Journal of Biomedical Science and Engineering
Vol.6 No.9(2013), Article ID:36946,11 pages DOI:10.4236/jbise.2013.69108
Macromolecular inhibitors of malarial cysteine proteases —An invited review
![]()
Host-Parasite Interaction Biology Group, Protein-Protein Interaction Unit, National Institute of Malaria Research, (ICMR) Sector-8, Dwarka, India
Email: kailashcp@mrcindia.org
Copyright © 2013 Kailash C. Pandey. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 28 May 2013; revised 29 June 2013; accepted 15 July 2013
Keywords: Malaria; Cysteine Protease; Hemoglobinase; Macromolecular Inhibitor; Protein-Protein Interaction
ABSTRACT
There are evidences indicating that cysteine proteases play an essential role in malaria parasites; therefore, an obvious area of investigation is the inhibition of these enzymes to treat malaria. Small cysteine protease inhibitors of malaria are well studied, but macromolecular nature of inhibitor is a new field to explore. In malarial cysteine proteases, there are macromolecular endogenous inhibitors playing important roles in regulation of the cysteine protease activity of parasite and host. Recent studies suggested that there are known and characterized endogenous inhibitors like falstatin present in P. falciparum, PbICP (inhibitor of cysteine protease in P. berghei), PyICP (inhibitor of cysteine protease in P. yoelli), and other macromolecular inhibitors which are the prodomain of enzyme itself regulating the activity of the mature enzyme. All the known macromolecular endogenous inhibitors are using specific loop-like structure to interact with malarial cysteine proteases. The majority of macromolecular inhibitors are competitive in nature, and block access to the active site of their target protease, but do not bind in a strictly substrate-like manner. They rather interact with the protease subsites and catalytic residues in a non-catalytically competent manner. In future, designing inhibitors based on these protein-protein interactions will be a new approach in the field of malaria. Since macromolecular inhibitors can gain potency through the burial of a large surface area and specificity through contacts with secondary binding sites critical for inhibition, and could be less prone to drug resistant mutation.
1. INTRODUCTION
Proteolytic enzymes are present in all organisms and constitute ~2% - 4% of encoded gene products [1]. Endogenous cysteine protease inhibitors have been described in a number of eukaryotic systems. In mammalian and plant cells, endogenous polypeptide inhibitors from the cystatin superfamily regulate lysosomal cysteine proteases. One member of this family, Cystatin C, is known to regulate the cell surface expression of MHC class II molecules in dendritic cells. Another one, an endogenous inhibitor of calpain known as calpastatin, also regulates its calpain activity. Proteases are critical for diverse biological processes such as blood clotting, digestion, pathogenic infection, host defense, microbe defense, and viral replication, wound healing. Since proteases activate an irreversible event so their activity must be tightly controlled. Since cysteine proteases have broad specificity but viral cysteine proteases are exceptional. The TEV protease is a highly site-specific cysteine protease that is found in the Tobacco Etch Virus (TEV, [2]). It is important to regulate the activity of malarial cysteine proteases for the survival of the parasites. Misregulated proteolytic activity causes a disruption in lots of biological activity, and homeostatic balance, and other infections. For that purpose, nature has developed a number of strategies to control proteolysis, including zymogen activation, protease degradation and the inhibition of active proteases by its macromolecular inhibitors. These macromolecular inhibitors of proteolytic enzymes regulate proteolysis and prevent the pathological effects of excess endogenous or exogenous proteases.
Cysteine proteases are a large and diverse family of enzymes found throughout the plant and animal kingdoms, and represent the dominant protease family in invertebrates. Disturbance of the equilibrium between cysteine proteases and their natural inhibitors is a key event in the diseases like cancer, rheumatoid arthritis, osteoporosis, and emphysema. There is a recent general review regarding mechanism of macromolecular inhibitors [1], but this review will survey available information on macromolecular inhibitors of malarial cysteine proteases.
Plasmodium cysteine proteases (falcipains; P. falciparum, vivapains; P. vivax, berghepains; P. Berghei, yoelipains; P. yoelii) play important roles during activation of pro-enzymes, hemoglobin degradation and egress of parasites from red blood cells, erythrocytes rupture [3-7].
Cysteine protease inhibitors blocked the invasion of hepatocytes by P. falciparum [8,9] as well as blocked invasion of red blood cells [10] and the disruption of cysteine protease gene of Plasmodium berghei which prevented sporozoite egress from oocytsts [9] indicating that cysteine protease plays an important role in both erythrocytic stage and non-erythrocytic stage parasites. Since malarial cysteine proteases have broad specificity, it is important to regulate their activity for the survival of the parasite and host. In human malaria parasite, falcipain-2, falcipain-3, and vivapain-2, vivapain-3, vivapain-4 are major hemoglobinases [11]. This review will discuss the macromolecular inhibitors of these hemoglobinases.
2. MACROMOLECULAR INHIBITORS OF CYSTEINE PROTEASES
Endogenous macromolecule inhibitors are polypeptide in nature, which are generally present inside the organisms. These endogenous cysteine proteases inhibitors have been described in a number of eukaryotic systems. Cystatins inhibit a wide range of papain-family cysteine proteases with high affinity (Figure 1(a)). In case of prokaryote, chagasin is a cysteine protease inhibitor that was first identified in Trypanosoma cruzi as the physiological regulator of cruzain (or cruzipain), the major cysteine protease of this protozoan parasite [12]. Cystatin and chagasin both inhibit cysteine protease in a similar fashion (Figures 1(a) and (b)) [1,13,14]. It was found that falcipain-2 and cystatin (Source; chicken egg white), formed 1:1 complex, shown by solving the crystal structure of their complex [13,14]. The inhibitory constant (Ki) of cystatin for falcipain-2 and falcipain-3 are 6.5 and 100 nM, respectively [14]. It is noticeable that cystatin is more potent inhibitor (~14 times) of falcipain-2 than falcipain-3, which indicate that cystatin regulate both the falcipains with different rate. It might be important biologically; since their timing of expression is slightly different as discussed in earlier report [15,16]. The crystal structure of falcipain 2-chagasin also demonstrates 1:1 binding with falcipain-2 [13]. The protease binding loops (BC, DE, and FG) in chagasin form an aligned wedge that fills the active site groove of falcipain-2 to obstruct substrate binding [12] (Figures 1(b) and 4(a)). The structure study demonstrates that the BC loop is one of the
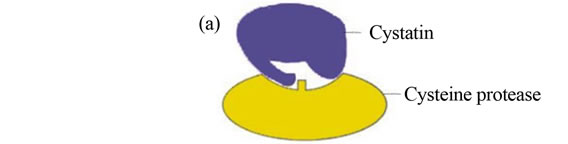
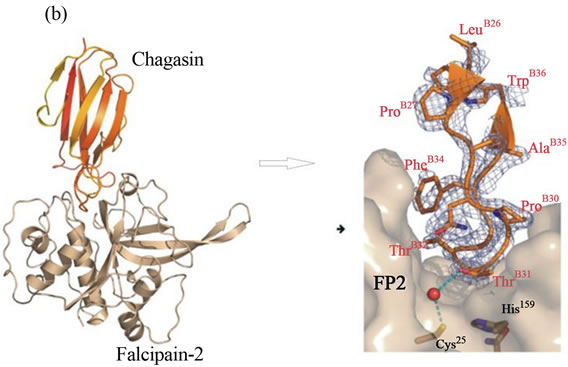
Figure 1. a) Active-site inhibitors of proteases (Competitive). Inhibitors bind in the active site, but not in a substrate-like manner. Peptide extensions of macromolecule inhibitor bind in specificity subsites, and can interact with the catalytic residues shown in rectangle [1]; b) Structure of falcipain-2 and chagasin complex. Structure of falcipain-2-chagasin complex: overall structure of chagasin with falcipain-2, chagasin in red and falcipain-2 in gold. Key binding interactions between chagasin and falcipain-2 can be seen in BC loop or L2 loop [13].
