Journal of Biomedical Science and Engineering
Vol.6 No.6(2013), Article ID:33672,30 pages DOI:10.4236/jbise.2013.66078
Systematic analysis of diabetesand glucose metabolism-related proteins and its application to Alzheimer’s disease
![]()
State Key Laboratory of Pharmaceutical Biotechnology, College of Life Sciences, Nanjing University, Nanjing, China
Email: *yangjie@nju.edu.cn
Copyright © 2013 Jie Yang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 19 April 2013; revised 22 May 2013; accepted 2 June 2013
Keywords: Codon Biases; Protein-Protein Interaction Network; Transcription Factors; BAD Complex; Bioinformatics
ABSTRACT
Alzheimer disease has been defined as Type 3 Diabetes due to their shared metabolic profiles. Like our previously research, results of Alzheimer’s disease and other neurodegenerative diseases, systematic analysis of diabetesand glucose metabolism-related proteins also provides help in the treatment of Alzheimer’s patients. Some interesting results indicate that diabetes-related proteins (DRPs) are rich in Lys and the content of Trp can distinguish between type 1 and type 2 diabetes mellitus in particular, while glucose metabolism-related proteins (GMRPs) possess Leurich and Trp-poor character. Moreover, the usage biases of codons depend on GC contents to a great extent, in concord with all codons of the highly expressed genes with the terminal of C/G. Especially, the deficit of CpG dinucleotides is largely attributed to the hypermutability of methylated CpGs to UpGs by the mutational pressure. Besides a common node insulin receptor, there are some similar node proteins, such as glucose transporter member, protein tyrosine phosphatase, and adipose metabolism signal protein. The sharing proteins involve glucagon, amylin, insulin, PPARγ, angiopoietin, PC-1/ENPP1, and adiponectin mediated signal pathway. Meanwhile, the gene sequences of node proteins contained the binding sites of 37 transcription factors divide into four kinds of superclasses. Additionally, BAD complex can integrate pathways of glucose metabolism and apoptosis by BH3 domain of BAD directly interacting with GK as well as GK binding with the consensus motif [G]-[1]-[K]-[2]-[S/T] or [L/M]-[R/K]-[2]-[T] of PP1 or WAVE1. This facilitates the therapies for diabetes mellitus as well as Alzheimer’s disease.
1. INTRODUCTION
Alzheimer’s disease (AD) and diabetes are both ageassociated diseases [1]. AD is a late-onset neurological disorder characterized by progressive loss of memory and cognitive abilities as a result of excessive neurodegeneration in the hippocampus and cortex [2], accompanied by the accumulation of intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein and extracellular amyloid beta (Aβ) peptides containing senile plaques [3]. Aβ peptides originate from an abnormal proteolytic processing of the amyloid precursor protein (APP) which is a large transmembrane type 1 (cytosolic C-terminal) glycoprotein. Cleavage of the APP ectodomain by β-secretase at the amino-terminus of Aβ is followed by cleavage of the β-secretase-generated carboxyl-terminal fragment (β-CTF, C99) at two sites of the carboxyl terminus of Aβ by γ-secretase [4], to produce either the Aβ residue 1-40 (Aβ 40) or a longer, more amyloidogenic form that contains the residues 1 - 42 (Aβ42) [99]. On the other hand, diabetes mellitus is a chronic metabolic disorder in the endocrine system and systemic underway disease with high blood glucose, specified to mammal and not to be effective cured until now [5]. Insulin-dependent diabetes mellitus (IDDM, type 1), accounting for 5% - 10% of diabetes, in which the body does not produce any insulin due to islet beta-cell apoptosis, most often occurs in children and young adults [6,7]. Noninsulin-dependent diabetes mellitus (NIDDM, type 2), accounting for 90% - 95%, in which the body does not produce enough, or properly use, insulin due to the damage of insulin gene, its receptor gene, glucokinase gene, and mitochondria gene, is nearing epidemic proportions, because of an increased number of elderly people, and a greater prevalence of obesity and sedentary lifestyles [8,9]. Diabetes mellitus is associated with decreased body insulin production than required (type 1) and impaired insulin signaling (type 2/T2DM). Diabetes especially T2DM and AD follow the same pathological mechanisms resulting in misfolded proteins, insulin impairment, abnormal glucose metabolism, abnormal fatty acid metabolism, mitochondrial dysfunction, and high oxidative stress [1]. These shared metabolic profiles and diabetes as extreme risk factor for AD lead to the assumption that AD may reflect type-3 diabetes.
We have previously finished construction of cell model for screening Alzheimer’s drugs [10], codon usage biases in AD and other neurodegenerative diseases [11], and protein-protein interactions related to AD based on complex network [12]. Similarly, we are to propose a computational analysis on codon biases of some proteins related to diabetes and glucose metabolism, hoping that the findings might be useful for developing new therapeutic treatment for diabetes as well as AD. Moreover, the proteins related to diabetes are located at signal transduction pathway [13,14], gene control pathway [15, 16], glucose metabolic pathway [17,18], etc. A large number of research work on these proteins and their structures, functions, pathways, networks, and relations to diabetes have been conducted yet [19,20]. It is strongly significant to pay close attention to the systematical integration of known diabetes-related proteins (DRPs) for treatment of diabetes mellitus and its complications, which helps to the treatment of Alzheimer’s patients. With the application of bioinformatics and functional proteomics to drug research and development, we try to systematically analyze the property of glucose metabolism-related proteins (GMRPs) and DRPs, involving the amino acid compositions, protein-protein interaction network, codon biases, and transcript factors binding sites, etc. Up to now, there have not been enough effective ways to treat diabetes thoroughly as well as its complications, while the control and treatment of diabetes and its complications mainly depend on the chemical or biochemical agents, namely insulin, insulin-like growth factor, alpha-glycosidase inhibitors, aldose reductase inhibitor, sulfonylureas, biguanide, and others.
Additionally, mitochondrial dysfunction, as one of the shared metabolic profiles between diabetes and AD, results in increased oxidative stress and induces glyceroldehyde-derived advanced glycation end products (AGEs) [21,22]. The progressive deposition of fibrillar Aβ in the brain can induce disruption of the mitochondrial membrane, resulting in the production of reactive oxygen species, the release of cytochrome c from mitochondria into the cytosol, and the activation of caspase-dependent apoptotic pathways [23]. The PI3K/Akt-mediated interaction between Bad and Bcl(XL) plays an important role in maintaining mitochondrial integrity [24]. Zeng KW et al. have demonstrated that hyperoside, a bioactive flavonoid compound from Hypericum perforatum, significantly inhibited Aβ(25-35)-induced cytotoxicity and apoptosis by reversing Aβ-induced mitochondrial dysfunction, including mitochondrial membrane potential decrease, reactive oxygen species production, and mitochondrial release of cytochrome c via PI3K/Akt/Bad/ Bcl(XL)-regulated mitochondrial apoptotic pathway [24]. Additionally, Danial and co-workers have indicated that human BAD (a pro-apoptotic Bcl-2 family member) resides in a mitochondrial complex together with protein kinase A (PKA) and protein phosphatase 1 (PP1) catalytic units, Wiskott-Aldrich family member WAVE-1, and glucokinase (GK, hexokinase D, HXK4), integrating pathways of glucose metabolism and apoptosis [25]. Glucose deprivation results in dephosphorylation of BAD and BAD-dependent cell death, while the phosphorylation status of BAD helps regulate glucokinase activity. In this study, we sought to discuss some proteins contributing to diabetes emergence and development. This present paper addresses the key attempts to propose ways for future discovery and validation of potential targets for diabetes as well as AD therapies.
2. METHODS
2.1. Data
The amino acid sequences of all proteins come from SWISSPORT (http://au.expasy.org/) and their coding sequences were extracted from EMBL-EBI (http://www.ebi.ac.uk) linked to GenBank database (http://www.ncbi.nlm.nih.gov). Firstly, there are five groups of diverse proteins in various species for analysis of amino acid composition, composed of 211 DRPs, 31 proteins related to diabetes type I (DRPsI), 66 to diabetes type II (DRPsII), 223 GMRPs, 100 human GMRPs (hGMRPs) and 123 mouse GMRPs (mGMRPs), respectively. Secondly, a series of complete genes (including initiation and stop codons) were analyzed and are available for codon usage biases, such as 211 DRPs, 100 hGMRPs and 123 mGMRPs. And then some indices of codon biases in the coding sequences of DRPs and GMRPs were calculated and the frequency of optimal codons (FOP) was analyzed. A correlation between codon usage for DRPs and gene expression level was built using correspondence analysis (COA) while the codon usage pattern in the two data sets was compared using chi square test. Thirdly, protein-protein interaction networks for DRPs and GMRPs were built using Osprey software. Finally, BAD complex was analyzed and evaluated.
2.2. Amino acid Composition
According to our methods [26,27], the accumulative times 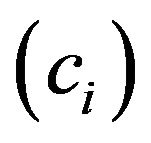 and the percentage
and the percentage  of each amino acid in 211 known DRPs, 100 hGMRPs and 123 mGMRPs (Table 1) were analysized and calculated by symbolic statistic method as follows:
of each amino acid in 211 known DRPs, 100 hGMRPs and 123 mGMRPs (Table 1) were analysized and calculated by symbolic statistic method as follows:
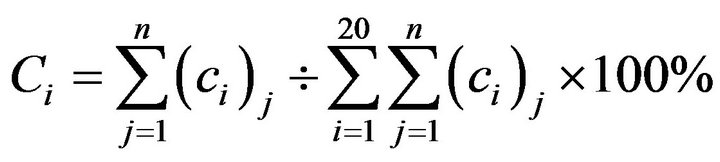 .
.
2.3. Relative Synonymous Codon Usage
Codon usage, COA, GC3s (the frequency of codons ending in C and G, excluding Met, Trp and stop codons), A3s, T3s, G3s, C3s (the ratio of A/T/G/C content at the third position in codon to total gene bases), 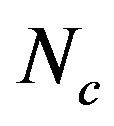 (the “effective number of codons”) [28], RSCU (relative synonymous codon usage, relative probability of certain codon to other synonymous codons coding the same amino acid) [29], and the frequency of optimal codons [30] were computed using the program CodonW version 1.4 (www.molbiol.ox.ac.uk/cu) and GCUA [31]. Furthermore, the correlation analysis and cluster analysis were carried out by SPSS version 13.0.
(the “effective number of codons”) [28], RSCU (relative synonymous codon usage, relative probability of certain codon to other synonymous codons coding the same amino acid) [29], and the frequency of optimal codons [30] were computed using the program CodonW version 1.4 (www.molbiol.ox.ac.uk/cu) and GCUA [31]. Furthermore, the correlation analysis and cluster analysis were carried out by SPSS version 13.0.
We estimated codon frequencies from the observed codon counts of these protein-coding sequences mentioned above. 64 codons are obtained using the sliding window method one by one from 5’-terminal of protein-coding sequences to 3’-teminal: a sequence of n residues gives rise to  units [32]. The accumulative times
units [32]. The accumulative times 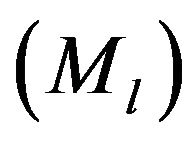 of each codon in 211 DRPs and 223 GMRPs were analysized and calculated as follows:
of each codon in 211 DRPs and 223 GMRPs were analysized and calculated as follows:
 .
.

Table 1. Content percentage of 20 types of standard amino acids in a series of proteins distributed over various species.
where 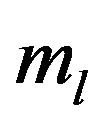 express the times of 64 codons of each protein. The usage frequency of 64 codons respectively in each protein was calculated by using C++ program written by us.
express the times of 64 codons of each protein. The usage frequency of 64 codons respectively in each protein was calculated by using C++ program written by us.
To normalize synonymous codon usage within dataset of different amino acid compositions, RSCU is calculated by dividing the observed frequency of a codon by the expected frequency assuming all synonymous codons are used equally [33]. The RSCU values of different codons in each sequence were calculated as follows:
 .
.
Here,  means relative usage probability of the codon at
means relative usage probability of the codon at  position in
position in  mRNA sequence;
mRNA sequence;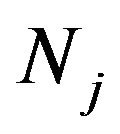 , the actual observed number of certain codon in some template sequence;
, the actual observed number of certain codon in some template sequence;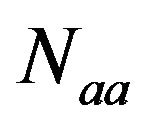 , the actual observed number of other synonymous codons coding the same amino acid coded by certain codon in the template sequence; k, the number of synonymous codons coding the same amino acid. It is obvious that RSCU values close to 1.0 indicate a lack of bias for the corresponding codon [34].
, the actual observed number of other synonymous codons coding the same amino acid coded by certain codon in the template sequence; k, the number of synonymous codons coding the same amino acid. It is obvious that RSCU values close to 1.0 indicate a lack of bias for the corresponding codon [34].
2.4. Indices of Codon Biases
As Ghosh and co-worker [35] mentioned, some indices of codon biases were introduced and calculated for each gene, such as GC3s, 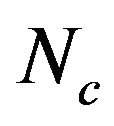 , A3s, T3s, G3s, and C3s.
, A3s, T3s, G3s, and C3s. 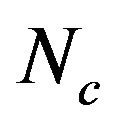 whose value ranges from 20 to 61 was used to quantify the codon usage bias of a gene. This is a measure of general non-uniformity of synonymous codon usage. The expected
whose value ranges from 20 to 61 was used to quantify the codon usage bias of a gene. This is a measure of general non-uniformity of synonymous codon usage. The expected 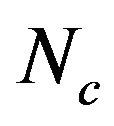 value under random synonymous codon usage can be calculated for any value of GC3s by the following formula:
value under random synonymous codon usage can be calculated for any value of GC3s by the following formula:
 .
