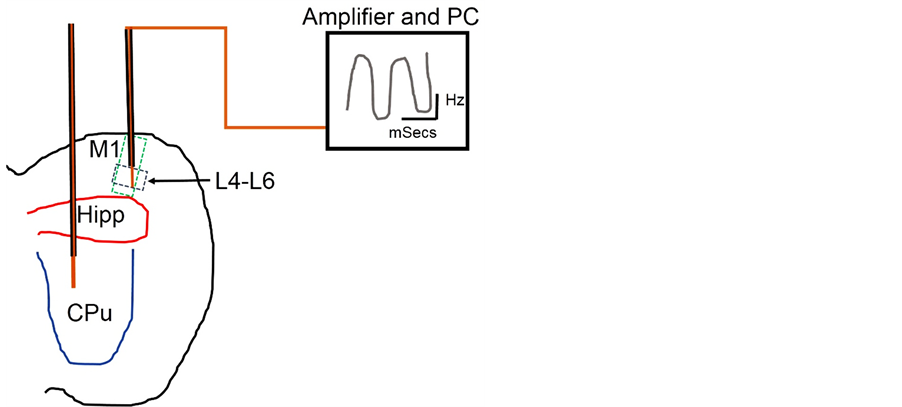 (F)
(F)
Figure 2. (A)-(C). Neural basis of motor function change in Haloperidol (−D2) and VD3RA treated BALB/c Mice. (A) The latency of fall (LOF) recorded in Rotarod test for motor function. A reduction in motor function was seen in the -D2 treated mice when compared with the control (P < 0.01). An improvement in motor function was observed in these mice after 7 days of VD3RA treatment (−D2/+VDR); this was shown by an increase in the LOF when compared with the −D2 treatment group. VD3RA treatment without a prior −D2 treatment also caused a significant decrease in LOF when compared with the control (P < 0.05). (B) Motor coordination was assessed in the treatment and control group in the parallel bar test to determine the latency of turning (LOT). Haloperidol treatment (−D2) caused a decline in motor coordination after 14 days of treatment. The decline was recorded as an increase in the LOT when compared with the control (P < 0.05). Interestingly, VD3R treatment post Haloperidol treatment improved motor coordination which was observed as a decrease in LOT when the −D2/+VDR group was compared with the control (P < 0.01). However, VD3RA (only) treatment increased motor coordination in this test. This was shown by the decrease in LOT when compared with the control and other treatment groups (C) The score of climbing attempts was recorded for the control and treatment groups after the treatment phase. Haloperidol treatment (−D2) decreased motor function (decreased climbing attempts score) when compared with the control (P < 0.05). After VD3RA treatment (−D2/+VDR) an increase in motor function was recorded as shown by the increased score of climbing attempts when compared with the −D2 treatment (P < 0.05). Similarly, mice treated with VD3RA only showed a decline in climbing attempts when compared with the control empirically. Figures 2(D)-(F). In vivo neural extracellular epoch recording from the L4-L6 of the M1 and CPu. (D) The root mean square (RMS) represents the extracellular neural calcium activity (Summation) in the M1 (thalamic projections and nigrostriatal afferents) recorded through a chronic electrode implant in immobile awake mice. Haloperidol treatment increased the RMS in M1 when compared with the control (mean) and corroborates a decline in motor function observed in behavioural tests (Figures 2(A)-(C)). However, no major change was observed in the CPu RMS when compared with the control. To support this, the spike pattern for the −D2 treatment showed an increase in the calcium mediated hyperpolarization potential (Figure 2(E)) and frequency of the potential. The CPu also showed an increase in signalling when compared with the control in these mice (Figure 2(E) −D2). VD3RA treatment after a phase of Haloperidol induced PD increased the RMS and summation activity when compared with the −D2 treatment group (Figure 2(E) −D2/+VDR). This is also characterized by a spike pattern similar to the control M1. In addition, the CPu recordings showed burst spike patterns and was associated with an increase in motor coordination versus the −D2 treatment group (Figure 2(E) +VDR). In mice treated with VD3RA only, we recorded an increase in RMS in the M1 and CPu. Similarly, spike train showed an increase in hyperpolarization for M1-CPu and was characterized by a decline in motor coordination observed in behavioural tests (Figures 2(A)-(C)). Figure 2(F) represents the schematic locations of chronic unilateral electrode implants into the M1 (L4-L6) and the CPu.
