Journal of Biomedical Science and Engineering
Vol.07 No.11(2014), Article ID:49807,29 pages
10.4236/jbise.2014.711088
Differences in Amino Acid Composition between α and β Structural Classes of Proteins
Hiroshi Nakashima*, Yuka Saitou, Nachi Usuki
Department of Clinical Laboratory Science, Graduate Course of Medical Science and Technology, School of Health Sciences, Kanazawa University, Kanazawa, Japan
Email: *naka@kenroku.kanazawa-u.ac.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 6 July 2014; revised 21 August 2014; accepted 7 September 2014
ABSTRACT
The amino acid composition of α and β structural class of proteins from five species, Escherichia coli, Thermotoga maritima, Thermus thermophilus, yeast, and humans were investigated. Amino acid residues of proteins were classified into interior or surface residues based on the relative accessible surface area. The hydrophobic Leu, Ala, Val, and Ile residues were rich in interior residues, and hydrophilic Glu, Lys, Asp, and Arg were rich in surface residues both in α and β proteins. The amino acid composition of α proteins was different from that of β proteins in five species, and the difference was derived from the different contents of their interior residues between α and β proteins. α-helix content of α proteins was rich in interior residues than surface ones. Similarly, β- sheet content of β proteins was rich in interior residues than surface ones. The content of Leu residues was very high, approximately 20%, in interior residues of α proteins. This result suggested that the Leu residue plays an important role in the folding of α proteins.
Keywords:
Amino Acid Composition, α and β Proteins, Interior/Surface Residue, Protein Domains, Species-Specific Amino Acid Composition

1. Introduction
Nearly 30 years ago, Nakashima et al. [1] reported that the amino acid composition of proteins is different among the four structural classes: α, β, α/β and α + β. Many studies [2] - [8] have confirmed that there is a correlation between the amino acid composition and structural class of proteins. However, the reason behind the differences in amino acid composition among different structural classes is not clearly understood.
It is known that bacteria have species-specific nucleotide compositions in their protein-coding genes [9] - [11] . Due to the biased nucleotide composition, the amino acid composition of bacteria is also biased. The accumulation of data of the three-dimensional (3D) structure of proteins has provided the opportunity to statistically analyze the amino acid composition of proteins of different structural classes from different species. Here, we compared the amino acid composition of proteins form five species; Escherichia coli, Thermotoga maritima, Thermus thermophilus, yeast (Saccharomyces cerevisiae), and humans (Homo sapiens), because these species have relatively large numbers of structural proteins. The species-specific nucleotide composition is largely dependent on G + C content. The G + C content in the whole genome is 50.8% in E. coli, 46.2% in T. maritima, 69.5% in T. thermophilus, and 38.3% in yeast. These four species have a constant G + C content in their genomes. By contrast, the human genome has a considerably variable G + C content according to their chromosomal locations [12] . The optimal growth temperatures of T. maritima and T. thermophilus are 80˚C and 85˚C, respectively, as they are hyperthermophiles. It is reported that proteins of thermophiles have a different amino acid composition compared to that of mesophiles [13] - [17] .
The amino acid composition of α proteins is mostly distinct from that of β proteins among the four structural classes [1] . Therefore, comparison of the amino acid composition of α and β proteins appears to be ideal approach to understand the differences in amino acid composition between any two structural classes of proteins. As there is a correlation between amino acid composition and structural classes, we hypothesized that there might be some basic features that facilitate the organization of α or β proteins and that such features might be conserved. The purpose of this study was to identify such basic features from various sequences. We compared the amino acid composition of α or β proteins from five species, and analyzed their interior or surface residues to identify the common features as well as the differences.