three signature motifs that contribute mainly in inhibiting the cysteine protease (Figures 1(b) and 4(a)).
2.1. Inhibition of Falcipains by Macromolecules
There are two major classes of cysteine protease inhibitor, small inhibitors like leupeptin, vinyl sulfones, E-64, and another class known as macromolecular inhibitor. Published literature suggested that malarial cysteine proteases have broad specificity, it is important to regulate their activity for the survival of the parasite; therefore parasite harbors these macromolecular inhibitors. These endogenous cysteine protease inhibitors have been described in a number of eukaryotic systems. This review will discuss known macromolecular inhibitors, and the recent update of four (prodomain, falstatin, PbICP, PyICP) endogenous inhibitors of cysteine proteases of malaria parasites and their mode of regulation.
2.1.1. Prodomain as a Macromolecular Inhibitor
Prodomain act as an inhibitor and does not alter the catalytic behavior of falcipain-2 [17,18]. Earlier it had been shown that prodomain involved in folding of cysteine proteases of higher animal. But in case of malarial cysteine proteases, prodomain act as an inhibitory domain of catalytic enzyme, and not involved in folding of enzyme, unlike human cysteine proteases, cathespsin K, L, B [18]. Later Pandey et al., identified a C-terminal segment (Leu155-Asp243) of the prodomain, including two conserved motifs (ERFNIN and GNFD, and conserved Phe residues) that are conserved in cathepsin L sub-family and papain family proteases, as the mediator of prodomain inhibitory activity [19]. Modeling of the falcipain-2 prodomain suggests that the prodomain covers the enzyme active site, and thereby inhibits activity by preventing substrate access [19].
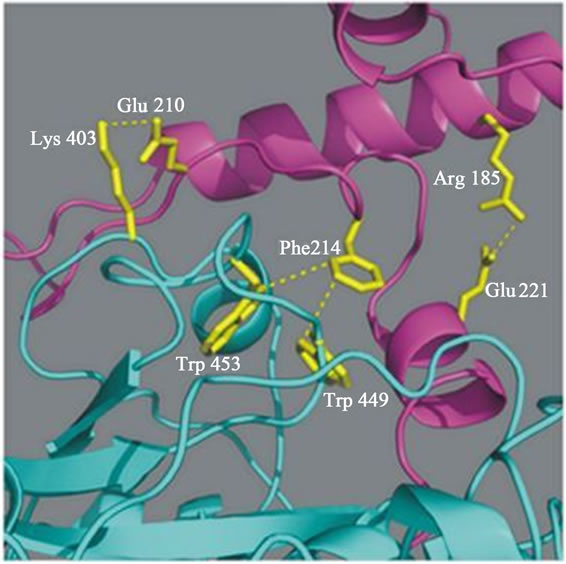
Our recent data suggests that the prodomain-mature domain of falcipain-2 (Figure 2(a)) and falcipain-3 (Figure 2(b)) interacts via salt bridges and hydrophobic interactions [20]. Mutations of specific residues of falcipain-2 and falcipain-3, which are involved in these interactions are evaluated and tested for their ability to undergo auto processing. Mutagenesis result showed that two salt bridges (Arg185-Glu221, Glu210-Lys403) in falcipain-2, and one salt bridge (Arg202-Glu238) in falcipain-3, play crucial roles in the activation of these enzymes. Further study revealed that hydrophobic interactions present both in falcipain-2 (Phe214, Trp449 Trp453) and falcipain-3 (Phe231 Trp457 Trp461) also play important roles in the activation of these enzymes (Figure 2) [20]. The prodomain-mature domain interactions are necessary for the auto-activation of falcipain-2 and falcipain-3 [20]. Disruptions of these forces may be allowed to block the processing and activity of enzyme, and this study may be helpful towards the development of anti-malarial chemothepeutic target.