burst activity (increased RMS) was observed in the CPu; this translated into an improvement in motor function in the behavioural tests (Figures 2(A)-(C) and Figure 2(E) and Figure 2(F)). VD3RA treatment (+VDR) increased the RMS in the M1 while an irregular tonic activity (reduced RMS) was recorded in the CPu for this treatment (Figure 2(D) and Figure 2(E)). This was characterized by a decline in motor function in behavioural tests (Figures 2(A)-(C)). From these findings, we deduced that haloperidol induced PD (−D2) caused hyperactivity in the M1 and CPu, thus, leading to a decline in motor function. Subsequent VD3RA intervention reduced the M1 activity and created a burst signal in the CPu; thus improving motor function when compared with the parkinsonian mice (−D2; Figures 2(A)-(C)).
3.2. Memory Function Tests
Y-Maze: Memory function was expressed as the frequency of arm alternation between the three arms of the maze. No significant change was observed in the memory index when the haloperidol treated mice were compared with the control (−D2); although an empirical reduction in memory index was seen in this group. Similarly, no significance was recorded in the VD3RA intervention group (−D2/+VDR) when compared with the −D2 and the control (Figure 3(A)). To verify these findings, the Y-Maze was re-adapted for a one place trial recognition test to determine the spatial working memory in the novel arm of the maze. The frequency of all arm entries showed that the control and the treatment groups had equal exposure to the novel arm as no significance was seen for all treatments and control groups (NS; Figure 3(C)). Despite the frequency of novel arm exposure, the haloperidol treatment recorded a decrease in novel arm duration (decreased spatial working memory) when compared with the control (P < 0.05); further supporting the empirical decrease in memory index observed in Y-Maze (−D2; Figure 3(A)). Similarly, the VD3RA intervention (−D2/+VDR) group recorded an increase in novel arm duration (increased spatial working memory) when compared with the parkinsonian (−D2) mice and the control (P < 0.05; Figure 3(B)). VD3RA treatment of control mice (+VDR) caused an empirical decrease in memory index when compared with the control (Y-Maze; Figure 3(A)), however, no significant change in novel arm duration was seen when +VDR was compared with the control. In contrast to observations in motor function, VD3RA treated (+VDR) mice recorded a novel arm duration (spatial working memory) higher than the −D2 (P < 0.05; Figure 3(B)) in the one place trial recognition test. We deduced from these findings that VD3R treatment did not reduce spatial working memory but reduced motor function in control mice.
3.3. Memory Epoch Neural Activity (Spatial Working Memory)
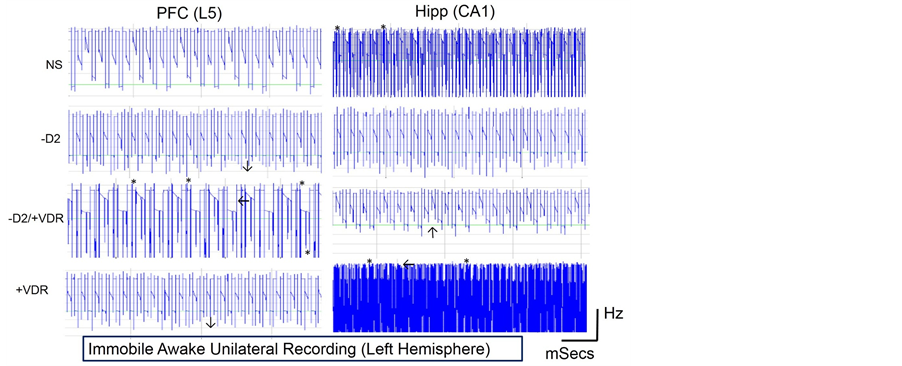
Haloperidol treatment (−D2) increased the frequency of PFC neural activity but reduced the Hippocampal (CA1) burst frequency in immobile awake mice. The increase in PFC spiking activity was also associated with an increase in RMS while loss of CA1 burst (high frequency) was linked with a reduction in CA1 RMS when the parkinsonian (−D2) mice was compared with the control. Interestingly, these variations in PFC and CA1 neural activities translated to a decline in spatial working memory in Y-maze and one place trial recognition test (Figures 3(A)-(C)). After VD3RA intervention, the −D2/+VDR treatment showed an increase in PFC activity (increased RMS; Figures 3(E)-(G)) and a reduction in CA1 spike frequency (reduced RMS) when compared with the control (Figures 3(E)-(G)). In behavioural tests for spatial working memory, the −D2/+VDR recorded an increase in spatial working memory when compared with the −D2, thus, the increased PFC activity, after VD3RA intervention, facilitated an increase in memory function despite a reduction in CA1 burst frequency.