2. Materials and Methods
2.1. Amino Acid Sequences
The amino acid sequences of known 3D structures were obtained from the structural classification of proteins (SCOP) [18] sequence database 1.75 released on the web site http://scop.mrc-lmb.cam.ac.uk/scop/. Each sequence has a Protein Data Bank (PDB) [19] entry code and a SCOP structural classification code, which represents the structural class, fold, superfamily, and family. The amino acid sequences in SCOP are divided into protein domains and the structural class is depicted for individual sequences. The amino acid sequences that constitute protein domains are not always sequential, as some sequences are composed of two fragments from separate regions. Sequences longer than 100 amino acid residues were selected for this analysis. The sequences of E. coli, T. maritima, T. thermophilus, yeast, and humans were selected, and then the sequences of α or β structural class proteins were further selected. The selected sequences were analyzed for their sequence similarity using the BLAST program [20] . The proteins that had more than 25% sequence identity over 100 residues were excluded to avoid any bias. Sequences with identical SCOP structural codes were included when the sequences had less than 25% sequence identity over 100 residues. The collected data included 77 sequences of E. coli, 22 of T. maritima, 23 of T. thermophilus, 45 of yeast, and 154 of humans for α protein domains, and 90 sequences of E. coli, 17 of T. maritima, 28 of T. thermophilus, 35 of yeast, and 187 of humans for β protein domains. The number of total protein domains was 321 for α and 357 for β proteins. The protein names, number of residues, and the SCOP and PDB codes of the sequences used in this study are listed in the supplementary data.
2.2. Classification of Interior/Surface Residues
The amino acid residues in the analyzed sequences were classified into two types, interior or surface residues, based on their relative solvent accessibility. Solvent accessibility and secondary structure calculated using the DSSP program [21] with the coordinate data of PDB were obtained from the European Bioinformatics Institute web site (http://www.ebi.ac.uk/). Amino acid residues with relative solvent accessibility greater than 25% were regarded as surface residues, and those with relative solvent accessibility less than 25% were considered as interior residues as described by Fukuchi and Nishikawa [14] . Secondary structures of a residue were classified into three states: α-helix, β-sheet, and coil. α-helices and β-sheets were categorized according to the definition of the DSSP program and residues other than an α-helix or β-sheet were considered as coils. The intrinsically disordered residues were not included in the DSSP sequences. The SCOP amino acid sequences were aligned with DSSP sequences, and the solvent accessibility and secondary structure were given for the corresponding residues between the two sequences.
2.3. Comparison of Amino Acid Composition
The average amino acid composition was compared between α and β proteins, and interior and surface residues, among the five species. To analyze the amino acid composition of a protein, amino acid composition space, introduced by Nishikawa and Ooi [22] - [24] , was employed. The number of each of the 20 types of amino acid residues in a protein sequence was counted and the composition was expressed in a normalized scale using the equation:
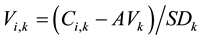
where
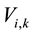 and
and
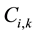 are the normalized and real composition of amino acid residues of the k-th component in a sequence i, respectively.
are the normalized and real composition of amino acid residues of the k-th component in a sequence i, respectively.
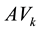 and
and
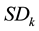 are the average composition and the standard deviation of the k-th component for the whole dataset, respectively.
are the average composition and the standard deviation of the k-th component for the whole dataset, respectively.
The amino acid sequence of a protein was converted to an amino acid composition vector of 20 components and it was plotted as a point in a 20-dimensional composition space. The distribution of proteins was visualized by projecting them onto a two-dimensional (2D) plane, which was defined by two axes of the principal component analysis. A principal component analysis was conducted for the total proteins. The x-coordinate for a given sequence was calculated using the scalar product of the unit vector of the first principal component and the vector of the sequence. The y-coordinate was calculated using the scalar product of the unit vector of the second principal component and the vector of the sequence. The origin of the x-y coordinate system in the composition space was set at the average amino acid composition of all analyzed sequences.
3. Results
3.1. Amino Acid Composition
The average and standard deviation values of the amino acid composition of total proteins analyzed in this study are indicated in Table 1. The average amino acid compositions of interior, surface, and whole residues of α and β structural classes of proteins are listed in Table 1. The number of domains used in the calculation is shown in the last row in Table 1. The hydrophobic Leu, Ala, Ile, and Val residues were rich in interior residues, and hydrophilic Glu, Lys, Asp, and Arg were rich in surface residues, both in α and β proteins. This trend was observed commonly in all five species. The ratios of the interior residues against surface residues clearly indicated that Cys, Phe, Ile, Trp, and Leu were favored as interior residues, and Lys, Glu, Asp, Arg, and Gln were favored as surface residues both in α and β proteins. This trend was also observed commonly in all five species. This result indicated that the location of a residue (interior or surface) in a protein is dependent on the character of the amino acid and independent of the structural class. The Leu residues content was very high, approximately 20%, in interior residues of α proteins. To show the difference between α and β proteins, the ratios of whole residues of α proteins to those of β proteins were calculated. The ratios indicated that Leu, Met, Ala, and Glu residues were predominant in the α proteins of the five species, while Gly, Pro, Val, and Thr residues were predominant in the β proteins of the five species. The favored amino acids in the two structural classes of proteins were consistent with a previous study [1] .