.
When all sense codons are used randomly,  takes a value of 61. Lower values of
takes a value of 61. Lower values of 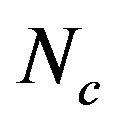 indicate stronger bias, with an extreme value of 20 when only one synonym is used for each amino acid. The values of
indicate stronger bias, with an extreme value of 20 when only one synonym is used for each amino acid. The values of 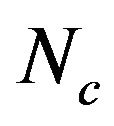 would fall on the continuous curve described by the formula, if the G + C composition at the synonymous third position is the only determinant factor shaping the codon usage [36].
would fall on the continuous curve described by the formula, if the G + C composition at the synonymous third position is the only determinant factor shaping the codon usage [36].
2.5. Statistical Analysis
COA was used to investigate the major trend in codon usage variation among genes [37]. In this study, the RSCU values of genes in each proteins related to diabetes and glucose metabolism were plotted in a multidimensional space of 59 axes, according to their usage of the 59 sense codons (excluding Met, Trp and stop codons), and COA identified a series of new orthogonal axes accounting for the greatest variation among genes. The analysis yielded the coordinate of each gene on each new axis, and the fraction of the total variation was accounted for by each axis. By this means, the axis that contributes most to the total variation among different genes as well as the primary trend of codon usage can be identified. The analysis was performed using the RSCU values in order to minimize the effects of amino acid composition [38]. COA of RSCU values was carried out to determine the major source of variation among genes. Hence, each sequence is described by a vector of 59 variables, which is the number of codons for which there are synonyms. COA plots these genes in a multidimensional space of 59 axes. Then a certain number of axes are identified.
2.6. The Relative Abundance of Dinucleotides Based on DRPs
16 dinucleotide relative abundances (DRA) in DRPs’ open reading frames (ORFs) were obtained using the method described by Karlin and Burge [39]. The odds ratio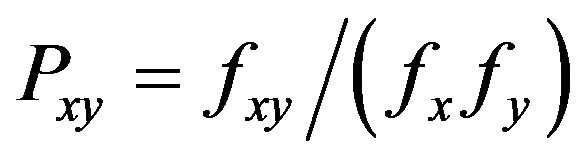 , where fx denotes the frequency of the nucleotide X and fxy the frequency of the dinucleotide XY, etc., for each dinucleotide were calculated. As a conservative criterion, for
, where fx denotes the frequency of the nucleotide X and fxy the frequency of the dinucleotide XY, etc., for each dinucleotide were calculated. As a conservative criterion, for  (or < 0.78), the XY pair is considered to be of high (or low) relative abundance compared with a random association of mononucleotides.
(or < 0.78), the XY pair is considered to be of high (or low) relative abundance compared with a random association of mononucleotides.
2.7. Protein-Protein Interaction Network
Many properties of complex system are decided by interaction compositions not by single. Life activity is a complex system while the research is dependent upon protein interaction network, which is beneficial to identifying and validating the drug targets and prediction of new functional proteins [40]. Protein-protein interaction networks are mostly large network and actually a scalefree network, which follows power-law principle:  [41]. Here, the network consists of nodes and edges, which express proteins and their interactions, respectively. The mark k donates link degree, namely the linking numbers of certain node protein.
[41]. Here, the network consists of nodes and edges, which express proteins and their interactions, respectively. The mark k donates link degree, namely the linking numbers of certain node protein.  displays the distributing coefficient of link degree that means the link degree of single node protein, namely the link degree divided by the numbers of nodes. The sign
displays the distributing coefficient of link degree that means the link degree of single node protein, namely the link degree divided by the numbers of nodes. The sign
2.8. Cluster Analysis of Codon Usages of DRPs Based on Protein Interaction Network
Cluster analysis refers to assign a set of samples into groups according to the sample characteristics and the adjacency and similarity of pattern space. The basic operation process of hierarchial cluster analysis is as follows. First, respectively classified the n samples as a category, calculated the distances between samples, and then selected a couple of samples with the minimum distance into a new class. Second, calculated the distances between the new class and other classes and merged the two classes with the minimum distance into another new class. This moved in circles until all the samples are combined into one category. Here, the variable group composed of different RSCU value of a signal mRNA sequence of 11 node proteins in DRPs were regarded as a point in the multidimensional space. Tryptophan (Trp) and methionine (Met) because of their codons UUG and AUG with degeneracy of 1 were not considered as well as 3 termination signals (UGA, UAA and UAG). So, the space dimension is 59. Every gene sequence of 11 node proteins was viewed as the space vector consisting of 59 variable components. The Euclidean distance coefficient of codon usage bias between two genes i and k is calculated as follows [43]:
 .
.
Here, the group average linkage method was used for clustering by SPSS software.
11 node proteins in DRPs are listed as follows: O43707 (ACTN4_human), P13987 (CD59_human), Q64521 (GPDM_mouse), P19357 (GTR4_rat), P20823 (HNF1A_human), P02545 (LMNA_human), P06213 (INSR_human, Q05329), P06858 (LIPL_human), P62845 (RS15_rat), Q16849 (PTPRN_human), and Q14191 (WRN_human).
2.9. Analysis of Transcription Factors at DNA Level Based on DRPs’ Node Proteins
The regulation of a gene depends on the binding of transcription factors (TFs) to specific sites located in the regulatory region of the gene. Transcription regulation is carried out by the interactions between TFs and their binding sites in DNA. Transcription factor binding to specific DNA sequence of its gene promoter activates or represses transcription of genes, which plays a critical role in regulation of specific gene expression and specific stress response. Transcription factor binding sites (TFBS), short, degenerate nucleotide sequences (usually 6 - 20 bps), are very important in drug research and development taking transcription factors as targets. Here, we search for the TFBS of DRPs interaction with transcription factors using TRANSFAC 7.0-Public database (http://www.gene-regulation.com/pub/databases.html).
2.10. Prediction of BAD Complex Proteins Based on the Theoretical Model
Analyze and calculate a series of parameters of BAD complex and assess whether these proteins might participate in diabetes pathogenesis in comparison with the results mentioned above.
Molecular Modeling of BAD Complex
According to our methods [44,45], molecular modeling of the three-dimensional (3D) structure of human BAD and its complexes were performed on a Silicon Graphics Iris O2 (SGI Inc., Silicon, CA, USA) workstation using the Homology modules of the commercial software packages InsightII 2000 (MSI, St Louis, Mi, USA). Molecular dynamics and molecular mechanics were used to optimize the model. The final model was assessed by PROCHECK (a protein structure validation program).
3. RESULTS
3.1. Diabetes-Related Proteins and Glucose Metabolism-Related Proteins
Diabetes-related proteins compiled here are composed of 211 kinds of proteins, which originated from mammalians, namely human, mouse, rat, Rhesus macaque, Guinea pig, Fat sand Rat, Bovine, Chinese hamster, Nutria, Golden hamster, Dog, Cat, Lowland gorilla, Chimpanzee, and Pig-tailed macaque, in turns. Among these proteins, 66 are related to NIDDM, 31 to IDDM, 17 to its complications, 10 to MODY1-6 type of diabetes, 7 to BBS syndrome 1-8 type, and other. These proteins are divided into six classes based on their function as follows: a) regulation of immunity processes, involving T-cell activating signal transduction, recognization of cytotoxin reacted with MHC-I or restrained T-cell, T-cell activation, transcript factors, adjustment of immunity cell function, etc; b) metabolic regulation, including synthesis of fatty acids, acidification of sugars, leptin receptor, signal transduction pathways related to obesity, energy balance, avoirdupois balance, etc; c) insulin effect, related to ATP-sensitive potassium channels and insulin release, related protein phosphate kinases, hormone balances, synthesis of cyclic ADP-ribose, activation of insulin excretion, adrenalin excretion, transcript factor binding with insulin gene E-box, insulin receptor, pancreas islet β-cell, insulin signal transduction, etc; d) blood glucose control, associated with glucose balance, fat metabolism, insulin sensitivity, related enzymes, glucose transport factors, synthesis of glycogen, etc; e) hormone regulation, relevant to calcium-related hormones excretion, pancreas-related regulating factors, hormone processes, differentiation and growth of islet β-cell, growth of islet endothelium cell, etc; and f) other, such as regulation of transcript, oxidation-reduction, retina, kidney, and blood vessel.
Similarly, glucose metabolism-related proteins consist of 223 kinds of proteins, involving total 54 kinds of protein families, namely 20 common families, 27 humanonly families and 7 mouse-only families. The former includes ADIPOR family, AGC Ser/Thr protein kinase family, aldose epimerase family, calcitonin family, galactose-1-phosphate uridylyltransferase type 1 family, glucagons family, glucose-6-phosphate dehydrogenase family, glycogen phosphorylase family, glycosyl hydrolase 30 family, glycosyltransferase 2 family, glycolsyltransferase 31 family, hexokinase family, inositol monophosphatase family, insulin family, myo-inositol- 1-phosphate synthase family, non-lysosomal glucosylceramidase family, PDK/BCKDK protein kinase family, phosphohexose mutase family, short-chain dehydrogenases/reductases (SDR) family, and sugar epimerase family; the latter contains 5’-AMP-activated protein kinase gamma subunit family, cyclic nucleotide phosphodiesterase family, FAD-dependent glycerol-3-phosphate dehydrogenase family, peptidase S10 family, PEPutilizing enzyme family, TORC family, and UDP-glycosyltransferase family. The rest is mainly 3-hydroxyacyl-CoA dehydrogenase family, 6-phosphogluconate dehydrogenase family, apolipoprotein A2 family, CALCOCO family, class I fructose-bisphosphate aldolase family, dTDP-4-dehydrorhamnose reductase family, FGGY kinase family, glucosamine/galactosamine-6-phosphate isomerase family, glycogen debranching enzyme family, glycogenin family, glycosyl hydrolase 1 family, glycosyl hydrolase 31 family, glycosyl hydrolase 37 family, GPI family, G-protein coupled receptor 2 family, LDLR family, mammalian/fungal glycogen synthase family, nuclear hormone receptor family, nucleotide pyrophosphatase/phosphodiesterase family, phosphofructokinase family, protein-tyrosine phosphatase family, SLC 37A family, Sugar transporter (TC 2.A.1.1) family, transketolase family, Tyr protein kinase family, UDP-glucose/GDP-mannose dehydrogenase family, and UDPGP type 1 family.
In addition, motif is some similar steric shape or topology structure occurring in local region of various proteins, which displays structure conserved regions (SCR) of proteins during evolution. The important motifs of DRPs involve the immunity-related motifs (i.e. Ig-like, Ig_c1, and Ig_MHC), the transport-related (e.g. Sub_transporter, MFS, and Sugar_transpt), the transcriptand adjustment-related (such as Homeobox and Homeodomain-re), the phosphorylation-related (including TYR_phosphatase, Tyr_PP, and Prot_kinase), and the insulin-related (i.e. Ins/IGF/relax and Insulin-like). On the other hand, there are NAD (P)-bd motif and Glc-6-P_DHase motif with a percentage of 5.47% and 2.57%, respectively, in human GMRPs, while G6PDH motif, Glyco_trans_35 motif, Glycg_phsphrylas motif, and Znf_ring motif with a percentage of 9.62%, 6.54%, 6.54%, and 3.85% in turn, exist in mouse GMRPs.
Furthermore, there are 14 kinds of same proteins between DRPs and GMRPs as follows: P01308 (INS_human, insulin), P01325 (INS1_mouse, insulin-1), P01326 (INS2_mouse, insulin-2), P06213 (INSR_human, insulin receptor), P10997 (IAPP_human, islet amyloid polypeptide), P12968 (IAPP_mouse), P22413 (ENPP1_human, ectonucleotide pyrophosphatase/phosphodiesterase 1), P31749 (AKT1_human, RAC-alpha Serine/threonineprotein kinsae, protein kinase B, PKB), P35557 (HXK4_ human, hexokinase D), P37231 (PPARG_human, peroxisome proliferator-activated receptor), P47871 (GLR_ human, glucagon receptor), P43428 (G6PT_rat, glucose- 6-phosphatase), P52789 (HXK2_human, hexokinase type II), Q15848 (ADIPO_human, adiponectin), and Q9BY76 (ANGL4_human, angiopoietin-related protein 4).
3.2. Amino Acid Composition
20 standard kinds of amino acids in 211 known DRPs are ranged by their percentages as follows: Lys, Trp, His, Asn, Leu, Ser, Ala, Gln, Gly, Glu, Pro, Val, Arg, Thr, Asp, Ile, Phe, Tyr, Met, and Cys in turn (Table 1). NIDDM is the most familiar diabetes whose composition of amino acid residues is consistent with that of the whole. Comparison NIDDM with IDDM, the obvious difference is the percentage of Trp that the former is 9.52% and the latter is 1.00%, which is supported by the results that the maladjustment of Trp could lead to type 2 diabetes because aromatic residues have been identified as crucial in formation and stabilization of amyloid structures. Similarly, 20 amino acids in 100 hGMRPs are listed in descending order of their percentages as follows: Leu, Ala, Gly, Val, Ser, Glu, Arg, Pro, Asp, Thr, Lys, Ile, Phe, Gln, Asn, Tyr, His, Met, Cys, and Trp. The amino acids in 123 mGMRPs are arranged by descending as follows: Leu, Val, Gly, Glu, Ala, Ser, Lys, Asp, Arg, Ile, Pro, Thr, Phe, Asn, Gln, Tyr, Met, His, Cys, and Trp.