Although PFC frequency increased in −D2, the threshold (RMS) was less than that recorded in the −D2/+VDR PFC. In addition, the −D2 and −D2/+VDR treatment showed a decline in CA1 RMS when compared with the control, however, the −D2/+VDR decline was more prominent versus −D2. Comparing these results with the outcome of behavioural tests, we deduced that PFC burst activity seen in the −D2/+VDR was linked with an increase in spatial working memory (Figure 3(E) and Figure 3(F)). Ultimately, VDRA intervention, after haloperidol induced PD, improved memory function through stimulation of PFC burst activities in cortical layers L4-L6.
VDRA, administered to control mice, had no significant effect on spatial working memory when compared with the control (untreated) mice (Figures 2(A)-(C)). This treatment raised PFC activity (frequency and RMS) to a threshold similar to −D2 treatment (Figure 3(F)) and increased the CA1 burst frequency when compared with the control. Despite the density of the CA1 burst pattern, the spatial working memory was empirically lower than control frequency and insignificant versus −D2/+VDR. Considering the spike pattern for the −D2/+VDR and −D2, the PFC frequency depicts the spatial working memory when compared with the CA1 activity. In support of this proposition, a prominent increase in CA1 burst activity without a corresponding increase in PFC neural activity, seen in +VDR, did not significantly increase memory function when compared with the −D2/+VDR (characterized by a prominent increase in PFC activity and a reduction in CA1 frequency) (Figure 3(E) and Figure 3(F)).
Novel Object Recognition Test (NOR): Object recognition non-spatial working memory was assessed in the parkinsonian (−D2) and the VDRA intervention group (−D2/+VDR) to determine the memory index. Although, the spatial working memory was significantly reduced in the −D2 (Y-Maze; Figures 3(A)-(C)), no significance was recorded for non-spatial working memory (Figure 3(D)) when the −D2 treatment was compared with the
 (F) (G)
(F) (G)
Figure 3. Neural Cognitive function in PD and after VD3RA intervention in Mice. (A) The memory index (M.I) was determined by the percentage alternation between the arms a, b and c of the Y-maze. Haloperidol treatment reduced spatial working in mice after 14 days. This was shown by an empirical reduction in memory index for this treatment when compared with the control. Subsequent treatment with VD3RA (−D2/+VDR) caused an increase in spatial working memory when compared with the −D2 and the control group. VD3RA treatment (only) caused no prominent change in the memory index versus the control. (B)-(C) The Spatial working memory was further investigated using the Y-maze one place trial recognition test. Similar to our observations in Figure 3(A), the Haloperidol treated mice showed a decline in spatial working memory when compared with the control (P < 0.05). Similarly, VD3RA treatment after the PD phase caused an improvement in cognitive function recorded as an increase in the spatial working memory when compared with the −D2 treatment (P < 0.05). However, VD3RA treatment caused no significant change when compared with the control. In addition, we analyzed the number of arm entries into the novel arm and observed no significance when the treatment groups were compared with the control. This implied that all groups had equal exposure to the novel and other arms (Figure 3(C)). (D) NOR; the memory index showed that haloperidol induced PD (−D2) and VD3RA intervention (−D2/+VDR) did not significantly alter non-spatial object recognition memory when the treatment groups were compared with the control. (E)-(F) After the treatment phase, the −D2 recorded an increase in PFC neural activity, a decline in hippocampal (CA1) activity (Figures 3(A)-(D)). The spike train for this treatment indicates an increase in neural activity in the PFC of these animals and was accompanied by a decrease in the CA1 summation. This was associated with a decrease in memory function in behavioral tests. VD3RA treatment after the PD phase increased the RMS in the PFC when compared with the control and the −D2 treatment group. However, a decrease in CA1 activity (Figure 3(F)) was observed in this treatment and corresponded to an increase in memory function in behavioral tests. VD3RA (only) treatment increased memory function and was associated with an increase in RMS in the PFC and CA1 when compared with the control, −D2 and −D2/+VDR. Thus, VD3RA intervention improved PFC but not CA1 activity when administered singly. Similarly, VD3RA treatment administered in PD, increased the activity of the PFC but not the CA1 region. Ultimately, the increase in PFC activity seen after VD3RA treatment in −D2 animals significantly improved memory function without a major change in CA1 activity. (G) The schematic locations of chronic unilateral electrode implants into the PFC (L4-L6) and the Hippocampus (CA1).