The falcipain-2 prodomain also efficiently inhibited other papain family proteases, including cathepsin K, cathepsin L, cathepsin B, and cruzain, but it did not inhibit cathepsin C and dipeptidyl aminopeptidase, because these enzymes have exo-peptidases activity [20]. A structural model of pro-falcipain-2 was constructed by homology modeling based on crystallographic structures of mature falcipain-2, procathepsin K, procathepsin L, and procaricain, offering insights into the nature of the interaction between the prodomain and mature domain of falcipain-2 as well as into the broad specificity of inhibitory activity of the falcipain-2 prodomain [20]. Many cathepsin L sub-family propeptides act in trans to inhibit related proteases [21]. However, selectivity has been observed, and it has been demonstrated that the prodomain of cathepsin L and cathepsin K are unable to inhibit cathepsin B [22]. Explanations for this observation include the following: First, cathepsin B lacks the ERFNIN motif, so that the protease lacks most of the α2 helix found in cathepsin L sub-family proteases. Second, cathepsin B contains a large occluding loop insertion, conferring dipeptidase activity, but preventing propeptides containing the ERFNIN motif from binding due to a steric clash between the occluding loop and the prodomain residues connecting α1 and α2. Interestingly, selectivity for prodomain inhibition was broader for falcipain-2, as the prodomain of falcipain-2 markedly inhib-
 (a)
(a)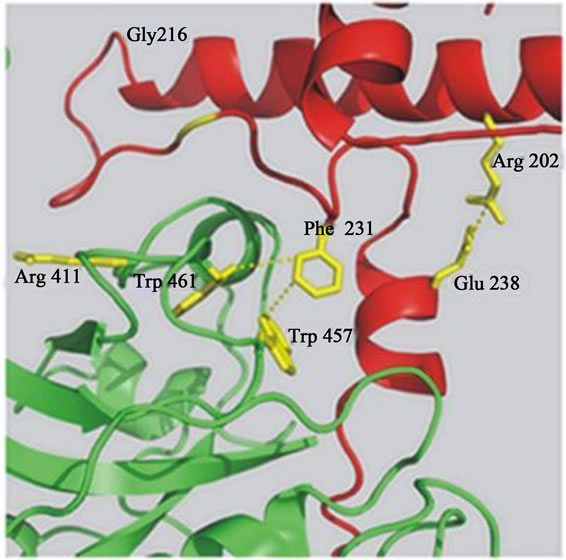 (b)
(b)
Figure 2. a) Predicted interactions between the prodomain and the mature domain of falcipain-2 and falcipain-3. Close up of predicted interactions between the mature enzyme and the ERFNIN and GNFD motifs of the prodomain (Arg185-Glu221, and Phe214-Trp449/Trp453, Glu210-Lys403). Blue dashed lines indicate presumed stabilizing interactions between residues in falcipain-2 [20]; b) Blue dashed lines indicate presumed stabilizing interactions (Arg202-Glu238 and Phe231-Trp457/Trp461) between the residues in falcipain-3 [20].
ited cathepsin B.
2.1.2. Falstatin as a Macromolecular Inhibitor
In case of malaria, falstatin has been recognized as a first endogenous cysteine protease inhibitor in P. falciparum [23]. Erythrocytic P. falciparum parasites express falstatin, a potent inhibitor of falcipains and many other cysteine proteases [23]. But it is unknown how falstatin regulate the P. falciparum cysteine proteases. It has recently been shown that falstatin is unique compare to its homologues, and only BC loop is required for inhibition of malarial cysteine proteases (Srinivasan et al., 2013, pers. comm.). The stage-specificity of falstatin expression and the effects of anti-falstatin antibodies on parasite development suggest that this inhibitor facilitates a process that also requires proteolytic activity, the invasion of erythrocytes by P. falciparum merozoites. Falstatin is a competitive (Ki = 0.028 nM ) and reversible inhibitor of falcipains, as demonstrated by increasing calculated Km values but similar Vmax values with increasing concentrations of falstatin [23]. It can also inhibit other cysteine proteases of malaria and human, except cathepsin B, cathepsin C and dipeptidyl aminopeptidase 1. To evaluate the mechanism of inhibition of cysteine proteases by falstatin, Pandey et al. tested the ability of active site-inhibited falcipain-2 to compete with active falcipain-2 for binding with falstatin. In contrast to results with the prodomain, the inhibitory effect of falstatin was not affected by the presence of active site inhibited falcipain-2 [23]. Thus, the binding of falstatin to falcipain-2 appears to be via interaction with the enzyme active site. Both prodomain and falstatin seems to inhibit falcipain-2 by getting closer to the active site and block the access of substrate, but their binding pattern look like different. Since there is no solved structure of falstatinfalcipain-2, but structure of chagasin and PbICP (berghei homologue of falstatin) with falcipain-2 explained that BC loop as well as other loops play crucial role in binding [24]. Our mutation data also suggests that the BC loop interact with active site of falicipain-3 and vivapain- 2 via hydrophobic and salt interactions (Srinivasan et al., 2013, pers. comm.). It may be possible that DE and FG loops are not required for direct inhibition but play important role in stability of inhibitor-cysteine protease complex. Unlike PbICP (Inhibitor of cysteine protease of P. berghei), falstatin blocks proteolytic activity of caspase and calpain in micro molar range concentration. Possible reasons may be besides it multimeric form falstatin structure is closer to serpins family and falstatin seems to have more flexible loops compare to PbICP. Since falstatin structure appears as different form compare to PbICP, and may interact with caspases and calpain differently. Inhibition of caspases and calpain-1 by falstatin may be helpful in programmed cell death and to protect the exposed merozoite from host proteases like caspases and calpain.
In contrast to both cystatin and chagasin, however, falstatin did not inhibit three human cysteine proteases with exopeptidase activity, cathepsin B, cathepsin C, and dipeptidyl aminopeptidase 1. Further, Hansen et al., 2011, suggests that there is steric repulsion between the two loops (L3 and L4) of PbICP and the occluding loop of cathepsin B, which inhibit the interaction between protease and inhibitor [24].
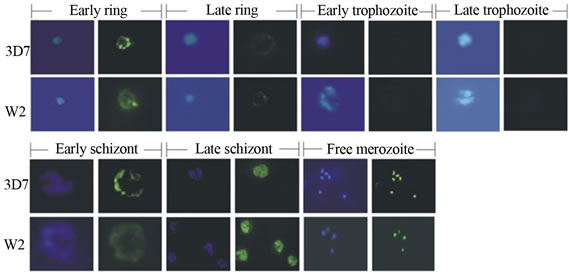
It has been showed that an active falcipain-2 lacking a C-terminal hemoglobin-binding domain was nearly as well inhibited by falstatin as the wild type enzyme, indicating that, unlike the inhibitory prodomain, falstatin does not require the C-terminal domain of falcipain-2 for interaction [23,25]. The maximal expression of falstatin was demonstrated in late schizonts, and no apparent expression in trophozoites (Figure 3(a)). This expression data suggested that there may be a strong regulation, because when the maximal expression of cysteine proteases that time presence of falstatin is absent and vice-
 (a)
(a)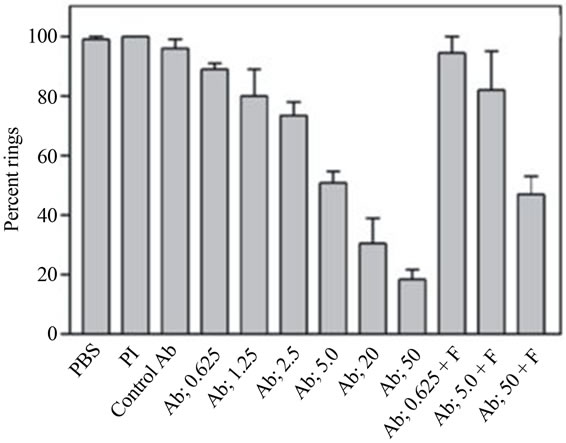 (b)
(b)
Figure 3. a) Immunolocalization of falstatin; Immunofluorescence microscopy of erythrocytes infected with synchronized 3D7 or W2 parasites were collected every 8 h, stained with DAPI and anti-falstatin antibodies and FITC-second antibody [23]; b) Effect of anti-falstatin antibodies on cultured parasites; Purified schizonts were incubated for 20 h with PBS, control pre immune serum (50 µg/ml), rat anti-serum against P. falciparum farnesyl pyrophosphate synthetase (control Ab; 50 µg/ml), or the indicated concentrations of antibodies (Ab; µg/ml) in PBS with or without preincubation for 10 min with 2 µg falstatin (F). Errors bars indicate standard deviations from means of two different assays, each done in triplicate. PI, pre-immune serum [23].