On the basis of amino acid composition, percentages of histidine (His), asparagine (Asn) and tryptophan (Trp) in DRPs are different from those of GMRPs (Figure 1). The percentages of His and Asn in DRPs are more than those of GMRPs whileas the percentage of Trp in DRPs is less than that of GMRPs. The Trp percentage of type I diabetes is similar to GMRPs. Table 2 also revealed that the content percentages of UGG coding Trp in DRPs, hGMRPs and mGMRPs were 1.74%, 1.57% and 1.46%, respectively, and more than those of UGG in healthy people. This means tryptophan disorders associated with diabetes mellitus by insulin, which is supported by Oxenkrug GF’s results that tryptophan-kynurenine metabolism might be a new target for prevention and treatment of metabolic syndrome, age-associated neuroendocrine disorders [46].
3.3. Synonymous Codon Usage Biases
About codon usage biases, their percentage trendlines of 64 types of codons of various proteins in various species are basically consistent with each other except Ser, Pro, Thr, Ala, Cys, Gly, Arg and Asp residues of mGMRPs (Table 2). Especially Arg contains six codons showing diversity, and four codons, such as CGC, CGG, AGG and AGA, show higher usage biases. The codon composition of those hGMRPs and DRPs are more close to those of normal person from reference [47]. Comparison of RSCU values of 64 types of codons of all various proteins with their percentage reveals that all RSCU are adjacent to each other with the exception of the eight residues of mGMRPs mentioned above. On the other hand, the genes of most DRPs are GC-rich and the average of GC3s is 61.8%. Due to base composition constraints, it is expected that G and/or C containing codons will predominate in the coding regions.

Figure 1. Content percentage of 20 standard amino acids in sixteen types of proteins possessing various biology activities, including diabetes-related proteins and glucose metabolism-related proteins.
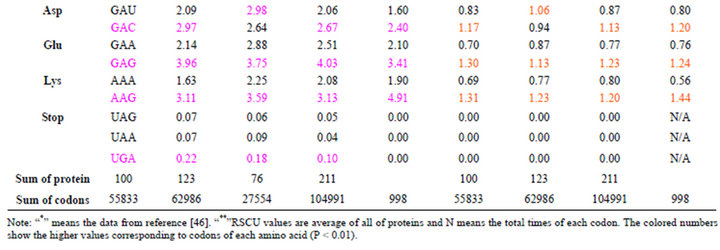

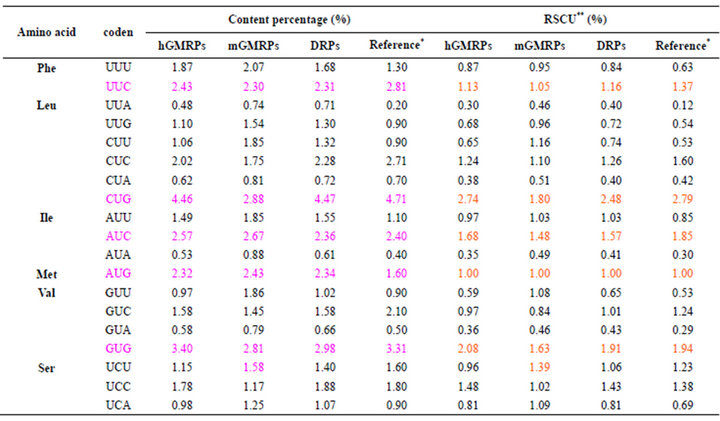
Table 2. Content percentage and RSCU of 64 types of codens in a series of proteins possessing various biology activities.
Although the overall RSCU values in a genome could unveil the codon usage pattern of a whole genome, there may be potential heterogeneity of codon usage among genes in DRPs: GC3s values both range from 24.6% to 92.2% with a mean of 61.8% and a standard deviation (S.D.) of 1.53%; while  values both range from 31.37 to 61.0 with a mean of 48.11 ± 7.06. Similarly, in human GMRPs: GC3s values both range from 23.75% to 87.73% with a mean of 61.21% and a standard deviation (S.D.) of 1.62%; while
values both range from 31.37 to 61.0 with a mean of 48.11 ± 7.06. Similarly, in human GMRPs: GC3s values both range from 23.75% to 87.73% with a mean of 61.21% and a standard deviation (S.D.) of 1.62%; while 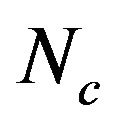 values both range from 34.35 to 61.19 with a mean of 47.94 ± 6.60; in mouse GMRPs: GC3s range from 25.6% to 95.7% with a mean of 62.1% ± 1.76% while
values both range from 34.35 to 61.19 with a mean of 47.94 ± 6.60; in mouse GMRPs: GC3s range from 25.6% to 95.7% with a mean of 62.1% ± 1.76% while  from 31.25 to 61.0 with a mean of 47.25 ± 7.33. The plot of
from 31.25 to 61.0 with a mean of 47.25 ± 7.33. The plot of  against GC3s can be effectively used to explore the heterogeneity (Figure 2), and most genes fall within a restricted cloud, near the area of the axes with GC3s between 0.40 and 0.85 and
against GC3s can be effectively used to explore the heterogeneity (Figure 2), and most genes fall within a restricted cloud, near the area of the axes with GC3s between 0.40 and 0.85 and 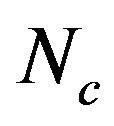 from 40 to 60. The points in the plot are quite spreaded out and the bulk of genes appear to be following a less slope than that of the theoretical curve, which suggests that there are possibly other contributors to the codon usage pattern in DRPs, hGMRPs, and mGMRPs besides the genomic composition. If the codon usage pattern of the genes has some influence other than the GC content, the comparison of the actual distribution of genes with the expected distribution under no selection could be indicative. In other words, if GC3s is the only determinant factor shaping the codon usage pattern, the values of
from 40 to 60. The points in the plot are quite spreaded out and the bulk of genes appear to be following a less slope than that of the theoretical curve, which suggests that there are possibly other contributors to the codon usage pattern in DRPs, hGMRPs, and mGMRPs besides the genomic composition. If the codon usage pattern of the genes has some influence other than the GC content, the comparison of the actual distribution of genes with the expected distribution under no selection could be indicative. In other words, if GC3s is the only determinant factor shaping the codon usage pattern, the values of  would fall on a continuous curve, which represents random codon usage [48].
would fall on a continuous curve, which represents random codon usage [48].
3.3.1. A Correlation between Codon Usage and Gene Expression Level
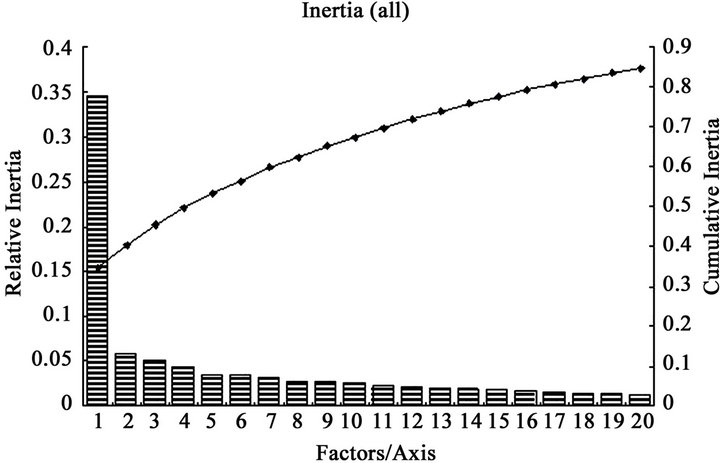
A more extensive and quantitative analysis of the sources of codon usage variation among genes can be achieved using multivariate statistical analysis [49]. In the present work, COA of codon usage in DRP genes which was performed on RSCU values shows that the first axis account for 34.59% of all variation among DRP genes and 43.65% of variation among human DRP genes, whereas the rest of the axes account for no more than 5.06% and 5.74% among all DRP and human DRP genes, respectively (Figure 3). The first principle trend apparently describes a quite large weight of the codon usage variation, which suggests that the major trend in codon bias is as strong as in other species [50]. The plots of genes on the first twenty axes show the relative inertia and cumulative inertia. From the plots, it can be seen that most genes falling within the first axis, demonstrating that there is a single major trend in codon usage in genes related to diabetes, especially human DRPs. According to two-way chi square  contingency test, codon usage differences in the two data sets for diabetes genes at between high and low expresses level was compared
contingency test, codon usage differences in the two data sets for diabetes genes at between high and low expresses level was compared
 (a)
(a)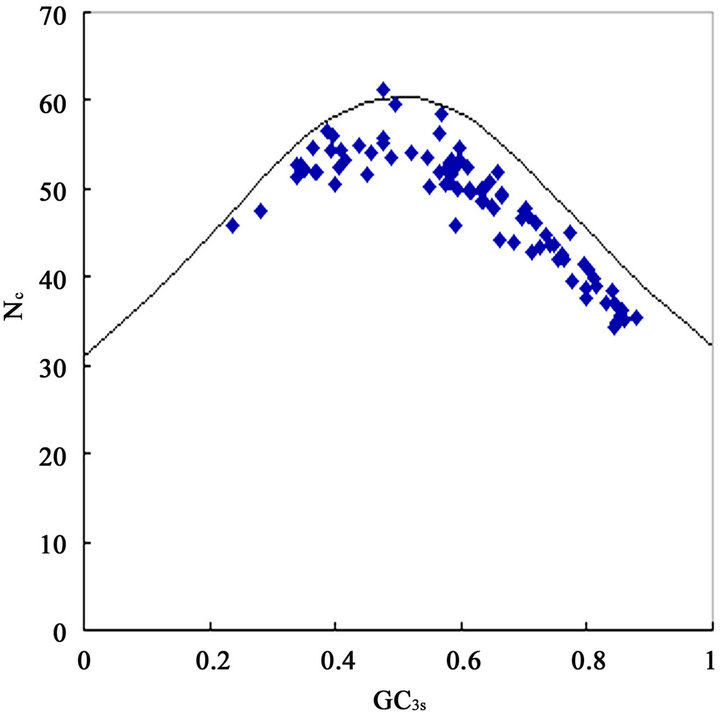 (b)
(b) (c)
(c)
Figure 2. The effective number of codons (Nc) used in a gene plotted against the G+C content at the synonymously variable third position (GC3s) for some genes. a) DRPs; b) human GMRPs; c) mouse GMRPs. The solid curve indicates the expected Nc value if bias is due to GC3s alone when codens are random used; the dispersed points the corresponding actual Nc value at certain GC3s value.
 (a)
(a)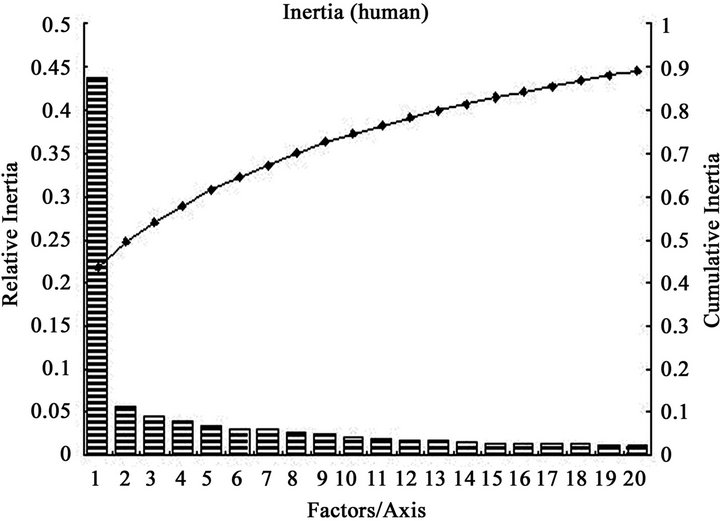 (b)
(b)
Figure 3. The relative inertia and cumulative inertia of codon usage biases plotted against explanation of the variation in the first twenty axes for some genes using correspondence analysis (COA). a) various species DRPs; b) human DRPs. The columns indicate relative inertias, whereas the curve indicates the trend of cumulative inertia.
with the criterion of  to assess significance, respectively. In order to find out whether translational selection is acting on the codon usage in diabetes, a new COA of the RSCU values of the genes located on the leading strand was conducted, since most of the highly expressed sequences are located on that strand, namely the first axis where synonymous codons usages of these genes exist. To investigate the expression trend in codon usage on axis 1, we selected 10 genes from the bottom of axis 1 (the High data set, mainly consisted of genes known to be expressed at high levels), and 10 genes from the other extreme (the Low data set, mainly including genes known to be expressed at low levels) (Figure 3). Statistical analysis showed that the position of gene on the first axis was high positive correlated with the GC3s content (
to assess significance, respectively. In order to find out whether translational selection is acting on the codon usage in diabetes, a new COA of the RSCU values of the genes located on the leading strand was conducted, since most of the highly expressed sequences are located on that strand, namely the first axis where synonymous codons usages of these genes exist. To investigate the expression trend in codon usage on axis 1, we selected 10 genes from the bottom of axis 1 (the High data set, mainly consisted of genes known to be expressed at high levels), and 10 genes from the other extreme (the Low data set, mainly including genes known to be expressed at low levels) (Figure 3). Statistical analysis showed that the position of gene on the first axis was high positive correlated with the GC3s content (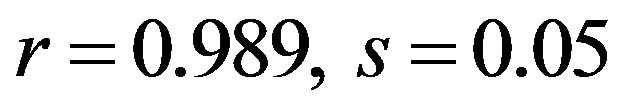 ; human
; human ). Similarly, the position of gene on the first axis was also positive correlated with A3s (
). Similarly, the position of gene on the first axis was also positive correlated with A3s (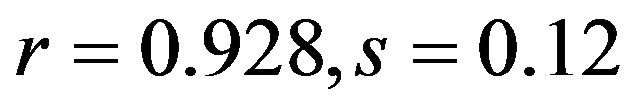 ; human
; human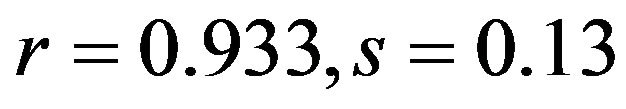 ), T3s (
), T3s (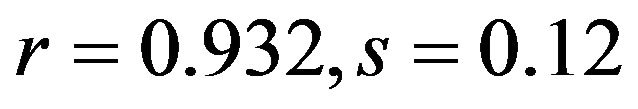 ; human
; human ), G3s (
), G3s (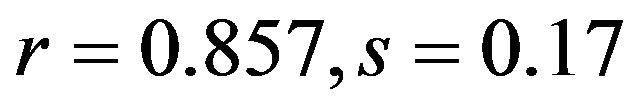 ; human
; human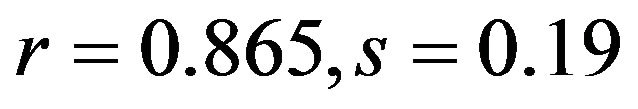 ), and C3s (
), and C3s (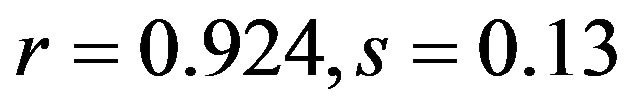 ; human
; human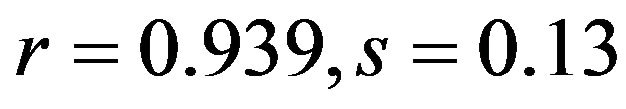 ) content, respectively. Thus, the value of GC3s plays a decisive influence in synonymous codon usage variation of DRPs.