control. Similarly, the VDRA intervention in parkinsonian mice (−D2/+VDR) caused no change in memory index compared with the −D2 and the control group.
3.4. Memory Epoch Neural Activity (Non-Spatial Working Memory)
Despite the changes induced in PFC and CA1 neural activities for −D2, −D2/+VDR and +VDR (Figure 3(E) and Figure 3(F)), no significant change was recorded in the object recognition memory when these groups were compared with the control. However, RMS scores and spike train tracing suggest that either an increase in either PFC or CA1 activity might be responsible for the observed effect on memory index. It was observed that alltreatment groups and control showed an increase in activity for either the PFC or CA1 (Figure 3(F)). Specifically, −D2 and −D2/+VDR treatments showed an increase in PFC activity while the VD3R treatment recorded an increased CA1 frequency similar to the control. From these findings, we deduced that haloperidol induced PD significantly reduced spatial working memory (Figures 3(A)-(C)) but did not affect non-spatial working memory (Figures 3(D)-(F)).
4. Discussion
Our results show that Dopaminergic D2 inhibition facilitated melanosome turnover observed as an increased white fluorescence intensity in the cell body and cellular processes. In addition, D2R inhibition blocked calcium signalling as no green fluorescence (calcium signal) was seen (Figure 1(C)). This suggests that vesicle accumulation and melanosome turnover increased after calcium signalling was blocked as a result of Dopaminergic D2 inhibition. As expected, VD3RA activation facilitated calcium signalling (green fluorescence; Figure 1(D)) and increased vesicle clearance in the cell body and cellular processes after 60 minutes of incubation in calcium free Ringer’s solution. Interestingly, co-incubation of melanocytes with VD3RA and haloperidol (D2 Antagonist) (Figure 1(E)) caused a reduction in both calcium signalling (compared with VD3RA only; Figure 1(D)) and melanosome turnover (reduced vesicle traffic versus D2 inhibition only; Figure 1(C)). From these findings, we hypothesize that D2 inhibition-induced Parkinsonism might be as a result of calcium signalling blockade and accumulation of melanosomes (vesicles) in the cell body and cellular processes of pigmented adrenergic neurons of the nigrostraitum (CPu). Thus, activation of VD3R, after D2 inhibition, restored calcium signalling and vesicle clearance from the cell body and cellular processes. To test this hypothesis, we set up an in vivo model to investigate the physiological effects of D2R blockade induced Parkinsonism in adult BALB/c mice [21] .
4.1. VD3RA Treatment Improved Motor Function in −D2 Parkinsonian Model
Haloperidol induced PD caused a decline in motor function when compared with the control. This was observed as a decrease in LOF (Rotarod; Figure 2(A)), climbing attempts (cylinder test; Figure 2(C)) and a decline in motor coordination on the parallel bar (Figure 2(B)). In vivo neural recording in the M1 (L4-L6) and the CPu showed differential effects of this treatment when the spike train frequency (Hz) for the parkinsonian model (−D2) was compared with the control (untreated). The observed decline in motor function was attributed to change in extracellular calcium hyperpolarization current in the M1 and CPu after haloperidol induced PD. The most significant change in −D2 was observed as an increase in the CPu hyperpolarisation calcium current which translated into the observed motor deficits (Figure 2(D) and Figure 2(E)). Similarly, the reports by Dragicevic and co-workers suggest that an alteration in D2R activation is associated with abnormal calcium currents in the CPu [22] . This was linked to the role of specific receptors, associated with DA neuronal activities, in the pathophysiology of the distinct neural activity observed in dopaminergic neurons [22] [23] .