versa. Falstatin contains a typical signal sequence, but the importance of this sequence for its targeting is uncertain. Control of papain-family cysteine protease activity is important, as this class of enzymes typically exhibits rather non-specific activity [26]. Indeed, the prototype for this family, papain is used in many industrial applications due to its nonspecific action against protease substrates. Activity against a range of peptide bonds seems a desired feature of enzymes responsible for bulk proteolysis, as exemplified by hemoglobin hydrolysis by plasmodial trophozoites. It seems logical that falstatin functions to limit the activities of parasite and/or host cysteine proteases. The principal cysteine protease hemoglobinases, falcipain-2 and falcipain-3, vivapain-2, vivapain-3, vivapain-4 are expressed by trophozoites, and hydrolyze hemoglobin in the acidic food vacuoles of human malaria parasites [3,4,10]. Importantly, falstatin expression is not apparent in trophozoites. Presumably, control of the action of trophozoite food vacuole cysteine proteases by a parasite inhibitor is not needed.
There was experimental evidence indicated that falstatin released from infected erythrocytes [22]. Falstatin was not detected in media from intact mature schizonts, but it was detected after schizont rupture. Since it was secreted during rupture of schizonts, antibodies raised against falstatin blocked the inhibitory action of this molecule. Inhibition of falcipain-2, falcipain-3, and the cysteine protease activity of trophozoite extracts was blocked by purified antibodies in a dose-dependent fashion [22]. The blocking effects of the antibodies were overcome by increasing concentrations of falstatin (Figure 3(b)). The biological role of falstatin was further demonstrated by using it antibody in invasion assay (Figure 3(b)). When incubated with erythrocytic parasites at the ring, trophozoite, or schizont stage, no effects of the antibodies were seen during the course of a single erythrocytic cycle. This result is not surprising, as antibodies would not be expected to access intracellular falstatin. It was of greater interest to assess the impact of anti-falstatin antibodies on the rupture of mature infected erythrocytes or the subsequent invasion of erythrocytes by merozoites. This effect was reversed by pre-incubation of the antibodies with falstatin (Figure 3(b)). Thus, falstatin activity is required for efficient invasion of erythrocytes by merozoites (Figure 3(b)). Falstatin homologues are present in P. vivax and P. berghei, and presumably involved in the same functions. Recent study suggests that falstatin is confined to the parasitophorous vacuole during liver-stage development, and may also have important role in liver stage [27].
2.1.3. PbICP as a Macromolecular Inhibitor
As discuss earlier, P. falciparum expresses an endogenous cysteine proteases inhibitor, falstatin, suggesting a role for control of parasite and/or host protease activity, and invasion of red blood cells. Similarly, an endogenous inhibitor has also been found in P. berghei (PbICP). PbICP, homologue of falstatin, is known to play a crucial role in sporozoite invasion and regulates the programmed cell death by cysteine proteases in liver stage [9]. The expression profile of PbICP suggested that this inhibitor expressed in all stage of parasite (blood stage, sporozoites, liver stage), and can potentially control parasite and host cell derived proteases [9]. The structure studies of chagasin-falcipain-2 and PbICP-falcipain-2 explained that all three loops BC and DE and FG get close to the active site of enzyme (Figure 4) [13,24]. But structure of chagasin-falcipain-2 suggests that BC loop is more important for inhibition, although two other loops also make wedge like structure and inserted close to the active site, and important for stability of enzyme-inhibitor complex [13].
PbICP is secreted by sporozoites prior to and after hepatocyte invasion, localizes to the parasitophorous vacuole as well as to the parasite cytoplasm in the schizont stage and is released into the host cell cytoplasm at the end of the liver stage [9]. Evidences are there to support that PbICP is secreted by P. berghei sporozoites to facilitate host cell invasion [9]. Similar to chagasin, PbICP also have three loops, L2 (BC), loop L4 (DE) and L6 loop (FG) (Figure 4(a)). The C-terminal of chagasin, PbICP, falstatin is required for inhibition, and all these loops are present at C-terminal (Figure 4(a)). At C-terminal the overall architecture of PbICP and falstatin were different from chagasin, the insertion of ~28 aa is missing in chagasin. This insertion polypeptide segment is highly flexible and not defined by electron density, hence it role is not clear yet. Kinetic studies show that PbICP, falstatin, chagasin are effective competitive inhibitor (Ki values in the nano molar range) of falcipain-2 and falcipain-3 and other homologues of malaria cysteine proteases. The structure of PbICP-falcipain 2 indicated that protease-binding loops (L2, L4 and L6) in PbICP form a well-aligned wedge that fills the active site groove of falcipain-2 to obstruct substrate binding (Figure 4(b)) [24]. According to chagasin-falcipain-2 structure the BC loop is one of the three signature motifs that contribute motely to the inhibition of the cysteine protease [13]. This is confirmed by a synthetic peptide corresponding to the BC loop of the chagasin like protein from E. histolytica, which specifically blocked the activity of cysteine proteases [28].