3.2. Distribution of α and β Proteins on a 2D Plane
The proteins of α and β structural classes of E. coli were plotted on a 2D plane by using the first principal component as the x-axis and the second component as the y-axis (Figure 1). The distribution of α proteins of E. coli was roughly separated from that of β proteins of E. coli. A similar plot indicated that α proteins were roughly separated from that of β proteins in the other four species, similar to that observed in E. coli. However, the plot of total α and total β proteins from the five species together indicated an overlapped distribution.
The variance of the first principal component was 12.6% and that of the second component was 12.1%. Asn, Ile, Phe, and Ser residues were largely shifted toward the positive direction, while Ala, Arg, Leu, and Glu resi-

Table 1. Average and standard deviation (SD) of amino acid composition (%) of total proteins. Average of interior, surface, and whole residues in α and β proteins.
dues were largely shifted toward the negative direction along the x-axis. Asn, Ile, and Phe residues have A + T-rich codons, and Ala and Arg residues have G + C-rich codons. This result suggested that the x-axis reflects the character of the G + C content of the residues. Along the y-axis, Gly, Pro, Val, and Thr residues showed a larger positive coefficient, and Lys, Glu, Ile, and Met residues showed a larger negative coefficient. The residues with positive coefficients were the favored residues in β proteins, and those with negative coefficients were the residues that were mostly detected in α proteins. These findings indicated that the y-axis reflects the frequency of the occurrence of amino acid residues between α and β proteins. This is consistent with the distribution of E. coli proteins in Figure 1, where most of the α proteins showed negative values of y-coordinates, and most of the β proteins had positive y-coordinates.
3.3. Differences in Interior and Surface Compositions among the Five Species
The average amino acid compositions of interior and surface residues in α proteins from the five species were plotted, and the results are shown in Figure 2 and Figure 3, respectively. To clearly demonstrate the differences in the amino acid content among the five species, the differences between the maximum/minimum and average composition were plotted. Figure 4 shows the plot indicating the differences in the interior composition of α proteins. Ala and Ile residues showed large differences. The Ala residue content in interior composition was highest (16.04%) in T. thermophilus and lowest (8.51%) in yeast in α proteins (Figure 2). Since the average Ala

Figure 1. Distribution of α (filled circles) and β proteins (open circles) of E. coli. The x- and y-axes represent the first and second axes determined by principal component analysis.

Figure 2. The average amino acid composition of interior residues of α proteins of the five species.
composition in the five species was 11.58%, the deviation was +4.46% and −3.07% (Figure 4). The Ile residue content was lowest (4.77%) in T. thermophilus and highest (11.30%) in yeast. Since the average Ile composition in the five species was 9.19%, the deviation was +2.55% and −4.42% (Figure 4). Taken together, the total of Ala and Ile content was 20.81% in T. thermophilus and 19.81% in yeast. This result indicated that the increase or decrease of Ala content is compensated by the decrease or increase of Ile, and their contents were adjusted in the species with different G + C content. The variation in Ala and Ile contents was consistent with previous reports [25] -[29] , which indicated that amino acids composed of G + C-rich or A + T-rich codons are related to the genomic G + C content of the species.
The differences in the surface composition of α proteins are shown in Figure 5. The Glu residue showed a large difference. The average Glu residue content in the five species was 14.57%. The maximum composition was 19.92% in T. maritima and the minimum composition was 11.42% in yeast. Therefore, the difference in the distribution of Glu was +5.35% and −3.15% (Figure 5). The Glu residue has GAA and GAG codons, which are neutral with respect to G + C content. Therefore, the large differences in the Glu residue content are not explained by the G + C content. It has been reported that thermophiles have a higher content of charged residues in

Figure 3. The average amino acid composition of surface residues of α proteins of the five species.