Subsequent VD3RA intervention (−D2/+VDR) relieved motor impairments by decreasing M1 neural output, and by increasing CPu burst frequencies. Such burst activities have been described in the dopaminergic neurons in vivo and are known to be different from the tonic irregular activity seen in the control (Figure 2(E)) [22] - [24] . In order to investigate the role of the CPu bursts in improved motor function after VD3RA intervention (−D2/+VDR), a separate set of control mice were treated with VD3RA (+VDR) without prior haloperidol treatment. After this treatment, an increase in M1 and CPu activities were recorded (Figure 2(E)), but did not significantly change motor function as no CPu burst was observed. From these findings we deduced that M1 hyperactivity (decreased motor function) was mostly associated with haloperidol induced PD and VD3RA (only treatment). By contrast, VD3RA intervention improved motor function by facilitating a CPu burstfrequency while reducing the activity of the M1.
4.2. VD3RA Intervention Improved Spatial and Not Non-Spatial Memory Function in −D2 Parkinsonian Model
Haloperidol induced PD (−D2) was characterized by a decrease in CA1 burst frequency, reduced PFC activity threshold (Figure 3(E) and Figure 3(F)) and a decline in memory function when compared with the control. Although the parkinsonian mice (−D2) showed a decline in spatial working memory (Figures 3(A)-(C)), no significant change in non-spatial memory was associated with the PFC-CA1 frequency (Figure 3(D)). A similar effect of haloperidol on CA1 neural activity was described by Buhusi and Schmajukas an induction of long term potentiation; this was also found to be associated with memory deficits in real time behaviour [25] . Subsequent VD3RA intervention (−D2/+VDR) increased the PFC activity (burst pattern), reduced CA1 neural activity and the mice recorded an improvement in spatial working memory when compared with the parkinsonian group (−D2; Figures 3(A)-(C)). Thus, VD3RA intervention in parkinsonian mice (−D2) improved spatial working memory by increasing cortical (PFC) neural outputs.
To verify the role of PFC outputs in memory function, a separate control group received VD3RA (+VDR) without prior haloperidol treatment. As a result of this treatment, a decline in PFC and an increase in CA1 dense burst patterns was observed (Figure 3(F)) but had no effect on memory function when compared with the control. Thus, we deuced that a reduction in PFC neural outputs was linked with a decline in memory function seen in parkinsonian mice (−D2) while its increase was associated with VD3RA intervention and improved spatial working memory (−D2/+VDR). However, PFC and CA1 cortical neural outputs did not significantly affect non- spatial memory in the parkinsonian mice and intervention group. Similar to our findings, other reports have implicated PFC outputs in memory function associated with dopamine-glutamate circuits. Activation or inactivation of these receptors had varying effects on PFC output projections which in turn regulates dopamine and acetylcholine release in guided behaviour [26] .
Previous studies have shown the importance of VD3R on motor-cognitive function in knockout mice model (VDR−/−) and observed that the absence of these receptors affected motor function and not memory function (Burne et al., 2005). VDR−/− mice showed a decline in motor function characterized by a short gait and a reduced LOF in Rotarod test. In addition, no significant change in cognition was recorded when the animals were examined in the Y-maze [27] . However, we observed a similar behavioural pattern after VD3RA intervention as the mice showed an improvement in spatial working memory but did not record any significant change in non-spatial memory when compared with the control or the parkinsonian group (−D2). Interestingly, in the Y- Maze test, the parkinsonian mice (−D2) showed no change in memory function when compared with the control. However, a modification to the Y-Maze (one place trial recognition) revealed memory deficits in this group. Although, the mice showed no significant change in arm exploration frequency when the treatments were compared with the control (Figure 2(C), the parkinsonian mice (−D2) spent less time exploring the novel arm of the Y-Maze in this test; thus depicting a decline in spatial memory function associated with site recognition rather than exploratory function.