) content, respectively. Thus, the value of GC3s plays a decisive influence in synonymous codon usage variation of DRPs.
Moreover, optimal codons are defined as those occurr significantly more often in highly expressed genes than they do in lowly expressed genes. 27 codons for 18 amino acids in diabetes genes were identified as significantly 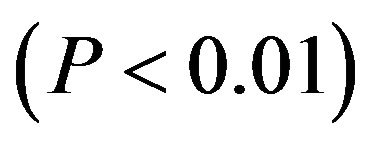 more frequent in the high set (Table 3). These codons are thought to be optimal for translation. 16 codons of them terminate in C (59.3%), 11 in G (40.7%), but no in A/U. The contrast indicates that codon usage biases of DRPs are induced by the composing restriction of those high-expressed GC-rich genes to a great extent. This is consistent with the result that DRP genes generally full contain GC, which hints the compositional restriction plays an important role in boosting GC3s usages of DRP codons. Here, the highest frequent codon are Phe (UUC), Leu (CUG), Ile (AUC), Met (AUG), Val (GUG), Ser (AGC), Tyr (UAC), Pro (CCC), Thr (ACC), Ala (GCC), His (CAC), Trp (UGG), Cys (UGC), Gln (CAG), Asn (AAC), Gly (GGC), Arg (CGG), Asp (GAC), Glu (GAG), Lys (AAG), and STOP (UGA), in turn. Ser and Arg don’t show certain bias but have lowest frequent codon, namely Ser (UCG) and Arg (CGU). COA results also revealed that the bigger RSCU value is, the higher codon usage bias is. The codon with RSCU value greater than 1.5 was Leu (CUG), Val (GUG), Ile (AUC), Thr (ACC), Ser (AGC), and Ala (GCC). Especially, RSCU values of Leu (CUG) and Val (GUG) were significantly higher than their synonymous codons, while the two amino acids had very high codon usage preference in diabetes for further study. Moreover, GC contents are evidently more than AU contents and the usage biases of codons depend on GC contents to a great extent. In high-expression gene, all codons show the terminal of C/G, which is consistent with the usage biases of codons, namely all biased codons with the terminal of C/G. This helps to treat and control diabetes at gene level by site mutant.
more frequent in the high set (Table 3). These codons are thought to be optimal for translation. 16 codons of them terminate in C (59.3%), 11 in G (40.7%), but no in A/U. The contrast indicates that codon usage biases of DRPs are induced by the composing restriction of those high-expressed GC-rich genes to a great extent. This is consistent with the result that DRP genes generally full contain GC, which hints the compositional restriction plays an important role in boosting GC3s usages of DRP codons. Here, the highest frequent codon are Phe (UUC), Leu (CUG), Ile (AUC), Met (AUG), Val (GUG), Ser (AGC), Tyr (UAC), Pro (CCC), Thr (ACC), Ala (GCC), His (CAC), Trp (UGG), Cys (UGC), Gln (CAG), Asn (AAC), Gly (GGC), Arg (CGG), Asp (GAC), Glu (GAG), Lys (AAG), and STOP (UGA), in turn. Ser and Arg don’t show certain bias but have lowest frequent codon, namely Ser (UCG) and Arg (CGU). COA results also revealed that the bigger RSCU value is, the higher codon usage bias is. The codon with RSCU value greater than 1.5 was Leu (CUG), Val (GUG), Ile (AUC), Thr (ACC), Ser (AGC), and Ala (GCC). Especially, RSCU values of Leu (CUG) and Val (GUG) were significantly higher than their synonymous codons, while the two amino acids had very high codon usage preference in diabetes for further study. Moreover, GC contents are evidently more than AU contents and the usage biases of codons depend on GC contents to a great extent. In high-expression gene, all codons show the terminal of C/G, which is consistent with the usage biases of codons, namely all biased codons with the terminal of C/G. This helps to treat and control diabetes at gene level by site mutant.
3.3.2. Effect of Relative Abundance of Dinucleotide and CpG Suppression Based on DRPs
It has been reported that dinucleotide biases can affect codon bias. Using the calculating method of a previous research about CpG under-representation in classical swine fever virus (CSFV) [51], we are supposed to determine whether the relative abundances of dinucleotides in human diabetes related genes affects codon usage. As a conservative criterion told above, for  (or
(or ), the XY pair is considered to be of high (or low) relative abundance compared with a random association of mononucleotides. The frequencies of occurrence for 16 dinucleotides in DRPs were not randomly distributed as follows (Figure 4):
), the XY pair is considered to be of high (or low) relative abundance compared with a random association of mononucleotides. The frequencies of occurrence for 16 dinucleotides in DRPs were not randomly distributed as follows (Figure 4):
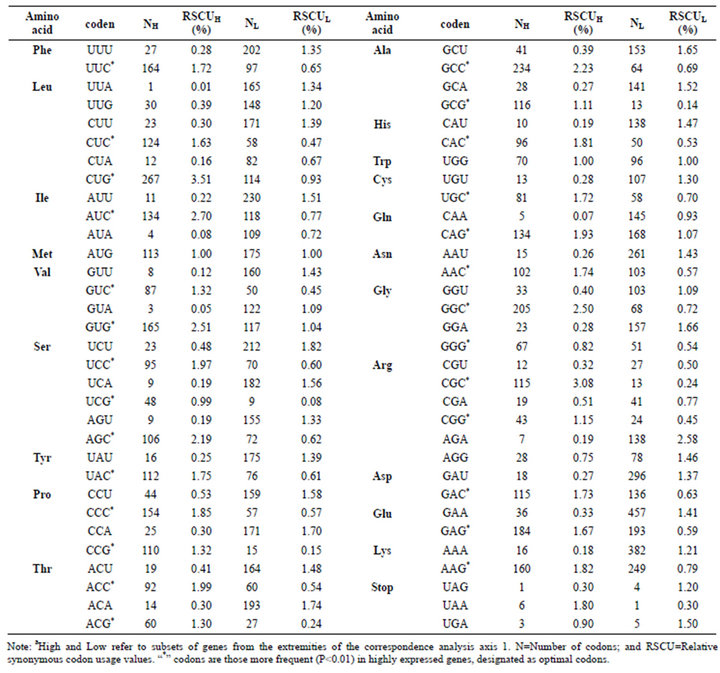
Table 3. Codon usages of high and low expression genes of diabetes-related proteins.a

Figure 4. Relative abundance of the 16 dinucleotides in diabetes related proteins. P < 0.01.
ApA , ApC
, ApC ApG
ApG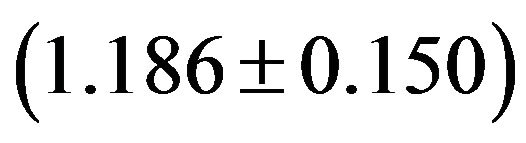 , ApU
, ApU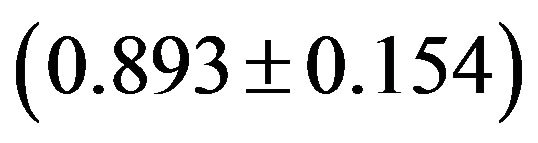 CpA
CpA , CpC
, CpC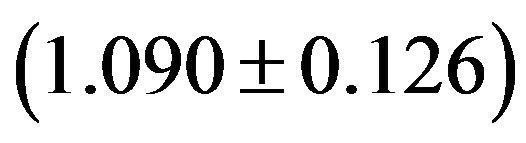 CpG
CpG , CpU
, CpU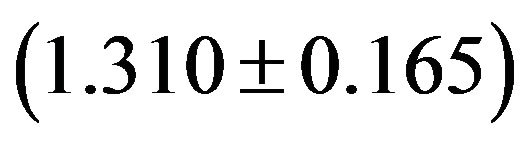 GpA
GpA , GpC
, GpC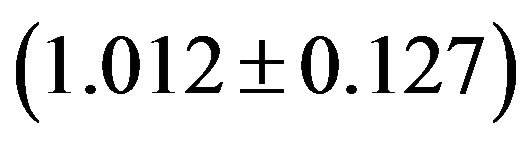 GpG
GpG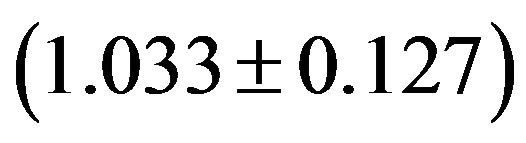 , GpU
, GpU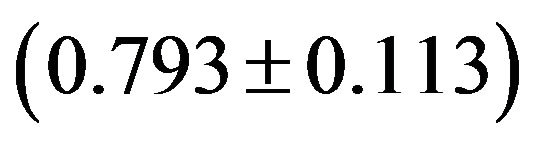 UpA
UpA , UpC
, UpC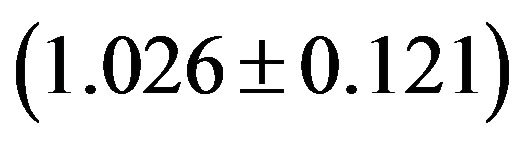 UpG
UpG , UpU
, UpU .
.
The relative abundances of CpG and UpA are lower than the “normal range” compared with a random association of mononucleotides, while those of UpG, CpU and CpA are higher than the “normal range”. These observations indicated that the composition of dinucleotides, which are independent of the overall base composition but still the result of differential mutational pressure, also determines the variation in synonymous codon usage among different diabetes related proteins ORFs.
To explore the possible effects of CpG under-representation on codon usage bias, RSCU values of the eight CpG-rich codons (CCG, GCG, UCG, ACG, CGC, CGG, CGU, and CGA) were analyzed. Six codons, CCG with mean RSCU value of 0.52, GCG (mean 0.45), UCG (mean 0.36), ACG (mean 0.53), CGU (mean 0.47), and CGA (mean 0.62), were more or less suppressed, while only two codons, CGC (mean 1.31) and CGG (mean 1.36), were over-represented. Likewise, to study the possible effects of UpG and CpU over-representation on codon usage bias, five UpG-rich codons (UUG, CUG, GUG, UGU and UGC) and seven CpU-rich codons (CUU, CUC, CUA, CUG, UCU, CCU and GCU) were analyzed. Three UpG containing codons, CUG (mean 2.59), GUG (mean 1.97) and UGC (mean 1.19), were over-used, while two codons, UUG (mean 0.71) and UGU (mean 0.81), were suppressed. CUC (mean 1.25) and CUG (mean 2.59) were over-used, while CUU (mean 0.70) was slightly suppressed. In the rest four CpU containing codons, GCU (mean 0.95), UCU (mean 0.99) and CCU (mean 1.05) were almost equally used while only CUA (mean 0.37) was highly suppressed.