Figure 4. The differences in amino acid content between the maximum and average composition (in blue) and that between the minimum and average composition (in red) of interior residues of α proteins among the five species.
surface composition than mesophiles [14] - [17] . The total content of charged residues of Asp, Glu, Lys, and Arg in the surface composition of α proteins was 52.79% in T. maritima, 45.42% in T. thermophilus, 39.44% in E. coli, 39.45% in humans, and 40.21% in yeast. The total content of charged residues was higher in thermophiles than in mesophiles, and it was higher in α proteins than in β proteins. There were more differences in the composition of surface residues in α proteins than interior residues (Figure 4 and Figure 5). Similar results were observed in β proteins. It is empirically known that the interior residues are more conserved than the surface residues in homologous proteins. Therefore, it is reasonable that the interior composition should show less difference than the surface composition.
4. Discussion
The amino acid residues were classified into interior or surface residues based on their relative solvent accessibility. The amino acid composition of interior and surface residues was dependent on the classification. In this

Figure 5. The differences in amino acid content between the maximum and average composition (in blue) and that between the minimum and average composition (in red) of surface residues of α proteins among the five species.
study, an average of surface residues of α proteins of the five species was 51%, and the average of interior residues was 49%. In β proteins, the average compositions of both surface and interior residues of the five species were 50%. Since we intended to classify the residues into interior and surface residues in equal proportions, the obtained result met our intended criterion.
The average content of secondary structures in α and β proteins is shown in Table 2. The interior residues in α proteins had a higher α-helix content, 73%, than the surface residues. Similarly, the interior residues in β proteins had a higher β-sheet content, 54%, than the surface residues.
The hydrophobic residues, such as Leu, Ala, Ile, and Val were enriched in interior residues of both α and β proteins. However, the percentages of α-helices and β-sheets in interior residues were quite different. The reason behind such a big difference is not clear. Since the Ala residue has G + C-rich codons and the Ile residue has A + T-rich codons, their contents were dependent on the G + C content of the species. Therefore, the differences in Ala and Ile compositions were large compared to those of Val and Leu among the five species (Figure 4). The Val residue has GTN (N stands for all four nucleotides) codons, which are neutral with respect to G + C content. This might be the reason for the smaller differences observed in Val content. The Leu residue has neutral CTN codons and A + T-rich TTA and TTG codons. The Leu residue showed smaller deviations among species even though its content was consistently high.
In E. coli, the distribution of whole residues of α proteins was roughly distinct from its β proteins. Similarly, the interior residues between α and β proteins showed a distinct distribution. However, the plot of surface residues in E. coli was overlapped. Similar results were also observed in other species. This indicates that the difference in amino acid composition of whole residues between α and β proteins was derived from the differences in the interior residues. This result was obtained by the analysis of the distribution of α and β proteins in the amino acid composition space. However, no clear differences were noticed in the initial observations of amino acid composition of interior residues between α and β proteins. The interior residues were rich in hydrophobic Leu, Ala, Ile, and Val residues both in α and β proteins. To clearly demonstrate the differences in interior residues between α and β proteins, the ratios of their amino acid composition were calculated. This indicated that Met, Leu, Glu, and Ala residues were favored in α proteins and Gly, Pro, Val, and Thr were favored in β proteins. This trend was observed commonly in all five species. Therefore, we concluded that the existence of favorable residues is a basic common feature of α and β proteins. The residues favored in α proteins were consistent with the residues favored in α-helix [30] , however, Gly and Pro residues favored in β proteins were reported as unfavorable in β-sheet [30] . The Gly and Pro residues contents were not large compared to those of the hydrophobic residues, however, they were commonly favored in β proteins in the five species. This suggested that Gly and
Table 2. Average content of secondary structures (%).
Pro residues are essential interior residues in the organization of β proteins outside of the β-sheet regions. The average Leu residue content was 17% in yeast, and was 20% in the other four species. This outcome suggested that the interaction between Leu residues is likely to be very large in the interior region and that it might play an important role in the folding of α proteins.