4.3. VD3R Activation and Motor-Cognitive Motor
Taken together, an intrinsic relationship was observed between Haloperidol (−D2) induced decline in motor and memory functions in parkinsonian mice. This was characterized, mostly, by a reduction of CPu output (burst) and the absence of PFC burst patterns in −D2 treatment. On the other hand, VD3R targeted intervention (−D2/+VDR) improved motor-cognitive functions by reversing the effects of −D2 treatment on the PFC and CPu. The implication in behavioural studies implies that the role of haloperidol in motor-cognitive dysfunction was linked to its effect on the corticostriatal projections from the CPu to the PFC; suggesting cortical modulation of striatal functions. In support of these findings, Scatton and co-workers described the effect of bilateral or selective bilateral ablation of the frontal cortex on striatal neural activities. Both forms of lesions prevented the cataleptogenic effects of haloperidol in rats. Subsequent surgical restoration of PFC functions facilitated the motor dysfunction associated with haloperidol treatment; thus supporting the role of PFC-CPu corticostriatal outputs in haloperidol induced motor-cognitive impairments [28] .
5. Conclusion
The primary targets of haloperidol induced motor-cognitive function decline are the PFC and CPu. The associated neural changes in parkinsonian mice involved formation of abnormal calcium currents that are reversed after VD3RA intervention (−D2/+VDR). Thus, VD3RA intervention improved spatial working memory function and motor function. No significant change was observed in non-spatial working memory when the treatment groups were compared with the control.
Grants
This work was supported by the IBRO-ARC Bursary (2014) and ISN-CAEN 1B (2013). Both grants were awarded to OOM.
Author Contributions
OOM and AOI initiated and designed the study. BJL, MUS, OOB, DEO, WGB, AA, and AEC participated in the implementation, design of apparatus for electrophysiology and behavioural tests. OOM, IDA and AOI analysed the data. OOM, BJL, OOB and AOI compiled the reports and interpreted the data for write up.
Conflict of Interest
The Authors hereby declare there is no conflict of interest associated with this study or any of the procedures and materials used for the purpose of the study.
Cite this paper
Azeez O.Ishola,Babafemi J.Laoye,Damilola E.Oyeleke,Oluwamolakun O.Bankole,Mujittapha U.Sirjao, Ansa E.Cobham,Wasiu G.Balogun,AminAbdulbasit,Ibukun D.Akinrinade,Olalekan M.Ogundele,11, (2015) Vitamin D3 Receptor Activation Rescued Corticostriatal Neural Activity and Improved Motor-Cognitive Function in -D2R Parkinsonian Mice Model. Journal of Biomedical Science and Engineering,08,601-615. doi: 10.4236/jbise.2015.89056
References
- 1. Seeman, P. and Tallerico, T. (2003) Link between Dopamine D1 and D2 Receptors in Rat and Human Striatal Tissues. Synapse, 47, 250-254.
http://dx.doi.org/10.1002/syn.10171 - 2. Karl, T., Duffy, L., O'brien, E., Matsumoto, I. and Dedova, I. (2006) Behavioural Effects of Chronic Haloperidol and Risperidone Treatment in Rats. Behavioural Brain Research, 171, 286-294.
http://dx.doi.org/10.1016/j.bbr.2006.04.004 - 3. Graff-Guerrero, A., Mamo, D., Shammi, C.M., Mizrahi, R., Marcon, H., Barsoum, P., Rusjan, P., Houle, S., Wilson, A.A. and Kapur, S. (2009) The Effect of Antipsychotics on the High-Affinity State of D2 and D3 Receptors: A Positron Emission Tomography Study with [11C]-(+)-PHNO. Archives of General Psychiatry, 66, 606-615.
http://dx.doi.org/10.1001/archgenpsychiatry.2009.43 - 4. Iderberg, H., Maslava, N., Thompson, A.D., Bubser, M., Niswender, C.M., Hopkins, C.R., Lindsley, C.W., Conn, P.J., Jones, C.K. and Cenci, M.A. (2015) Pharmacological Stimulation of Metabotropic Glutamate Receptor Type 4 in a Rat Model of Parkinson’s Disease and L-DOPA-Induced Dyskinesia: Comparison between a Positive Allosteric Modulator and an Orthosteric Agonist. Neuro-pharmacology, 95, 121-129.
http://dx.doi.org/10.1016/j.neuropharm.2015.02.023 - 5. Shirayama, Y., Mitsushio, H., Takahashi, K. and Nishikawa, T. (2000) Differential Effects of Haloperidol on Phencyclidine-Induced Reduction in Substance P Contents in Rat Brain Regions. Synapse, 35, 292-299.
http://dx.doi.org/10.1002/(SICI)1098-2396(20000315)35:4<292::AID-SYN7>3.0.CO;2-3 - 6. Samad, N. and Haleem, D.J. (2014) Haloperidol-Induced Extra Pyramidal Symptoms Attenuated by Imipramine in Rats. Pakistan Journal of Pharmaceutical Sciences, 27, 1497-1501.