In case of PbICP-falcipain 2 complex, BC loop binds to the primed subsites S1 and S2 of the substrate binding cleft of falcipain 2 that blocks access to the active site [24]. S1 is occupied by residues Ala230 and Gly231, and S2 subsite accommodates the side chain of Thr232 (Figure 4(b)). In PbICP-falcipain 2, BC is place by a hydrogen bond between the Gly231 and the Trp206 indol nitrogen. Comparing both the structure of PbICP and cha-
 (a)
(a)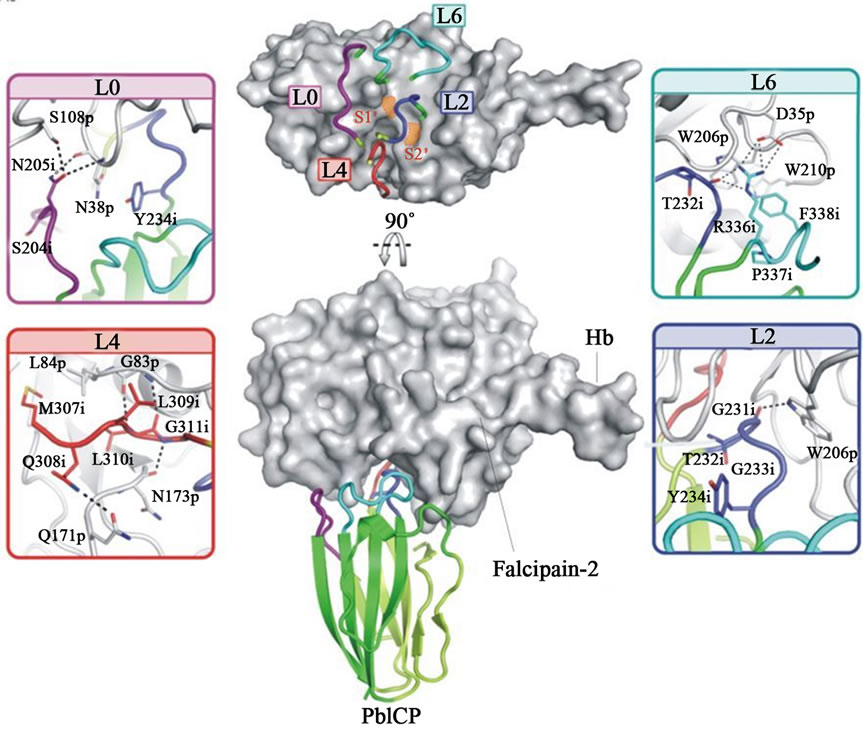 (b)
(b)
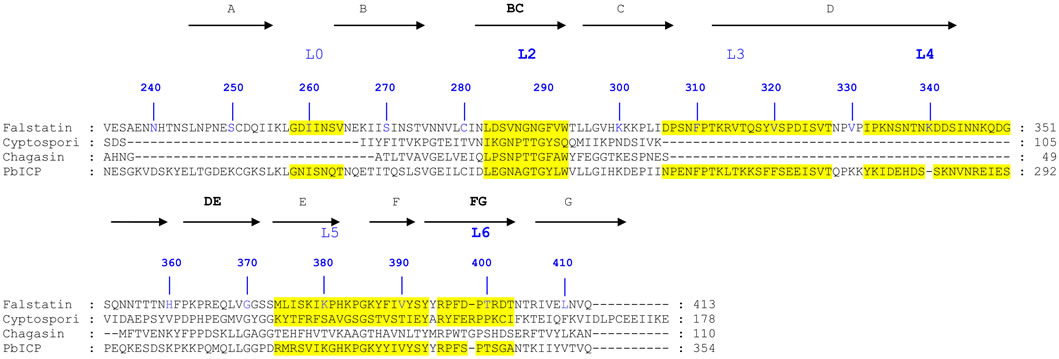
Figure 4. a) Alignment of macromolecular inhibitors of malaria parasite; A multiple sequence alignment of falstatin was performed using ICPs from other parasites as template. This alignment has predicted three important regions; BC (L2), DE (L4) and FG loops (L6); b) Structure of the PbICP-falcipain-2 Complex; The complex of PbICP (C terminal with ribbon representation) and falcipain-2 (with surface representation, gray). The top panel displays the L0, L2, L4, and L6 loops on the surface of falcipain-2 with S1 and S2 sites indicated (orange). The interactions between L0, L2, L4, L6, and falcipain-2 are shown as insets. The Interacting residues of PbICP and falcipain-2 are indicated and shown as sticks; hydrogen bonds are shown as dashed lines (24).
gasin, DE loop is highly mobile like structure in E-64, strong irreversible inhibitor of cysteine protease. The C-terminal inhibitory domain of PbICP binds with falcipain-2 in a 1:1 complex like chagasin, and the solved structure suggests that PbICP is a member of the I42 class of chagasin-like protease inhibitors. The BC loop can adopt two alternative confirmations that both enable efficient inhibition of various target proteases. The PbICP-falcipain-2 indicates that MQLLGG (DE loop) bind to the active site cleft of the protease (Figure 4(b)). The Gly311 of PbICP interacts with Asn173 of enzyme by hydrogen bond, and the side chain of Leu310 of inhibitor is located in the hydrophobic S2 specific pocket, which forms two hydrogen bonds with Gly 83 of enzyme (Figure 4(b)). The structure study further suggests that the FG loop of PbICP is dominated by Arg336, which reaches to the active site and accommodates P4 or P5 residues of substrates. The Arg336 of FG loop served the two important functions; it forms strong salt bridge with Asp35 of enzyme and contribute overall stability of complex (Figure 4(b)) [24]. Although the mutagenesis study of falstatin FG loop, where conserved Phe397 had no effect on inhibition, further suggest that it might be important for overall stability of the complex only and not for inhibition. The peptides study based on DE and FG loops of falstatin suggests that only BC loop is required for the inhibition of malarial cysteine proteases (Srinivasan et al., 2013, pers. comm.).