3.4. Protein-Protein Interaction Network
3.4.1. Diabetes-Related Proteins
Searching for the interaction in IntAct database (http://www.ebi.ac.uk/intact/index.jsp) of EMBL-EBI, 36 DRPs directly or indirectly possess interaction between each other and 4 proteins, such as P52789 (HXK2, hexokine type II), P40424 (PBX1, pre-B-cell leukemia transcription factor 1), Q03518 (TAP1, antigen peptide transporter 1), and P35557 (HXK4, hexokine D), can interact with other proteins. The indirect interactions of DRPs are connected by less than three other proteins during building of the interaction network. There are 86 node proteins and 148 links in the network and its mean link degree
Based on the Euclidean distance coefficients (dik) of codon usage biases, cluster analysis (Figure 6) show that the 11 node proteins in DRPs mentioned above were divided into two groups. Group I contains six node proteins, O43707, P20823, P02545, P06213, P62845, and Q16849, while group II includes five proteins, P13987, Q64521, P19357, P06858, and Q14191. Here, a class of O43707 and P20823 hints cytoskeletal protein association with hepatocyte nuclear factor. The former related to anchor with diabetes nephropathy as a good marker protein to examine the relation between O-linked N-acetylglucosamine (O-GlcNAcylation) and diabetic nephropathy [52]. The latter is a hepatocyte nuclear factor as key metabolic regulators of energy homeostasis pathways including GnT-4a (glycosyltransferase) glycosylation and glucose transporter expression [53]. Elevated free fatty acid (FFA) concentrations impair the expression and function of FOXA2 and HNF1A transcription factors sufficiently in beta cells to deplete GnT-4a glycosylation and glucose transporter expression. FFAs induce the activation of one or more beta cell G protein-coupled FFA receptor such as GPR40, while the chronic elevation of FFAs increases mitochondrial oxidation and reactive oxygen species. The resulting dysfunction of beta cells leads to impaired glucose tolerance and failure of GSIS (glucose-stimulated insulin secretion) and further contributes to hyperglycemia, hepatic steatosis and systemic insulin resistance. The molecules that impinge upon this pathway to sustain beta cell GnT-4a activity and glucose transporter expression may suggest new therapeutic targets to achieve effective prevention and treatment of
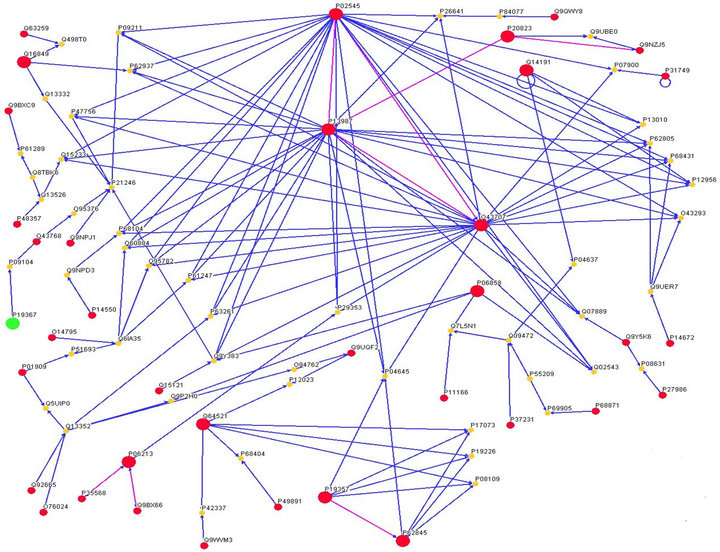
Figure 5. Interaction network of HXK 1 (green dot) and the diabetes-related proteins by Osprey 1.2.0 (91). HXK1 interacts with O43768 (ENSA_HUMAN, Alpha-endosulfine) by P09104 (ENOG_HUMAN, gamma enolase, Neuron-specific enolase). Here, red dots donate the proteins related to diabetes and the bigger ones do hub proteins of network, whilst yellow dots express the proteins whose anti-diabetes activities have not been reported until now, they nevertheless exist in the network. However, the smaller red dots at right corner under the figure don’t exist in the above network although they are related to diabetes. Moreover, the blue lines show the interaction among diabetes-related proteins by the other proteins while the purple lines figure the direct interactions between diabetes-related proteins. And the indirect interactions of the diabetes-related proteins are connected by less than three other proteins during building of the interaction network.
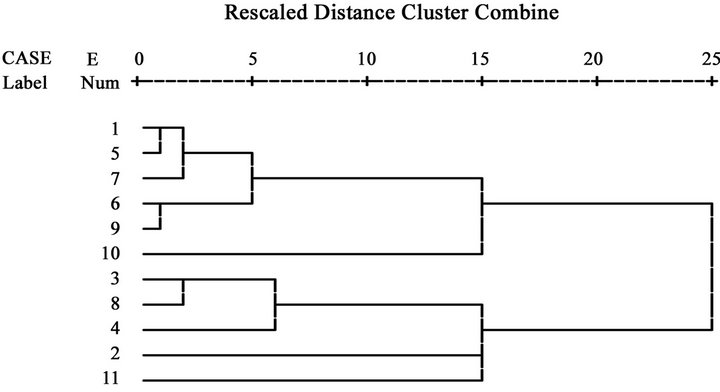
Figure 6. Cluster analysis. Here, number 1~11 displays O43707 (ACTN4_human), P13987 (CD59_human), Q64521 (GPDM_ mouse), P19357 (GTR4_rat), P20823 (HNF1A_human), P02545 (LMNA_human), P06213 (INSR_human, Q05329), P06858 (LIPL_human), P62845 (RS15_rat), Q16849 (PTPRN_human), and Q14191 (WRN_human), in turn.
diabetes [53]. A class of Q64521 and P06858 related to glycerin metabolism, indicating that adipose tissue dysfunction correlate with systemic insulin resistance and type 2 diabetes. Rogers C and co-worker have found that only ErbB1 expression was correlated with insulin sensitivity using ELISA and real-time PCR technology [54]. Additionally, EGF receptor (ErbB1) levels correlated positively with PPARγ and several PPARγ-regulated genes including acyl-coenzyme A synthetase long-chain family member 1 (ACSL1), adiponectin, adipose tissue triacylglycerol lipase (ATGL), diacylglycerol acyl transferase 1 (DGAT1), glycerol-3- phosphate dehydrogenase 1 (GPD1), and lipoprotein lipase (LPL), but negatively with CD36 and fatty acidbinding protein 4 (FABP4). These findings demonstrate a key role for ErbB1 in adipogenesis and suggest that lower ErbB1 protein abundance may lead to adipose tissue dysfunction [54]. A class of P02545 and P62845 hints ribosome protein may play a role in diabetes-related protein synthesis. The former related to Familial partial lipodystrophy (FPLD), an autosomal dominant disorder caused due to missense mutations in the lamin A/C (LMNA) gene encoding nuclear lamina proteins [55]. The latter related to insulinoma [56]. A class of P13987 and Q14191 concerned with diabetes by enzyme signal pathway. P13987 as membrane attack complex (MAC) inhibition factor plays an important role in diabetic macrovascular diseases associated with protein tyrosine kinase signal pathway [57]. Q14191 as a ATPand Mg2+-dependent DNA helicase participates in DNA repair, replication, recombinetion and telomere maintenance, which isolated from Werner’s syndrome, a rare human autosomal recessive segmental progeroid syndrome clinically characterized by atherosclerosis, cancer, osteoporosis, type 2 diabetes mellitus and ocular cataracts [58].
3.4.2. Glucose Metabolism-Related Proteins
There are 330 node proteins and 296 links in the protein-protein network related to GMRPs (Figure 7) and its mean link degree

Figure 7. Protein-protein interaction network of GMRPs by Osprey 1.2.0. Here, red dots donate the proteins related to human glucose metabolism; bule dots don’t exist in the above network.
P31749 (RAC-alpha serine/threonine-protein kinase, protein kinase B, PKB) is one of closely related serine/ threonine-protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase, and which regulate many processes including metabolism, proliferation, cell survival, growth and angiogenesis. This is mediated through serine and/or threonine phosphorylation of a range of downstream substrates. AKT is responsible of the regulation of glucose uptake by mediating insulin-induced translocation of the SLC2A4/GLUT4 glucose transporter to the cell surface. PTP1B is a protein tyrosine phosphatase that negatively regulates insulin sensitivity by dephosphorylating the insulin receptor [59]. Phosphorylation of PTPN1 by AKT at Ser50 negatively modulates its phosphatase activity creating a positive feedback mechanism for insulin signaling, namely preventing dephosphorylation of the insulin receptor and the attenuation of insulin signaling. AKT phosphorylation of TBC1D4 (the Rab GTPase-activating protein, 160-kDa Akt substrate) triggers GLUT4 translocation and the binding of this effector to inhibitory 14-3-3 proteins, which is required for insulin-stimulated glucose transport [60]. AKT regulates also the storage of glucose in the form of glycogen by phosphorylating GSK3A at Ser21 and GSK3B at Ser9, resulting in inhibition of its kinase activity.
P35568 (insulin receptor substrate 1, IRS1) functions as one of the key downstream signaling molecules in both the insulin receptor and the insulin-like growth factor-1 receptor signaling pathways (IGF-1R). Thus genetic changes in IRS-1 may potentially contribute toward the development of insulin resistance [61]. After autophosphorylation of the insulin receptor, the receptor kinase is activated and phosphorylates IRS-1 and other intracellular substrates. This signaling molecule then acts as a docking protein for multiple Src homology-2 domain (SH2)-containing proteins, including phosphatidylinositol 3-kinase (PI3-k), Grb-2, and SHP2. The G972R polymorphism is found near the C terminus of IRS-1 flanked by two tyrosine phosphorylation consensus sites (EY941MLM and DY989MTM), which are known binding sites for the p85α regulatory subunit of PI 3-kinase. The G972R polymorphism impairs the ability of insulin to stimulate glucose transport, glucose transporter translocation, and glycogen synthesis by affecting the PI3K/ AKT1/GSK3 signaling pathway. The polymorphism at G972R may contribute to the in vivo insulin resistance observed in carriers of this variant. G972R could contribute to the risk for atherosclerotic cardiovascular diseases associated with non-insulin-dependent diabetes mellitus (NIDDM) by producing a cluster of insulin resistance-related metabolic abnormalities [62]. In insulinstimulated human endothelial cells from carriers of the G972R polymorphism, genetic impairment of the IRS1/ PI3K/PDPK1/AKT1 insulin signaling cascade results in impaired insulin-stimulated nitric oxide (NO) release and suggested that this may be a mechanism through which the G972R polymorphism contributes to the genetic predisposition to develop endothelial dysfunction and cardiovascular disease. The G972R polymorphism not only reduces phosphorylation of the substrate but allows IRS1 to act as an inhibitor of PI3K, producing global insulin resistance [61].
3.5. Analysis of Transcription Factors at DNA Level Based on DRPs
The genes of 25 DRPs exist in TRANSFAC database but the matching TF binding information of only 15 proteins can be found, including CP2E1_Rat (cytochrome P450, family 2, subfamily E, polypeptide 1; gene name: Cyp2e1; pretein/genomic DNA code: P05182/M20131), GLUC_ human (glucagon, Gcg, P01275/K02808), GTR4_mouse (glucose transporter member 4, Glut4, Slc2a4, P14142/ M29660), GTR4_rat (Glut4, Slc2a4, P19357/L36125), DQB1_human (MHC class II histocompatibility antigen, DQ beta 1 chain, HLA-DQB1, P01920/K02405), HBB_ human (hemoglobin subunit beta, HBB, P68871/U01317), HNF4A_human (hepatocyte nuclear factor 4 alpha, HNF4A, P41235/Z49825), IAPP_human (human islet amyloid polypeptide, IAPP, P10997/M26650), INSR_ human (insulin receptor, INSR, P06213/J03466), INS_ human (insulin, INS, P01308/J00265), LIPL_human (lipoprotein lipase, LPL, P06858/X68111), NR0B2_ human (nuclear receptor subfamily 0 group B member 2, NR0B2, Q15466/AF044316), TNFA_mouse (tumor necrosis factor, Tnf, P06804/Y00467), IGF1_rat (insulinlike growth factor, Igf1, P08025/M84484), and PDGFB_ human (platelet-derived growth factor subunit B, PDGFB, P01127/M19719). Most of gene sequences of 15 proteins contain several TFBS, especially glucagon gene, hemoglobin subunit beta gene, etc. Moreover, a total of 37 TFs interact with DRPs, which cover four kinds of superclasses of transcription factors, such as basic domain (such as AP-1, C/EBP, USF, and NF-E2), beta-Scaffold factors with minor groove contact (i.e. CP2, NF-κB and p50), helix-turn-helix (HTH) (i.e. HNF, POU2F1, IPF, Pax, and RFX), and zinc-coordinating DNA-binding domains (i.g. Sp1, GATA-1, MAZ, and CACCC-binding factor), except the following CBF, BP1, CTF, and CAC-binding protein (Table 4). Sequence alignment shows that BP1 binding site presents a conserved motif of A-Py-AT-[0,1]-TA-Py-Pu-TA-A/T-ATA while the leucine zipper factor family C/EBP distinguishs a nine-nucleotide motif of T-[1]-C-[1]-G/C-[1]- A-Pu-T in Slc2a4 and INSR gene. Here, pyrimidines and purines are abbreviated as Py and Pu, respectively. The transcription factor BP1 is believed to be a repressor of the β-globin (HBB) gene. HNF bind with an adenine-rich motif, A-[5]-AA-[1]-A/T-G/C-[1]-A-[2]-[A/T]6-[2]-[A/T]2-C, in Igf1, Gcg, and Cyp2e1 genes, while Pax interacts with a thymine-rich motif of TT-T/A-TT-Py-AC-Pu-C/G-[1]-TGA and a A/T-rich motif of T-[1]-A-[1]-AT in Gcg and INS genes. Similarly, IPF binding site contains a seven-nucleotide motif of TAATGAC in LAPP and INS genes, while POU2F1 binding site involves in a conserved motif of ATT-[0,1]- T-Pu-CAT in HBB and LPL genes. Interestingly, Cys4 zinc finger family GATA-1 binding site displays two reverse symmetry motifs, such as Py-TATC-A/T and A/T-GATA-Pu, existing in HBB gene.
3.6. Prediction of BAD Complex Proteins
Danial and co-workers indicates an unanticipated role for BAD in integrating pathways of glucose metabolism and apoptosis using proteomics, genetics and physiology [25].