- 7. Kiyatkin, E.A. and Rebec, G.V. (1999) Striatal Neuronal Activity and Responsiveness to Dopamine and Glutamate after Selective Blockade of D1 and D2 Dopamine Receptors in Freely Moving Rats. The Journal of Neuroscience, 19, 3594-3609.
- 8. Martel, P., Leo, D., Fulton, S., Bérard, M. and Trudeau, L.E. (2011) Role of Kv1 Potassium Channels in Regulating Dopamine Release and Presynaptic D2 Receptor Function. PLoS One, 6, e20402.
http://dx.doi.org/10.1371/journal.pone.0020402 - 9. Hudson, C.J., Seeman, P. and Seeman, M.V. (2014) Parkinson’s Disease: Low-Dose Haloperidol Increases Dopamine Receptor Sensitivity and Clinical Response. Parkinson’s Disease, 2014, Article ID: 684973.
http://dx.doi.org/10.1155/2014/684973 - 10. Pifarré, P., Cuberas, G., Hernández, J., Lorenzo, C., Miquel, F. and Castell-Conesa, J. (2010) Cortical and Subcortical Patterns of I-123 Iodobenzamide SPECT in Striatal D(2) Receptor Parkinsonisms. Clinical Nuclear Medicine, 35, 228-233.
http://dx.doi.org/10.1097/rlu.0b013e3181d18cb3 - 11. Napier, T.C. and Maslowski-Cobuzzi, R.J. (1994) Electrophysi-ological Verification of the Presence of D1 and D2 Dopamine Receptors within the Ventral Pallidum. Synapse, 17, 160-166.
http://dx.doi.org/10.1002/syn.890170304 - 12. Shi, W.X., Smith, P.L., Pun, C.L., Millet, B. and Bunney, B.S. (1997) D1-D2 Interaction in Feedback Control of Midbrain Dopamine Neurons. The Journal of Neuroscience, 17, 7988-7994.
- 13. Perreault, M.L., Hasbi, A., O’Dowd, B.F. and George, S.R. (2014) Heteromeric Dopamine Receptor Signaling Complexes: Emerging Neurobiology and Disease Relevance. Neuropsychopharmacology, 39, 156-168.
http://dx.doi.org/10.1038/npp.2013.148 - 14. Zhang, S., Xie, C., Wang, Q. and Liu, Z. (2014) Interactions of CaMKII with Dopamine D2 Receptors: Roles in Levodopa-Induced Dyskinesia in 6-Hydroxydopamine Lesioned Parkinson’s Rats. Scientific Reports, 4, 6811.
http://dx.doi.org/10.1038/srep06811 - 15. Mahmoudi, A.R., Zarnani, A.H., Jeddi-Tehrani, M., Katouzian, L., Tavakoli, M., Soltanghoraei, H. and Mirzadegan, E. (2013) Distribution of Vitamin D Re-ceptor and 1α-Hydroxylase in Male Mouse Reproductive Tract. Reproductive Sciences, 20, 426-436.
http://dx.doi.org/10.1177/1933719112459235 - 16. Durk, M.R., Han, K., Chow, E.C., Ahrens, R., Henderson, J.T., Fraser, P.E. and Pang, K.S. (2014) 1α,25-Dihydroxy- vitamin D3 Reduces Cerebral Amyloid-β Accumulation and Improves Cognition in Mouse Models of Alzheimer’s Disease. Journal of Neuroscience, 34, 7091-7101.
http://dx.doi.org/10.1523/JNEUROSCI.2711-13.2014 - 17. Shinpo, K., Kikuchi, S., Sasaki, H., Moriwaka, F. and Tashiro, K. (2000) Effect of 1,25-Dihydroxyvitamin D3 on Cultured Mesencephalic Dopaminergic Neurons to the Combined Toxicity Caused by L-Buthionine Sulfoximine and 1-Methyl-4-phenylpyridine. Journal of Neuroscience Research, 62, 374-382.