2.2. PyICP as a Macromolecular Inhibitor
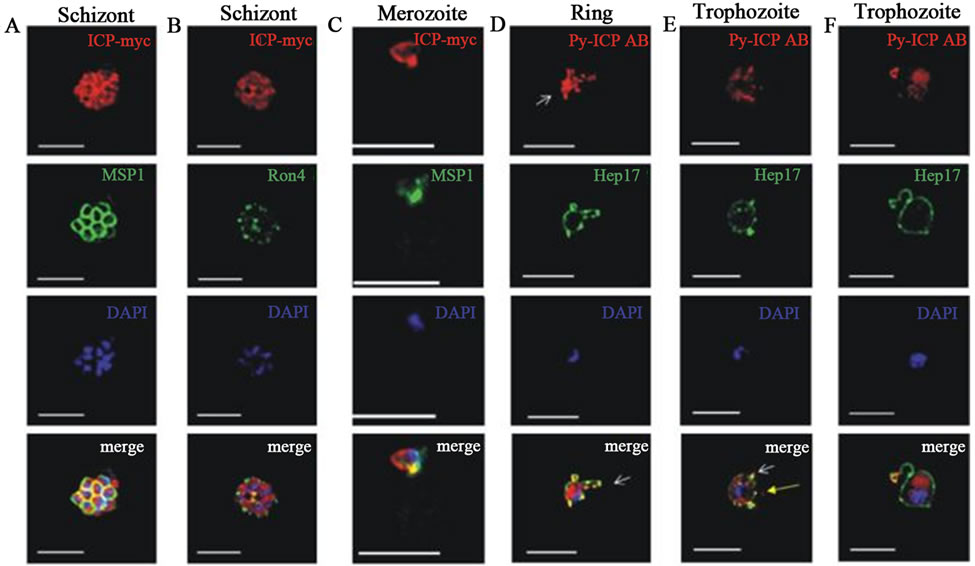
Macromolecular cysteine proteases inhibitor of P. yoelii (PyICP) localized to vesicles within the asexual bloodstage parasite cytoplasm, and the parasitophorous vacuole (Figure 5(a)) [27]. PyICP also express in sexual stage of sporozoite rhoptries (Figure 5(b)). The gene similarity between PyICP and PbICP is 85%, but very less (38%) with other orthologous, falstatin (Figure 4(a)). PyICP is proteolytically processed in the blood stage parasite, further, using PyICP antibodies, recombinant E.coli-expressed PyICP also identified as two protein species (55 kDa and 30 kDa) showing that PyICP is similarly processed during expression in E. coli [27]. It has been also shown that PbICP and falstatin are post-translationally processed in blood stage, but in case of recombinant falstatin, no processing has been seen [23]. The IFA also suggest that the expression of PyICP in mosquito stage parasites. The detail cell biology further confirmed that PyICP is expressed in sporozoite rhoptries and is not secreted prior to host cell invasion [27]. Ying Pei et al. showed that both falstatin and PyICP are exported from the parasite compartment into exo-membrane structures during intraerythrocytic development but are released into the infected hepatocyte only after PVM breakdown in late liver-stage infection. In vivo study using co-imumoprecipatation assay with PyICP antibody indicated that PyICP interacts with the cysteine protease of P. yoelii, yoelipain-2, the orthologue of falcipain-2 and other cysteine proteases. In pre-erythrocytic stages, PyICP differed from PbICP in that it was localized in sporozoite rhoptries and was not deposited in trails during gliding motility [27].
To study the importance of ICP during the parasite life cycle, PyICP gene was deleted in blood-stage parasites using a double-crossover recombination strategy. However, it was not successful to obtain transfected parasite. The other alternative is to disrupt the PyICP open reading frame using a single-crossover recombination strategy, but again this strategy was also unsuccessful and no recombinant parasites were obtained. Finally, the disruption of PyICP could not be achieved, but the gene was replaced with a tagged functional copy, indicating that this gene is essential for erythrocytic parasites. In the liver stage release of these macromolecule inhibitors to the host hepatocyte may allow inhibiting host cell proteases and thus saving from proteolytic attack once the parasite comes into direct contact with the host cell cytoplasm [27]. Our work on falstatin also shown that recombinant molecule can inhibit both host-derived cathepsins and caspases in vitro [23], suggested that these macromolecule inhibitors inhibiting host proteases, and may be protecting surface molecules of merozoite and hepatocyte, which are crucial for interactions. The other potential role of these macromolecule cysteine proteases inhibitors may be during late liver-stage development is in exo-erythrocytic merozoite egress from hepatocytes, which is similar like egress from red blood cell. The knock out and gene replacement studies suggested that ICP appears to be essential for blood-stage parasite survival by regulating protease activities in the parasitederived intra-erythrocytic membrane network. In case of liver-stage development, ICP remains within the PV, and ruling out a role in regulating host hepatocyte apoptosis. Once PV breakdown ICP released into the host hepatocyte cytoplasm, allowing it to potentially inhibit host proteases.
3. CONCLUSIONS AND OUTLOOK
In summary, all the known macromolecular inhibitors, falstatin, PbICP, PyICP, Chagasin and Cystatin, bind to almost similar region within the substrate binding cleft of falcipain-2. Like other cysteine proteases in various parasitic organisms, cysteine proteases of Plasmodium species play a crucial role in parasite development both at liver stage and blood stage. The sequence of events participated by these proteases is tightly regulated by a class of inhibitors known as inhibitor of cysteine proteases (ICPs). This class of cysteine protease inhibitor adopts an immunoglobulin β-fold structure sharing no significant sequence similarity with known classes of cysteine protease inhibitors including the cystatin family. The structure studies of chagasin-falcipain-2 and PbICP-falcipain-2 explained that all three loops BC and DE and FG make interactions with enzyme. But structure of chagasin-falcipain-2 explained that BC loop is more important for inhibition, although two other loops also make wedge-like structure and inserted close to the active site, and are important for stability of enzyme-inhibitor complex. Our mutagenesis study also confirmed that falstatin BC loop is crucial for inhibition, and further explained that DE and FG loops are not important for inhibition of malarial cysteine proteases (Srini et al., 2013, pers. comm.).
The PbICP shares very little sequence and structural
 (a)
(a)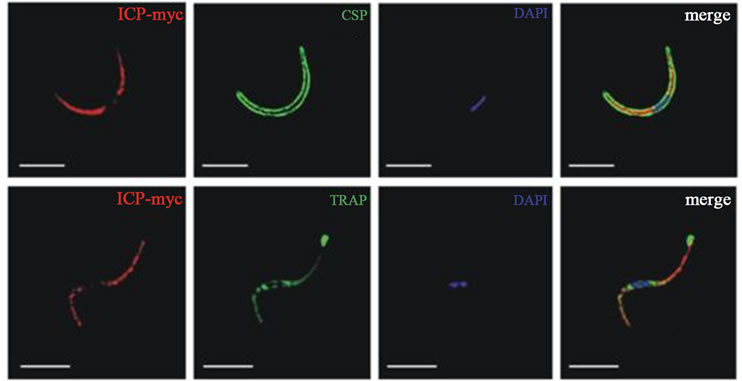 (b)
(b)
Figure 5. a) PyICP expression and localization in blood stages. The expression of PyICP in blood stages was visualized using IFA. PyICP-myc schizonts were imaged with antibodies to c-myc (red), MSP1 (A, green) or the rhoptry neck marker RON4 (B, green). (C) PyICP-myc merozoite invading a RBC. Merozoites were stained with antibodies against c-myc (red) and MSP1 (green). (D-F) WT blood-stage parasites at the ring (D) or trophozoite (E-F) developmental stages were imaged with antibodies to PyICP (red) and the PVM marker Hep17 (green). Vesicular structures associated with the PV (Hep17 positive) are indicated by white arrows (D-F), or in Hep17-negative structures with yellow arrow in (E). DNA was imaged with DAPI. Scale bar: 5 µm [27]; b) PyICP expression and localization in sporozoites; (A) Isolated sporozoites were fixed on slides and PyICP-myc was localized by IFA with an anti-myc antibody and sporozoites were co-stained with anti-CSP [26]. Microneme secretion. The secretion was induced by incubation of PyICP-myc sporozoites with RPMI with 20% FCS for 30 min prior to fixation. Sporozoites were then stained with antibodies to myc and the micronemal protein, TRAP [27].