In liver mitochondria, BAD (Q92934) resides in a functional holoenzyme complex together with PKA (Q4P0B3) and PP1 (Q9FSU8) catalytic units, WAVE-1 (Q92558), and GK (P35557). BAD complex includes phosphorylation motif (hexokinase and cAMP_kin), dephosphorylation motif (T_phtase_apaH), apoptosis regulator related motif (Bcl2_BH and BCL2_apoptsis), cyclic nucleotidebinding domain (cNMP_bd), cytoskeleton regulation related motif (WH2_actin_bd), and regulatory subunit portion of type II PKA R-subunit (RIIa). Comparison with the frequency of amino acids in BAD complex, these proteins basically accord with the amino acid composition, especially GK, WAVE1 with lowest Trp contents (Table 1). The Cys contents of BAD and PAK are the lowest but the Tyr content of PP1 is the lowest. Table 5 displays the usage biases of codons of BAD complex. Usage biases of codons reveal that GK are consistent with the results of DRPs while BAD and PKA nearly coincide with the codon biases. But WAVE1 and PP1 depart from the codon usage biases.
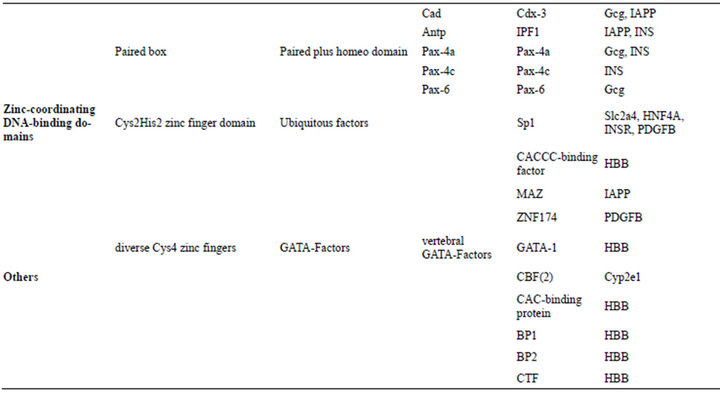
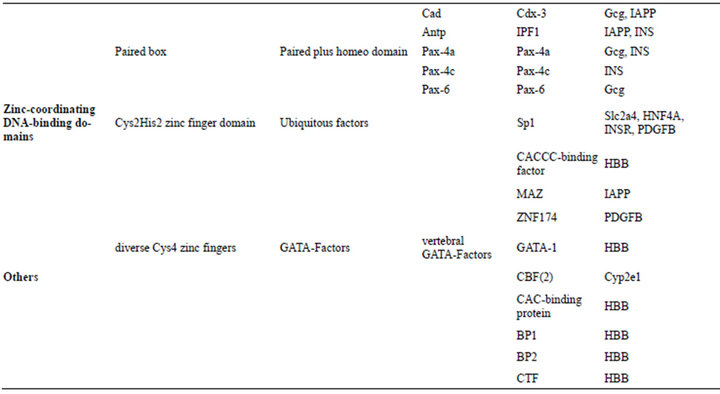
Table 4. Classification of transcription factors.
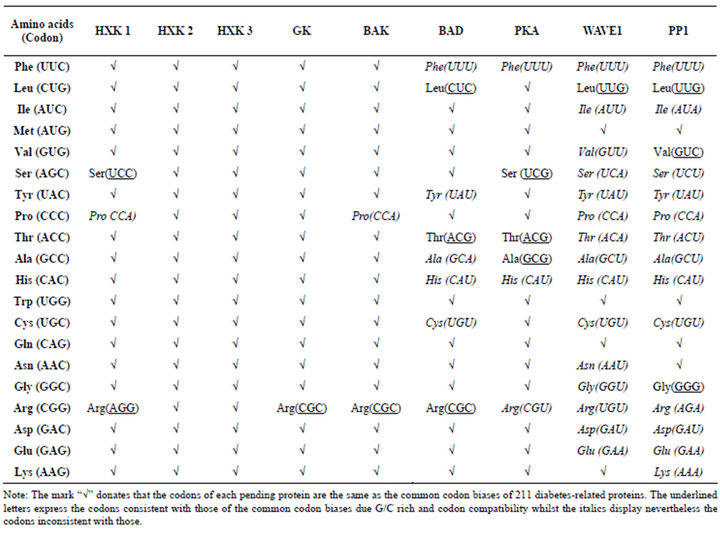
Table 5. Usage biases of Codons of BAD complex.
The interaction between BAD complex and other proteins was searched in IntAct database. The B cell leukemia-2 gene product (Bcl-2) family of proteins can be divided into three different subclasses based on conservation of BCL-2 homology (BH1–4) domains: multidomain anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1, BCL-W, and Bfl-1/A1), multidomain proapoptotic proteins (BAX and BAK), and BH3-only proapoptotic proteins (BID, BAD, BIM, PUMA, NOXA, and NBK/BIK) [44,63]. Notably, BH3-only proteins are not able to kill cells that lack BAX and BAK, indicating that BH3-only proteins function upstream of and are dependent on BAX and BAK [64]. IntAct database shows that BAD interacts with Bcl-2, Bcl-x, Bcl-w, RNA-binding protein EWS, and 14-3-3 protein sigma, respectively. BAK could contact the node protein Lamin-A/C by E1B protein of the following pathway, namely Q16611 (BAK_human, Bcl-2 homologous antagonist/killer)-P03247 (E1BS_ ADE02, E1B protein, small T-antigen)-P02545 (LMNA_ Human: Lamin-A/C).
On the other hand, gamma-enolase might associate the node protein alpha-endosulfine with HXK 1, viz. HXK 1 (P19367, Hexokinase-1)-P09104 (ENOG_human, Gammaenolase)-O43768 (ENSA_human, alpha-endosulfine). Figure 5 shows HXK1 (green dot) interaction with O43768 by P09104. O43768 is the endogenous ligand for sulfonylurea receptor, while endosulfine is the endogenous ligand for the ATP-dependent potassium channels that occupy a key position in the control of insulin release from the pancreatic beta cell by coupling cell polarity to metabolism. By inhibiting sulfonylurea from binding to the receptor, it reduces K+ channel currents and thereby stimulates insulin secretion. P09104, neuron-specific enolase, has neurotrophic and neuroprotective properties on a broad spectrum of central nervous system (CNS) neurons, binding to cultured neocortical neurons in a calcium-dependent manner to promote cell survival. It is normally expressed in neural tissue and involves a freely reversible cytosolic reaction in glycolysis where one molecule each of phosphoenolpyruvate and water react to form one molecule of 2-phosphoglycerate. It is of possible clinical interest as a marker of some types of neuroendocrine and lung tumors. Moreover, HXK2 interacts with ubiquilin-1, while GK interacts with two consensus peptides, EYLSAIVAGPWP of GCKR1 (Glucokinase regulatory protein) and HGMKVWTLPATS of PFKFB1 (F261, 6-phosphofructo-2- kinase/fructose-2,6-biphosphatase 1) by phage display, respectively. The homology of WAVE1 and PKA by alignment of F261 or GCKR is more than 35%, respectively. Our results show that GK might distinguish from PP1 or WAVE1 in a special pattern by binding with the consensus motif [G]-[1]-[K]-[2]-[S/T] or [L/M]-[R/K]-[2]-[T] of PP1 or WAVE1, which is supported by our previous results that GK distinguishing a nine-residue motif EGLKFYTNP (146-154) of WAVE1 construct the BAD-GK-PKAc-PP1c-WAVE1 complex [45]. Additionally, BAD and GK play key roles because of BAD as a substrate for the PKA-PP1 pair and by BH3 domain directly interacting with GK. Apparently, this may be the reason of the BAD complex existing in liver mitochondria, regardless of phosphorylated and dephosphorylated BAD, integrating glycolysis and apoptosis.
3.7. Molecular Modeling of BAD Complex
Knowledge, both from the 3D structures of homologous proteins and from the general analysis of protein structure, is of value in modeling a protein of known sequence but unknown structure. Many models are constructed by homologous modeling on graphics devices. Our group has even built molecular models of HIV-1 co-receptor CCR5 [65], fibrinogen receptor [66], ADP receptor [27], and recombinant SCF-MCSF (Stem Cell Factor-Macrophage Colony Stimulating Factor) fusion proteins and their receptors [67] using the same method, especially integrin GPIIB and purinergic receptor P2Y12 were embodied into PDB database, and their PDB codes were 1UV9 and 1VZ1, respectively. Here we mainly discuss the mechanism of BAD complex using homologous modeling and molecular dynamics. Firstly, human BAD model was built using NMR structure of mouse Bid (PDB code: 1DDB) [68] as a template. Secondly, based on the sequence alignments between BAD and CAPKI and between PKAc and GK, taking X-ray crystal structure of PKAc interaction with its inhibitor (CAPKI) (PDB code: 1CTP) [69] as a template, a complex model of BAD with GK was constructed using Biopolymer module. Similarly, a complex model of PKAc, BAD, and GK was built using a complex model of human BAD with Bcl-xL and PKAc [70] built by us as a template. Thirdly, based on the structure of PP1c-MYPT1 (myosin phasphatase targeting subunit 1) complex (PDB code: 1S70) [71], a structural model of PP1c-GK complex was constructed using Biopolymer module. Combined with BAD complex with GK and PKAc, a complex model of PP1c, BAD, GK, and PKAc was built. Finally, the fourmember complex by interaction with the dimerization domain of PKA regulatory subunit could also connect with WAVE1 to construct the nine-member complex, namely (BAD-GK-PKAc-PP1c)2-WAVE1 complex.
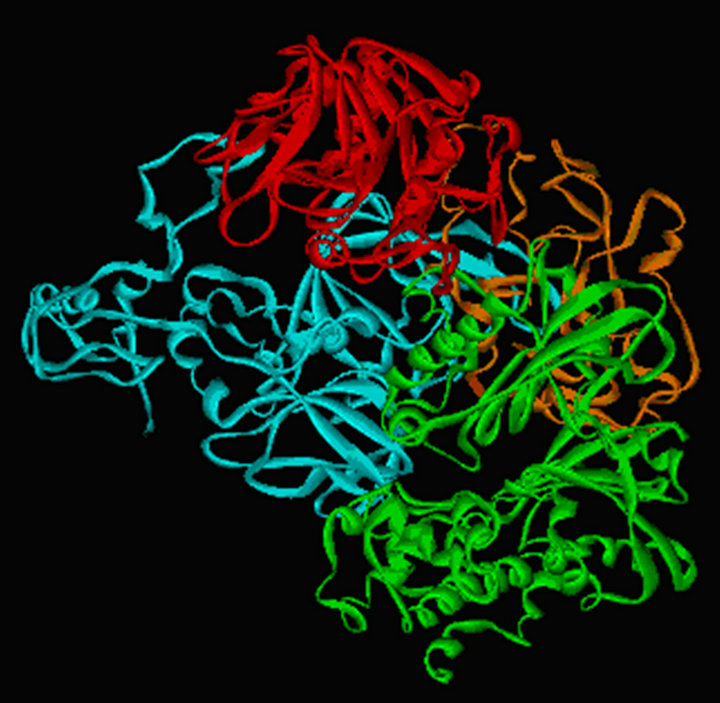
The survey for their interactions in the complex with human BAD and GK focuses on a helical region (98 to 126 residues) of BAD and a shallow pocket at surface of GK (Figure 8). The direct interatomic contacts are made between BAD and GK by van der Waals contacts, hydrophobic interaction, electrostatic interaction, hydrogen bond, and salt bridge, respectively. Residues in contact are congregated in five scattered regions of human BAD: Gln12-Ser16 (loop, pink), Ala27 (loop, pink), Leu44- Glu53 (turn, yellow), Arg98-Lys126 (BH3 domain, red), and Gln152-Ser167 (loop and helix, green); and they spread over six segments of GK: Asp78-Met87 at a turn, two loops (Ser100-Glu112 and Gly170), two helices (Glu128-Phe133 and Arg327-Gln347), and a span from 415 to 446. Besides the interaction between BAD and GK mentioned above, other interactions exist in the complex with BAD, GK, PP1c and PKAc. Especially the amidogen hydrogen of Lys81 and Lys83 of PKAc make hydrogen bridges with the carbonyl oxygen of Leu314 and Pro359 of GK, respectively. Besides, the direct interatomic contacts are made between 7 residues of BAD and 6 residues of PKAc as follows: the active sites of BAD distributing over three segments: two loops (Glu68-Ser71 and Glu84-Glu88) and a turn (Tyr76- Gly79); and the active sites of GK over three parts: a helix (Glu248-Ser252) and two loop regions (Gln242 and Lys254-Arg256). Additionally, the direct interatomic contacts are made between 5 residues of GK and 5 residues of PP1c. Residues in contact are concentrated in three dispersed regions of GK: Gln18 residue at a helix, Gln24-Leu25-Gln26 motif and Glu421 residue at loops; and they are distributed over three segments of PP1c: Asp179-Gln181, Arg188 residue, and Typ216-Glu218 segment.
Our research results reveal that BAD-GK-PKAc-PP1c complex by GK distinguishing a nine-residue motif EGLKFYTNP (146-154) of WAVE1 construct the BADGK-PKAc-PP1c-WAVE1 complex, which is supported by Danial NN’s results [25]. This is a case study of how human BAD was phosphorylated and inactivated at Ser75 by PKAc. Another for dephosphorylated and activated BAD on Ser75, maybe substitute PP1c for PKAc in the complex where PP1c interacts with BAD to result in the dissociation of PKAc and its binding to the regulatory subunit of PKA. Thus, PKA by the dimerization domain of the regulatory subunit could interact with N-terminal peptides of WAVE1, which is supported by Newlon MG’s results [72]. Additionally, we have built a complex model of double PKA with a peptide from WAVE1 [73]. Moreover, PKA by WAVE1 interacts with GK to form (BAD-GK-PKAc-PP1c)2-WAVE1 complex.