Cite this paper
HiroshiNakashima,YukaSaitou,NachiUsuki, (2014) Differences in Amino Acid Composition between α and β Structural Classes of Proteins. Journal of Biomedical Science and Engineering,07,890-918. doi: 10.4236/jbise.2014.711088
References
- 1. Nakashima, H., Nishikawa, K. and Ooi, T. (1986) Folding Type of a Protein Is Relevant to the Amino Acid Composition. The Journal of Biochemistry, 99, 153-162.
- 2. Klein, P. (1986) Prediction of Protein Structural Class by Discriminant Analysis. Biochimica et Biophysica Acta, Protein Structure and Molecular Enzymology, 874, 205-215.
http://dx.doi.org/10.1016/0167-4838(86)90119-6 - 3. Dubchak, I., Muchnik, I., Holbrook, S.R. and Kim, S.H. (1995) Prediction of Protein Folding Class Using Global Description of Amino Acid Sequence. Proceedings of the National Academy of Sciences of the United States of America, 92, 8700-8704.
http://dx.doi.org/10.1073/pnas.92.19.8700 - 4. Bahar, I., Atilgan, A.R., Jernigan, R.L. and Erman, B. (1997) Understanding the Recognition of Protein Structural Classes by Amino Acid Composition. Proteins, 29, 172-185.
- 5. Chou, K.C. (1999) A Key Driving Force in Determination of Protein Structural Classes. Biochemical and Biophysical Research Communications, 264, 216-224.
http://dx.doi.org/10.1006/bbrc.1999.1325 - 6. Ofran, Y. and Margalit, H. (2006) Proteins of the Same Fold and Unrelated Sequences Have Similar Amino Acid Composition. Proteins: Structure, Function, and Bioinformatics, 64, 275-279.
http://dx.doi.org/10.1002/prot.20964 - 7. Taguchi, Y. and Gromiha, M.M. (2007) Application of Amino Acid Occurrence for Discriminating Different Folding Types of Globular Proteins. BMC Bioinformatics, 8, 404.
http://dx.doi.org/10.1186/1471-2105-8-404 - 8. Rackovsky, S. (2009) Sequence Physical Properties Encode the Global Or-ganization of Protein Structure Space. Proceedings of the National Academy of Sciences of the United States of America, 106, 14345-14348.
http://dx.doi.org/10.1073/pnas.0903433106 - 9. Karlin, S. and Burge, C. (1995) Dinucleotide Relative Abundance Extremes: A Genomic Signature. Trends in Genetics, 11, 283-290.
http://dx.doi.org/10.1016/S0168-9525(00)89076-9 - 10. Karlin, S., Mrázek, J. and Campbell, A.M. (1997) Compositional Biases of Bacterial Genomes and Evolutionary Implications. Journal of Bacteriology, 179, 3899-3913.
- 11. Nakashima, H., Ota, M., Nishikawa, K. and Ooi, T. (1998) Gene from Nine Genomes Are Separated into Their Organisms in the Dinucleotide Composition Space. DNA Research, 5, 251-259.
http://dx.doi.org/10.1093/dnares/5.5.251 - 12. Bernardi, G., Olofsson, B., Filipski, J., Zerial, M., Salinas, J., Cuny, G., Meunier-Rotival, M. and Rodier, F. (1985) The Mosaic Genome of Warm-Blooded Vertebrates. Science, 228, 953-958.
http://dx.doi.org/10.1126/science.4001930 - 13. Kreil, D.P. and Ouzounis, C.A. (2001) Identification of Thermophilic Species by the Amino Acid Compositions Deduced from Their Genomes. Nucleic Acids Research, 29, 1608-1615.
http://dx.doi.org/10.1093/nar/29.7.1608 - 14. Fukuchi, S. and Nishikawa, K. (2001) Protein Surface Amino Acid Compositions Distinctively Differ between Thermophilic and Mesophilic Bacteria. Journal of Molecular Biology, 309, 835-843.