similarity with falstatin (30% and 38% respectively). Moreover, the secondary structure predicted indicated a highly flexible loop in falstatin compared to PbICP. This may be a reason for inhibiting caspases in micro molar range by falstatin unlike PbICP. The literature suggests that a wide range of cysteine proteases is inhibited by chagasin, falstatin and PbICP. The distinct concept of potent competitive inhibition due to a high degree of homology in the active site, substrate-like binding mostly leads to inhibitors that can potently inhibit more than one target protease. This indiscriminate nature is evidenced by the fact that there are more proteases than their inhibitors present in the parasites. Further, it may be possible that protein inhibitors of cysteine proteases may have evolved more than once on non-homologous scaffolds to inhibit a wide range of cysteine proteases. As mentioned before, unlike PbICP, falstatin blocks proteolytic activity of caspase and calpain in micro molar range concentration. Possible reasons may be that the falstatin structure is closer to serpins family and has more flexible loops compared to PbICP, and further unlike its homologues, falstatin is active in multimeric form. The significance of inhibition of caspases and calpain-1 by falstatin may be useful in programmed cell death, which further needs to be explored.
The majority of macromolecular inhibitors are completive inhibitor, and block access to the active site of their target protease, but do not bind in a strictly substrate-like manner. They interact with the protease subsites and catalytic residues in a non-catalytically competent manner. This mechanism differentiates them from standard inhibitors (small competitive inhibitor). However, like standard mechanism inhibitors, they get most of their potency from interactions within the protease active site, and tend to potently inhibit many related proteases. The classic example is like inhibition of cysteine proteases by cystatin (Figure 1(a)). The two hairpin loops of cystatin bind to the prime side of the active site of cysteine protease, which provides the majority of the binding energy for the interaction. Thus, the prime and nonprime sides of the active site are occupied, but still no interactions are actually made with the catalytic machinery of the enzyme.
The literature suggests that a wide range of cysteine proteases is inhibited by chagasin, falstatin and PbICP. It may be possible that protein inhibitors of cysteine proteases may have evolved more than once on non-homologous scaffolds to inhibit a wide range of cysteine proteases.
Small protease inhibitors tend to compete with substrate binding, either through direct competition or deformation of the protease active site, and they primarily gain potency through interactions with the catalytic machinery, while macromolecular inhibitors can gain potency through the burial of a large surface area and specificity through contacts with secondary binding sites outside the active site that are critical to inhibition.
4. FUTURE OF MACROMOLECULAR INHIBITORS
Designing inhibitors based on macromolecular inhibitorenzyme complex (protein-protein interactions; PPIs) is a new approach. Earlier this approach has deliberately been avoided by small molecule drug developers, largely because of technological hurdles. Daniela Cardinalea et al. suggested that protein-protein interface-binding peptides can inhibit the cancer therapy targeting enzyme and human thymidylate synthase [29]. Furthermore, researchers aimed to block the interactions of two monomers, and prevented it from forming the active dimer interface, and developed the first small-molecule inhibitor of a herpes virus protease that blocked PPIs [30]. Currently, macromolecular inhibitors have started to make their way into the clinic. The serpins α1-anti-trypsin and antithrombin III, which were purified from human plasma, are used as replacement therapies for patients deficient in those proteins [31].
Recently, our group also focused on interactions crucial for activation of enzyme [10]. Another target is the C-terminal insert of falcipain-2, which specifically binds to hemoglobin, a natural substrate of falcipains [22]. Since the C-terminal motif of falcipain-2 acts as a hemoglobin binding domain, and the disruption of C-terminal motif-hemoglobin interaction should offer a novel means of inhibition of parasite development. Continued research on macromolecular inhibitors and its role in Plasmodium development will help to further our understanding of protease and protease inhibitor functions during malaria parasite infection. A drug design viewpoint targeting in protein-protein interactions, which is opposed to an enzyme active site could be an interesting field in drug discovery. Targeting “hot-spot” in proteinprotein interaction could be less prone to drug resistant mutation. This is because acquiring a drug resistance point mutation will not be companied by the right compensatory mutations, which are less likely to be tolerated by a complex protein-protein interface.
5. ACKNOWLEDGMENTS
Work in the author laboratory was supported by Department of Biotechnology, Govt. of India. Thank to Rahul and Dhruv for the art work and formatting, and appreciate NIMR, ICMR for extramural funds for setting a new laboratory.
REFERENCES
- Farady, C.J. and Craik, C.S. (2013) Mechanisms of macromolecular protease inhibitors. ChemBioChem, 11, 2341- 2346.
- Phan, J., Zdanov, A., Evdokimov, A.G., Tropea, J.E., Peters, H.P., et al. (2002) Structural basis for the substrate specificity of tobacco etch virus protease. The Journal of Biological Chemistry, 277, 50564-50572. doi:10.1074/jbc.M207224200
- Pandey, K.C. and Dixit, R. (2012) Structure-function of falcipains: malarial cysteine proteases. Journal of Tropical Medicine, 2012, 345195. doi:10.1155/2012/345195
- Na, B.K., Shenai, B.R., Sijwali, P.S., Choe, Y., Pandey, K.C., et al. (2004) Identification and biochemical characterization of vivapains, cysteine proteases of the malaria parasite Plasmodium vivax. Biochemical Journal, 378, 529-538. doi:10.1042/BJ20031487
- Na, B.K., Bae, Y.-A., Zo, Y.-G., Choe, Y., Kim, S.-H., et al. (2010) Biochemical properties of a novel cysteine protease of Plasmodium vivax, vivapain-4. PLOS Neglected Tropical Diseases, 4, e849. doi:10.1371/journal.pntd.0000849
- Singh, A, Shenai, B.R., Choe, Y., et al. (2002) Critical role of amino acid 23 in mediating activity and specificity of vinckepain-2, a papain-family cysteine protease of rodent malaria parasites, Biochemical Journal, 368, 273- 281. doi:10.1042/BJ20020753
- Prasad, R., Soni, A.A., Puri, S.K., et al. (2013) Expression, characterization, and cellular localization of knowpains, papain-like cysteine proteases of the Plasmodium knowlesi malaria parasite. PLoS ONE, 7, e51619.