4. DISCUSSION
Diabetes is an important endocrine disorder whose pathogenesis is so complex and indefinite that it has never been reported that someone had recovered totally from diabetes. Genomic and proteomic data analysis is essential for understanding the underlying factors that are involved in human disease, such as AD previously reported by ourselves [11]. By extracting significant genomic and proteomic biomarkers in controlled experiments, scientists come closer to understanding how biological mechanisms contribute to human diseases such as neurodegenerative diseases and cardiovascular disorders, and how drug treatments interact with the nervous system. By integrating proteins associated with the pathology of AD into a database and mapped into a protein interaction network, we report a novel approach to predict protein-protein interactions and their potential interaction
 (a)
(a) (b)
(b)
Figure 8. Molecular modeling of BAD complex. a) a four-member complex model of human BAD (orange) with GK (green), PKAc (cyan), and PP1c (red); b) a nine-member complex model of double four-member complex with a peptide of WAVE1, here, BAD colored by orange and blue, respectively, GK (green and purple), PKAc (cyan and gray), and PP1c (red and white).
sites, based on the characteristics of the free-scale network [74]. These networks are constructed with the proteins being the “nodes” and an observation of interaction being an “edge”. The scale free nature of the protein interaction network indicates that a limited number of proteins have a large number of interactions, and function as “hubs” [74]. Thus, protein interaction networks provide an indication of which proteins are more likely to be critical for overall functioning, and to indicate groups of proteins sharing common functions.
Similarly, we analyzed and built protein-protein interaction networks for DRPs and GMRPs with a common node protein P06213 (insulin receptor, ISR). There are some similar node proteins in DRP interaction network (DRPIN) and GMRP interaction network (GMRPIN), such as glucose transporter members P19357 (GLUT4) and P11166 (GLUT1), protein tyrosine phosphatases (PTP) Q16849 (PTPRN) and P18031 (PTP1B, tyrosineprotein phosphatase 1B), and adipose metabolism signal proteins P06858 (LIPL, lipoprotein lipase) and P02652 (APOA2, apolipoprotein A2). Moreover, GMRPIN’s node protein P37231 (PPARγ) could regulate expression of the following genes: ACSL1 (acyl-coenzyme A synthetase long-chain family member 1), adiponectin, ATGL (adipose tissue triacylglycerol lipase), and DGAT1 (diacylglycerol acyl transferase 1), as well as the expression of DRPIN’s node proteins Q64521 (glycerol- 3-phosphate dehydrogenase) and P06858 (lipoprotein lipase) [54]. Our research results reveal that the motifs of these proteins mainly involve in the immunity, transferring, transcription, phosphonation, insulin metabolism and so on. The sharing proteins involve some signal pathway, such as glucagon receptor [75], amylin (islet amyloid polypeptide) receptor [76], insulin- [77], PPARγ- [78], angiopoietin-[79], PC-1/ ENPP1 (Plasma cell membrane glycoprotein-1 or ectonucleotide pyrophosphatase/phosphodieterase)- [80], and adiponectin- [81] mediated signal pathway.
Insulin is traditionally thought to reduce blood glucose levels by stimulating glucose uptake into skeletal muscle and adipose tissues via GLUT4 and by suppressing hepatic glucose production [82]. The insulin receptor (ISR) recruits adaptor protein IRS (insulin receptor substrates) to connect with downstream signalling pathways [77]. Insulin activates the ISR tyrosine kinase, which subsequently tyrosine phosphorylates IRS [83] (Figure 9). In the fed state, dietary carbohydrate increases plasma glucose and promotes insulin secretion from the pancreatic beta cells. In the skeletal muscle, insulin increases glucose transport, permitting glucose entry and glycogen synthesis. In the liver, insulin promotes glycogen synthesis and de novo lipogenesis while also inhibiting gluconeogenesis. In the adipose tissue, insulin suppresses lipolysis and promotes lipogenesis [83]. A family of IRS proteins was defined based on three major common structural elements: amino-terminal PH and PTB domains (PH/PTB) that mediate protein-lipid or protein-protein interactions, mostly carboxy-terminal multiple tyrosine residues (multi-Tyr) that serve as binding sites for proteins that contain one or more SH2 domains, and serine/ threonine-rich regions (Ser/Thr-yich) which may be recognized by negative regulators of insulin action [77]. Glucagon, a major insulin counterregulatory hormone, binds to specific Gs protein–coupled receptors (GPCRs) to activate glycogenolytic and gluconeogenic pathways via adenylyl cyclase and phospholipase C (PLC) signaling pathways leading to increased intracellular cAMP and [Ca2+]i, causing blood glucose levels to increase [75]. Inappropriate increases in serum glucagon play a critical role in the development of insulin resistance and target organ damage in type 2 diabetes. Xiao C et al. have demonstrated that glucagon activated specific receptors to increase rat mesangial cell proliferation by stimulating MAPK ERK1/2 phosphorylation and that glucagon-induced ERK 1/2 phosphorylation requires receptor-mediated activation of cAMP-dependent PKA and PLC/ [Ca2+]i-mediated signaling cascades [75]. Hepatic glucagon action increases in response to accelerated metabolic demands and is associated with increased whole body substrate availability, including circulating lipids [84]. The hypothesis that increases in hepatic glucagon action stimulate AMP-activated protein kinase (AMPK) signaling and peroxisome proliferator-activated receptor-α (PPARα) and fibroblast growth factor 21 (FGF21) expression in a manner modulated by fatty acids was tested in vivo. These findings demonstrate that glucagon exerts a critical regulatory role in liver to stimulate pathways linked to lipid metabolism in vivo and shows for the first time that effects of glucagon on PPARα and FGF21 expression are amplified by a physiological increase in circulating lipids [84]. Moreover, accumulating evidence indicates a role for metabolic dysfunction in the pathogenesis of AD, with the evidence of insulin resistance in the AD brain [85]. Thus, insulin-based therapies, especially targeting insulin signaling, have emerged as potential strategies to slow cognitive decline and to improve cognitive function in AD.
PC-1/ENPP1 has been shown to inhibit insulin signaling by inhibition of insulin-stimulated ISR tyrosine phosphorylation in cultured HEK293 cells in vitro and in transgenic mice in vivo when overexpressed [86]. Moreover, knockdown of ENPP1 with siRNA significantly increases insulin-stimulated AKT phosphorylation in HuH7 human hepatoma cells, supporting the proposition that ENPP1 inhibition is a potential therapeutic approach for the treatment of type 2 diabetes [79]. It is reported that disruption of Angiopoietin-1 (Ang-1)/Tie-2 signaling pathway by PTP contributes to the diabetes-associ-
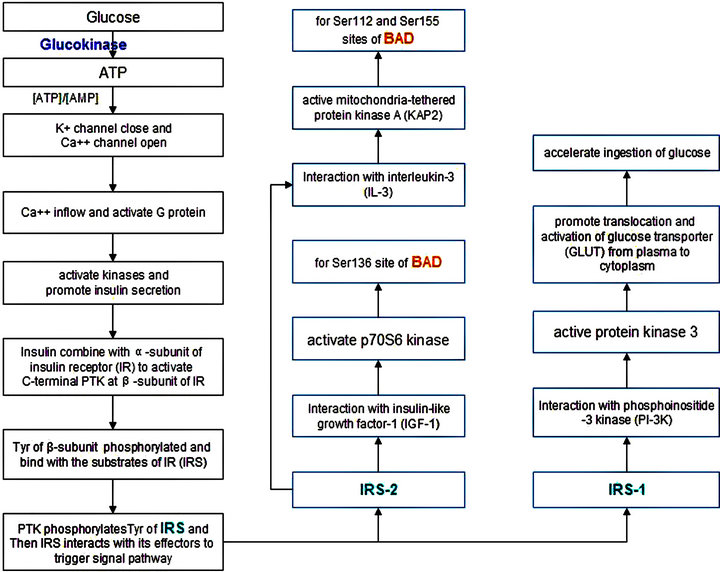
Figure 9. Potential signal pathway between glucose and BAD.
ated impairment of angiogenesis [78]. Chen JX et al have found that protein tyrosine phosphatase-1 (SHP-1) bond to Tie-2 receptor and stimulation with Ang-1 led to SHP- 1 dissociation from Tie-2 in mouse heart microvascular endothelial cell (MHMEC). Exposure of MHMEC to high glucose blunted Ang-1-mediated SHP-1/Tie-2 dissociation while treatment with PTP inhibitors restored Ang-1-induced AKT/eNOS phosphorylation and angiogenesis. Upregulation of Ang-1/Tie-2 signaling by targeting SHP-1 should be considered as a new therapeutic strategy for the treatment of diabetes-associated impairment of angiogenesis [78]. Similarly, microvessels isolated from the brains of AD patients express a large number of angiogenic proteins, such as hypoxia inducible factor 1-alpha (HIF-1α), angiogenic proteins, angiopoietin-2 (Ang-2), and matrix metalloproteinase 2 (MMP2), and survival/apoptotic proteins (Bcl-xL, caspase 3), accompanied by Ang-2, MMP2 and caspase 3 elevated and the anti-apoptotic protein Bcl-xL decreased [87]. Hypoxia is thought to contribute to AD pathogenesis and the cerebro-microvasculature is an important target for the effects of hypoxia in the AD brain.
The prevalence of type 2 diabetes increases rapidly due to the obesity epidemic. Obesity is the primary risk factor for insulin resistance, a hallmark and a driving factor for NIDDM progression [82]. The best-established connection between obesity and insulin resistance is the elevated and/or dysregulated levels of circulating free fatty acids that cause and aggravate insulin resistance, type 2 diabetes, cardiovascular disease and other hazardous metabolic conditions. Karki S and co-worker have indicated that palmitate, a major dietary saturated fatty acid, not only decreases adiponectin expression at the level of transcription by PPARγ and C/EBPα signal pathway and its intracellular metabolism via the acylCoA synthetase 1-mediated pathway, but may also stimulate lysosomal degradation of newly synthesized adiponectin by the intracellular sorting receptor sortilin [80]. INT131 is a potent non-thiazolidinedione (TZD)-selective PPARγ modulator being developed for the treatment of type 2 diabetes [77]. In preclinical studies and a phase II clinical trial, INT131 has been shown to lower glucose levels and ameliorate insulin resistance without typical TZD side effects. Lee DH and co-worker have suggested that a newly developed insulin-sensitizing agent, INT131, normalizes obesity-related defects in insulin action on PI3K signaling in insulin target tissues by a mechanism involved in glycemic control. If these data are confirmed in humans, INT131 could be used for treating type 2 diabetes without loss in bone mass [77]. Moreover, nsulin-sensitive neurons in the brain, especially hypothalamus, also play a key role in the maintenance of normal body weight and glucose homeostasis. Morris DL et al. recently demonstrated that SH2B1, an SH2 and PH domain-containing adaptor molecule, links to human obesity and type 2 diabetes [82]. Genetic deletion of SH2B1 results in severe leptin resistance, insulin resistance, hyperphagia, obesity, and type 2 diabetes in mice. Neuronal SH2B1 serves as an endogenous leptin sensitizer and directly enhances leptin signaling in hypothalamic neurons via a JAK2-dependent mechanism, whereas peripheral SH2B1 directly regulates glucose metabolism by enhancing insulin sensitivity. Furthermore, adipose SH 2B1 mediates insulin stimulation of GLUT4 trafficking and glucose uptake in adipocytes by a novel mechanism. SH2B1 appears to be evolutionally conserved to regulate body weight and glucose metabolism via the brain-adipose tissue axis [82]. Nuclear receptors are attractive targets for the treatment of AD due to their ability to facilitate degradation of Aβ, affect microglial activation and suppress the inflammatory milieu of the brain [88]. Countering neurovascular impairment with PPARγ agonists and acetylcholinesterase inhibitors (AChEi) may improve clinical outcome and delay progression to severe dementia [89]. PPARγ agonists that act by enhancing insulin sensitivity are believed to derive from their ability as nuclear receptor ligands to regulate transcription of a wide variety of oxidative, inflammatory, fibrotic, and neuronal survival genes, although transcription independent effects may also be involved. PPARγ agonists thus have the ability to improve neuronal, glial, and cerebrovascular networks in AD, and consequently to rescue brain hemodynamics [89].
Obviously, more than 60 percent of the glucose streaming through our blood is consumed by the brain. Uncontrolled diabetes can also be indirectly linked to numerous maladies by interfering with cerebral glucose metabolism to ultimately affect brain functioning. Impairment and damage of brain in lack of enough glucose supply will induce Alzheimer's disease (AD), considered as the “brain-type diabetes” or a third form of diabetes [90]. In fact, brain insulin has been shown to regulate both peripheral and central glucose metabolism, neurotransmission, learning, and memory and to be neuroprotective. Insulin signaling in central nervous system (CNS) has emerged as a novel field of research since decreased brain insulin levels and/or signaling were associated to impaired learning, memory, and age-related neurodegenerative diseases [90]. The formation of misfolded proteins, AGEs (advanced glycation end products), mitochondrial disorders, generation of oxidative stress, insulin resistance and abnormal glucose metabolism are main hallmarks of diabetes and AD [1]. Insulin may constitute a promising therapy against diabetesand age-related neurodegenerative disorders, as the potential missing link between diabetes and aging in CNS. By integration of the metabolic, neuromodulatory, and neuroprotective roles of insulin in two age-related pathologies: diabetes and AD, Duarte AI and co-worker have revealed that the two age prevalent diseases, AD and type 2 diabetes, share many common features including the deposition of amyloidogenic proteins, amyloid β protein (Aβ) and amylin (human islet amyloid polypeptide, hIAPP), respectively [90]. Recent evidence suggests that both Aβ and amylin may express their effects through the amylin receptor although the precise mechanisms for this interaction at a cellular level are unknown [91]. Aβ (1-42) and human amylin increase cytosolic cAMP and Ca2+, trigger multiple pathways involving the signal transduction mediators PKA, MAPK, Akt, and cFos. In the presence of human amylin, Aβ(1-42) effects on HEK293- AMY3 expressing cells are occluded suggesting a shared mechanism of action between the two peptides. Amylin receptor antagonist AC253 blocks increases in intracellular Ca(2+), activation of PKA, MAPK, Akt, cFos and cell death that occur upon amylin receptor-3 (AMY3) activation with human amylin, Aβ(1-42), or their coapplication. Fu W et al suggested that AMY3 plays an important role by serving as a receptor target for actions Aβ and thus may represent a novel therapeutic target for development of compounds to treat neurodegenerative conditions such as AD [91]. Additionally, hIAPP as another hallmark of diabetes leads to pancreatic β-cells dysfunction. Neprilysin, a metallopeptidase known to degrade amyloid in AD, decreases islet amyloid deposition by inhibiting hIAPP fibril formation, rather than degrading hIAPP [92]. Targeting the role of neprilysin in IAPP fibril assembly, in addition to IAPP cleavage by other peptidases, may provide a novel approach to reduce and/or prevent islet amyloid deposition in type 2 diabetes. The fact that some drugs developed to treat diabetes also have a positive effect on Alzheimer’s patients, such as metformin, supports the view that there is a common metabolic basis for both disorders [1].