http://dx.doi.org/10.1002/1097-4547(20001101)62:3<374::AID-JNR7>3.0.CO;2-7 - 18. Landfield, P.W. and Cadwallader-Neal, L. (1998) Long-Term Treatment with Calcitriol (1,25(OH)2 vit D3) Retards a Biomarker of Hippocampal Aging in Rats. Neurobiology of Aging, 19, 469-477.
http://dx.doi.org/10.1016/S0197-4580(98)00079-7 - 19. Baas, D., Prüfer, K., Ittel, M.E., Kuchler-Bopp, S., Labourdette, G., Sarliève, L.L. and Brachet, P. (2000) Rat Oligodendrocytes Express the Vitamin D3 Receptor and Respond to 1,25-Dihydroxyvitamin D3. Glia, 31, 59-68.
http://dx.doi.org/10.1002/(SICI)1098-1136(200007)31:1<59::AID-GLIA60>3.0.CO;2-Y - 20. Ogundele, O.M., Nanakumo, E.T., Ishola, A.O., Obende, O.M., Enye, L.A., Balogun, W.G., Cobham, A.E. and Abdulbasit, A. (2014) -NMDA R/+VDR Pharmacological Phenotype as a Novel Therapeutic Target in Relieving Motor-Cognitive Impairments in Parkinsonism. Drug and Chemical Toxicology, 4, 1-13.
http://dx.doi.org/10.3109/01480545.2014.975355 - 21. Cheng, J.T., Schallert, T., De Ryck, M. and Teitelbaum, P. (1981) Galloping Induced by Pontine Tegmentum Damage in Rats: A Form of “Parkinsonian Festination” Not Blocked by Haloperidol. Proceedings of the National Academy of Sciences of the United States of America, 78, 3279-3283.
http://dx.doi.org/10.1073/pnas.78.5.3279 - 22. Dragicevic, E., Schiemann, J. and Liss, B. (2015) Dopamine Midbrain Neurons in Health and Parkinson’s Disease: Emerging Roles of Voltage-Gated Calcium Channels and ATP-Sensitive Potassium Channels. Neuroscience, 284, 798- 814.
http://dx.doi.org/10.1016/j.neuroscience.2014.10.037 - 23. Ishida, Y., Kozaki, T., Isomura, Y., Ito, S. and Isobe, K. (2009) Age-Dependent Changes in Dopaminergic Neuron Firing Patterns in Substantia Nigra Pars Compacta. Journal of Neural Transmission, 73, 129-133.
http://dx.doi.org/10.1007/978-3-211-92660-4_10 - 24. Lee, C.R. and Tepper, J.M. (2009) Basal Ganglia Control of Substantia Nigra Dopaminergic Neurons. Journal of Neural Transmission, 73, 71-90.
- 25. Buhusi, C.V. and Schmajuk, N.A. (1996) Attention, Configuration, and Hippocampal Function. Hippocampus, 6, 621-642.
http://dx.doi.org/10.1002/(SICI)1098-1063(1996)6:6<621::AID-HIPO6>3.0.CO;2-J - 26. Del Arco, A. and Mora, F. (2008) Prefrontal Cortex-Nucleus Accumbens Interaction: In Vivo Modulation by Dopamine and Glutamate in the Prefrontal Cortex. Pharmacology Biochemistry and Behavior, 90, 226-235.
http://dx.doi.org/10.1016/j.pbb.2008.04.011 - 27. Burne, T.H., McGrath, J.J., Eyles, D.W. and Mackay-Sim, A. (2005) Behavioural Characterization of Vitamin D Receptor Knockout Mice. Behavioural Brain Research, 157, 299-308.
http://dx.doi.org/10.1016/j.bbr.2004.07.008 - 28. Scatton, B., Worms, P., Lloyd, K.G. and Bartholini, G. (1982) Cortical Modulation of Striatal Function. Brain Research, 232, 331-343.
http://dx.doi.org/10.1016/0006-8993(82)90277-3
NOTES

*Corresponding author.