- Aly, A.S. and Matuschewski, K. (2005) A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts. The Journal of Experimental Medicine, 202, 225-230.
- Rennenberg, A., Lehmann, C., Heitmann, A., et al. (2010) Exoerythrocytic Plasmodium parasites secrete a cysteine protease inhibitor involved in sporozoite invasion and capable of blocking cell death of host hepatocytes. PLoS Pathogens, 6, e1000825. doi:10.1371/journal.ppat.1000825
- Pandey, K.C., Singh, N., Arastu-Kapur, S., Bogyo, M., and Rosenthal, P.J. (2006) Falstatin, a cysteine protease inhibitor of Plasmodium falciparum, facilitates erythrocyte invasion. PLoS Pathogens, 2, e117. doi:10.1371/journal.ppat.0020117
- Pandey, K.C. (2011) Centenary celebrations article: Cysteine proteases of human malaria parasites. Journal of parasitic Diseases, 35, 94-103.
- Monteiro, A.C., Abrahamson, M., Lima, et al. (2001) Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. Journal of Cell Science, 114, 3933- 3942.
- Wang, S.X., Pandey, K.C., Scharfstein, J., et al. (2007) The structure of chagasin in complex with a cysteine protease clarifies the binding mode and evolution of an inhibitor family. Structure, 15, 535-543. doi:10.1016/j.str.2007.03.012
- Wang, S.X., Pandey, K.C., Somoza, J.R., et al. (2006) Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. PNAS, 103, 11503-11508. doi:10.1073/pnas.0600489103
- Sijwali, P.S., Shenai, B.R., Gut, J., Singh, A. and Rosenthal, P.J. (2001) Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochemical Journal, 360, 481-489. doi:10.1042/0264-6021:3600481
- Shenai, B.R., Sijwali, P.S., Singh, A., Rosenthal, P.J. (2000) Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. The Journal of Biological Chemistry, 37, 29000-29010.
- Rosenthal, P.J. (2004) Cysteine Proteases of malaria parasites. International Journal for Parasitology, 34, 1489-1499. doi:10.1016/j.ijpara.2004.10.003
- Sijwali, P.S., Shenai, B.R. and Rosenthal, P.J. (2002) Folding of the Plasmodium falciparum cysteine protease falcipain-2 is mediated by a chaperone-like peptide and not the prodomain. The Journal of Biological Chemistry, 277, 14910-14915. doi:10.1074/jbc.M109680200
- Pandey, K.C., Barkan, D.T., Sali, A. and Rosenthal, P.J. (2009) Regulatory elements within the prodomain of Falcipain-2, a cysteine protease of the malaria parasite Plasmodium falciparum. PloS one, 4, e5694. doi:10.1371/journal.pone.0005694
- Sundararaj, S., Singh, D., Saxena, A.K., Vashisht, K., Sijwali, P.S., Dixit, R. and Pandey, K.C. (2012) The Ionic and hydrophobic interactions are required for the auto activation of cysteine proteases of Plasmodium falciparum. PloS one, 7, e47227. doi:10.1371/journal.pone.0047227
- Wiederanders, B., Kaulmann, G. and Schilling, K. (2003) Functions of propeptide parts in cysteine proteases. Current Protein & Peptide Science, 4, 309-326. doi:10.2174/1389203033487081
- Groves, M.R., Coulombe, R., Jenkins, J. and Cygler, M. (1998) Structural basis for specificity of papain-like cysteine protease proregions toward their cognate enzymes. Proteins, 32, 504-514. doi:10.1002/(SICI)1097-0134(19980901)32:4<504::AID-PROT8>3.0.CO;2-F
- Pandey, K.C., Singh, N., Kapur, A.S., Bogyo, M., Rosenthal, P.J. (2006) Falstatin, a cysteine protease inhibitor of Plasmodium falciparum, facilitates erythrocyte invasion. PLoS Pathog, 2, e117. doi:10.1371/journal.ppat.0020117
- Hansen, G., Heitmann, A., Witt, T., et al. (2011) Structural basis for the regulation of cysteine-protease activity by a new class of protease inhibitors in Plasmodium. Structure, 19, 919-929.doi:10.1016/j.str.2011.03.025
- Pandey, K.C., Wang, S.X., Sijwali, P.S., Lau, A.L., McKerrow, J.H. and Rosenthal, P.J. (2005) The Plasmodium falciparum cysteine protease falcipain-2 captures its substrate, hemoglobin, via a unique motif. Proceedings of the National Academy of Sciences of the United States of America, 102, 9138-9143. doi:10.1073/pnas.0502368102
- Storer, A.C. and Menard, R. (1994) Catalytic mechanism in papain family of cysteine peptidases. Methods in Enzymology, 244, 486-500. doi:10.1016/0076-6879(94)44035-2
- Pei, Y., Miller, J.L., Lindner, S.E., et al. (2013) Plasmodium yoelii inhibitor of cysteine proteases is exported to exomembrane structures and interacts with yoelipain-2 during asexual blood-stage development. Cellular Microbiology, 15, 1508-1526. doi:10.1111/cmi.12124
- Riekenberg, S., Witjes, B., Saric, M., Bruchhaus, I. and Scholze, H. (2005) Identification of EhICP1, a chagasinlike cysteine protease inhibitor of Entamoeba histolytica. FEBS Letters, 579, 1573-1578.
- Cardinalea, D., Guaitolia, G., Tondia, D., et al. (2011) Inhibition of calpain by mimicking a natural proteinprotein interaction between Protein-protein interfacebinding peptides inhibit the cancer therapy target human thymidylate synthase. PNAS, 108, E542-E549.
- Shahian, T., Lee, G.M., Lazic, A., et al. (2009) Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nature Chemical Biology, 5, 640-646.
- Leader, B., Baca, Q.J. and Golan, D.E. (2008) Protein therapeutics: A summary and pharmacological classification. Nature Reviews Drug Discovery, 7, 21-39.