http://dx.doi.org/10.1006/jmbi.2001.4718 - 15. Chakravarty, S. and Varadarajan, R. (2002) Elucidation of Factors Responsible for Enhanced Thermal Stability of Proteins: A Structural Genomics Based Study. Biochemistry, 41, 8152-8161.
http://dx.doi.org/10.1021/bi025523t - 16. Nakashima, H., Fukuchi, S. and Nishikawa, K. (2003) Compositional Changes in RNA, DNA and Proteins for Bacterial Adaptation to Higher and Lower Temperatures. The Journal of Biochemistry, 133, 507-513.
http://dx.doi.org/10.1093/jb/mvg067 - 17. Yokota, K., Satou, K. and Ohki, S. (2006) Comparative Analysis of Protein Thermo-stability: Differences in Amino Acid Content and Substitution at the Surfaces and in the Core Regions of Thermophilic and Mesophilic Proteins. Science and Technology of Advanced Materials, 7, 255-262.
- 18. Murzin, A.G., Brenner, S.E., Hubbard, T. and Chothia, C. (1995) SCOP: A Structural Classification of Proteins Database for the Investigation of Sequences and Structures. Journal of Molecular Biology, 247, 536-540.
http://dx.doi.org/10.1016/S0022-2836(05)80134-2 - 19. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank. Nucleic Acids Research, 28, 235-242.
http://dx.doi.org/10.1093/nar/28.1.235 - 20. Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic Local Alignment Search Tool. Journal of Molecular Biology, 215, 403-410.
http://dx.doi.org/10.1016/S0022-2836(05)80360-2 - 21. Kabsch, W. and Sander, C. (1983) Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers, 22, 2577-2637.
http://dx.doi.org/10.1002/bip.360221211 - 22. Nishikawa, K. and Ooi, T. (1982) Correlation of the Amino Acid Composition of a Protein to Its Structural and Biological Characters. The Journal of Biochemistry, 91, 1821-1824.
- 23. Nishikawa, K., Kubota, Y. and Ooi, T. (1983) Classification of Proteins into Groups Based on Amino Acid Composition and Other Characters. I. Angular Distribution. The Journal of Biochemistry, 94, 981-995.
- 24. Nishikawa, K., Kubota, Y. and Ooi, T. (1983) Classification of Proteins into Groups Based on Amino Acid Composition and Other Characters. II. Grouping into Four Types. The Journal of Biochemistry, 94, 997-1007.
- 25. Lobry, J.R. (1997) Influence of Genomic G+C Content on Average Amino-Acid Composition of Proteins from 59 Bacterial Species. Gene, 205, 309-316.
http://dx.doi.org/10.1016/S0378-1119(97)00403-4 - 26. Singer, G.A.C. and Hickey, D.A. (2000) Nucleotide Bias Causes a Genomewide Bias in the Amino Acid Composition of Proteins. Molecular Biology and Evolution, 17, 1581-1588.
http://dx.doi.org/10.1093/oxfordjournals.molbev.a026257 - 27. Bharanidharan, D., Bhargavi, G.R., Uthanumallian, K. and Gautham, N. (2004) Correlations between Nucleotide Frequencies and Amino Acid Composition in 115 Bacterial Species. Biochemical and Biophysical Research Communications, 315, 1097-1103.
http://dx.doi.org/10.1016/j.bbrc.2004.01.129 - 28. Hu, J., Zhao, X., Zhang, Z. and Yu, J. (2007) Compositional Dynamics of Guanine and Cytochine Content in Prokaryotic Genomes. Research in Microbiology, 158, 363-370.
http://dx.doi.org/10.1016/j.resmic.2007.02.007 - 29. Nakashima, H., Yoshihara, A. and Kitamura, K. (2013) Favorable and Unfavorable Amino Acid Residues in Water-Soluble and Transmembrane Proteins. Journal of Biomedical Science and Engineering, 6, 36-44.
http://dx.doi.org/10.4236/jbise.2013.61006 - 30. Chou, P.Y. and Fasman, G.D. (1978) Empirical Predictions of Protein Conformation. Annual Review of Biochemistry, 47, 251-276.
http://dx.doi.org/10.1146/annurev.bi.47.070178.001343
Supplementary Data
NOTES

*Corresponding author.