Furthermore, mitochondrial abnormalities like over oxidative stress and impaired calcium homeostasis are reported in AD and diabetes [1]. Although mitochondria are capable of generating ROS/RNS in AD and diabetes by themselves, other sources/mechanisms like misfolded proteins (Aβ and hIAPP), accumulated NFTs (tau plaques), hyperglycemia and AGEs also promote ROS generation [93]. Abundant evidence indicates that direct neurotoxic effects of Aβ involve activation of apoptosis pathways [94]. Apoptosis is regulated by the B cell leukemia-2 gene product (Bcl-2) family (Bcl-2, Bcl-x, Bax, Bak and Bad) and the caspase family (ICH-1 and CPP32), with apoptosis being prevented by Bcl-2 and Bcl-x, and promoted by Bax, Bak, Bad, ICH-1 and CPP32 [95]. Members of the Bcl-2 family are pivotal regulators of the apoptotic process and include both proteins that promote cell survival (e.g., Bcl-2, Bcl-xL, and Bcl-w) and others that antagonize it (e.g., Bax, Bad, Bak, Bik, Bid, BNIP3, and Bim). Previous work suggests that Aβ-induced apoptosis is characterized by decreased expression of the antiapoptotic Bcl-2, Bcl-xL, and/or increased expression of the proapoptotic Bax, Bim. Furthermore, overexpression of antiapoptotic Bcl-2 and Bcl-xL or suppression of Bim can attenuate Aβ toxicity [94]. Kitamura Y et al. have indicated that Bak and Bad but not Bax may contribute to neuronal death in AD [95]. The mechanism of Aβ-induced neuronal apoptosis sequentially involves JNK (c-Jun N-terminal kinase) activation, Bcl-w downregulation, and release of mitochondrial Smac (second mitochondrion-derived activator of caspase, an important precursor event to cell death), followed by cell death [94]. Especially, the mitochondrial BAD complex composed of GK, PP1, PKA, WAVE-1 and BAD in integrating pathways of glucose metabolism and apoptosis [25]. GK activation in the liver results in reduction of glucose concentrations by increasing glycogen synthesis [84]. Dual role of BAD in insulin secretion and beta-cell survival is represented by the BH3 domain, which endows BAD with bifunctional activities to differentially control insulin secretion and apoptosis [86]. Phosphorylation of serine 155 in the BH3 domain instructs BAD to assume a metabolic role by activating GK. The metabolic function of BAD controls insulin secretion, hepatic glucose sensing and overall glucose tolerance. When dephosphorylated, BAD BH3 targets BCL-XL to induce apoptosis. Apoptosis, together with other negative and positive regulators of beta-cell growth, proliferation and survival, contributes to the physiological control of beta-cell mass homeostasis. In addition, exposure to perfluorononanoic acid (PFNA), an increasingly persistent organic pollutant, has been demonstrated to cause hepatotoxicity in animals because PFNA exposure changed the expression levels of several genes related to hepatic glucose metabolism [96]. The protein expression levels of GK, PI3Kca (phosphoinositide-3-kinase, catalytic, alpha polypeptide), phospho-insulin receptor substrate 1 (p-IRS1), p-PI3K, p-AKT and p-PDK1 (phosphoinositide-dependent kinase 1) were decreased in the livers of rats that received 5mg/kg/d PFNA, while that of glucose-6-phosphatase, GLUT2 and p-GSK3β (glycogen synthase kinase-3 beta) were increased, which explains the augment of hepatic glycogen [96]. Hyperoside can activate the PI3K/Akt signaling pathway, resulting in inhibition of the interaction between Bad and Bcl(XL), without effects on the interaction between Bad and Bcl-2, and further inhibited mitochondria-dependent downstream caspase-mediated apoptotic pathway, such as that involving caspase-9, caspase-3, and poly ADP-ribose polymerase (PARP) [24]. Hyperoside could be developed into a clinically valuable treatment for AD and other neuronal degenerative diseases associated with mitochondrial dysfunction.
The constructed model of BAD complex with GK, PKAc, PP1c and WAVE-1 integrating glycolysis and apoptosis using homology modeling method revealed that human BAD is phosphorylated and inactivated on Ser 75 in a BAD-Bcl-xL complex by PKA (targeted to mitochondria through association with WAVE1), resulting in the dissociation of BAD and its binding to GK [45]. Moreover, GK can interact with PP1c and also distinguish WAVE1. On the other hand, BAD is dephosphorylated and activated on Ser75 by PP1c, leading to the separation of PKAc and its binding to the regulatory (R) subunit of PKA which by the dimerization domain of its R subunit connects with WAVE1 linked with GK of the complex. This may be the reason of the complex existing in liver mitochondria, regardless of phosphorylated and dephosphorylated BAD. Additionally, GK like PKA may also prevent Bcl-xL from rebinding to BAD by phosphorylating BAD at Ser 118. The BAD complex model reveals that BAD and GK play key roles because of BAD as a substrate for the PKA-PP1 pair and by BH3 domain directly interacting with GK. This is helpful for our development and research of the molecular mechanism of BAD. Our previously results have shown that PKA, targeted to mitochondria through association with AKAPs, phosphorylated and inactivated human BAD on Ser75 in a BAD–Bcl-xL complex resulting in the dissociation of BAD and its binding to 14-3-3, whereas PKA may also prevent Bcl-xL from rebinding to BAD by phosphorylating BAD at Ser118 of BH3 domain in a BAD-14-3-3E complex [97]. The 14-3-3 family consists of homoand heterodimeric proteins representing a novel type of “adaptor proteins” modulating the interaction between components of signal transduction pathways, especially 14-3-3 isoforms gamma and epsilon (14-3-3E) increased in several brain regions of AD patients [98]. 14-3-3E may reflect impaired signaling and/or apoptosis in the brain as several kinases (such as protein kinase C, Ras, mitogen-activated kinase MEK) involved in signaling and apoptotic factors due to BAD and BAG-1 binding to 14-3-3 motifs [98].
The activation of Wnt signaling and Akt is the maintenance of mitochondrial membrane potential and the regulation of Bcl-xL, mitochondrial energy metabolism, and cytochrome c release [99]. Loss of Wnt signaling also appears to play a role in more widespread neurodegenerative disorders, such as AD. Neurotoxicity of *β- amyloid deposition in hippocampal neurons has been linked to increased levels of GSK-3β* and loss of *β-catenin [99]. Decreased production of Aβ* can occur during the enhancement of protein kinase C (PKC) activity which may be controlled by the Wnt pathway. In addition, PS1 has been shown to downregulate Wnt signaling and interact with β-catenin to promote its turnover [99]. Disheveled (a downstream transducer of Wnt signaling) can promote non-amyloidogenic alpha-secretase cleavage of APP to yield secreted APP (sAPP) and inhibit GSK-3β to reduce the phosphorylation of tau. Thus, disheveled may increase neuronal protection during neurodegenerative disorders through sAPP production and reduction in tau phosphorylation [99]. Wnt and Akt act upon downstream substrates, such as GSK-3β, Bad, and WISP-1 to block the induction of apoptotic cellular injury [99]. EPO decreases the toxic effect of Aβ on microgliain through up-regulation of Wnt1 and activation of the PI3-K/Akt1/mTOR/p70S6K pathway-integration of the PI3-K/Akt1 pathways, mTOR, and mitochondrial related signaling of Bad, Bax, and Bcl-xL. EPO may in turn increases phosphorylation and cytosol trafficking of Bad, reduces the Bad/Bcl-xL complex and increases the Bcl-xL/Bax complex, thus preventing caspase 1 and caspase 3 activation and apoptosis. This may foster development of novel strategies to use cytoprotectants such as EPO for AD and other degenerative disorders [100].
On the other hand, studies of synonymous codon usage in human genes can reveal some of human genome features, and the knowledge of codon usage pattern in diabetes or glucose metabolism related genes may assist mechanism researches of diabetes. Although the causes of codon usage bias are still under discussion, a lot of factors that influence the codon usage patterns are discovered in many researches: the GC content of the genome, especially the GC content at the third position of codons [101]; concentrations of corresponding acceptor tRNA molecules [102]; the functions or hydrophilicity of proteins expressed by a gene [103]; gene expression levels [104]; the amino acids composition of a protein [105]; the structure of proteins [106]; the mutational frequency and the method of mutation [107], etc. All these factors can be summarized as the influence of mutational pressure and translational selection (natural selection). Our results revealed that the bias of the codon usage in these proteins is primarily determined by the GC content and the third codons of the highly expressed genes are all G/C bases except part of mouse GMRPs. The synonymous codon usage biases in DRPs’ genes and human GMRPs’genes are relatively strong, using G or C ended codons, which is consistent with that of human genes (table 2). 12 amino acids prefer codons ended with C, including Asn (AAC), Asp (GAC), His (CAC), Tyr (UAC), Ala (GCC), Pro (CCC), Thr (ACC), Ser (AGC), Cys (UGC), Gly (GGC), Ile (AUC), and Phe (UUC), as well as Arg (CGC) for human genes. Besides Met (AUG) and Trp (UGG), 5 amino acids prefer codons ended with G, such as Gln (CAG), Glu (GAG), Lys (AAG), Leu (CUG), and Val (GUG), as well as Arg (CGG) for DRPs’gene. The compositional constraints (or mutational bias) and gene functions are the cause of the bias and the effect of natural selection is slight. All these analysis rendered some clue in discovering the mystery and curing the human diabetes disease. As analyzed in this study, CpG containing codons are markedly suppressed while UpG containing codons are over-represented. These results can be explained by mutational pressure. The frequencies of occurrence for dinucleotides were not randomly distributed and no dinucleotides were present at the expected frequencies. The codon usage in human diabetes related can be strongly influenced by underlying biases in dinucleotide frequencies. The global methylation pattern is a key feature of the methylation landscape of the human genome. Most of the gene bodies and intergenic sequences are globally methylated with the exception of regions called CpG islands (CGIs). CGIs are often unmethylated, but there are increasing number of CGIs being reported to be methylated in normal tissues [108]. In mammals, the methylated form of cytosine (5-methylcytosine) is hypermutable. 5-methylcytosine is formed by the enzyme DNA methyltransferase operating on a cytosine occurring immediately 5' of a guanine. One effect of methylation is to increase the rate of spontaneous deamination of 5-methylcytosine to form thymine. It has been estimated that transitions in the methylated CpG dinucleotide occur 8 - 16 times faster than non-CpG transitions [109]. The deficit of CpG dinucleotides in human diabetes related gene coding sequences is largely attributed to the hypermutability of methylated CpGs to UpGs (or CpAs in the complementary strand). Since, CpGs in CGIs are often unmethylated and their mutations are rare, the deficiency of CpG can be considered an influence of mutation, which is in accordance with the conclusion that the codon usage of these diabetes related genes are influenced by the cause mutational pressure. Our results also displayed that the synonymous codon usage biases in mouse GMRPs’ genes are ordered by the third codon position as follows: C > G > U > A, which is in accord with Zeeberg’s results [110]. 5 amino acids prefer codons ended with U, including Ala (GCU), Pro (CCU), Ser (UCU), Asp (GAU), and Cys (UGU). 3 amino acids prefer codons ended with A, such as Gly (GGA), Arg (AGA), and Thr (ACA). This appears to be a manifestation of an evolutionary strategy for placement of genes in regions of the genome with a GC content that relates synonymous codon bias and protein folding. Similarly, codon usage biases in AD and other neurodegenerative diseases indicated that GCrich codons are mainly in charge of forming contracted conformation, especially the first nucleotide of codons plays a dominant role in translating the genomic GC signature into protein sequences and structures [11]. Our previous research has revealed that conformation biases of amino acids are present in natural proteins and the corresponding biases of codons show an evident tendency in protein folding [26].
Meanwhile, accumulative evidences suggest that human virus infections can induce diabetes, including diabetes Type 1 and Type 2. The former is caused by progressive destruction of pancreatic β-cells, leading to insulin deficiency. There is extensive epidemiological evidence that viral infections, among other environmental agents, may contribute to the pathogenesis of Type 1 and several enteroviruses can infect human β-cells, resulting in functional impairment or cell death [111]. Likewise, insulin resistance, caused by HCV infection, evolves to type 2 diabetes when superimposed on a high-fat diet and obesity. That means there exists a correlation exists between HCV infection and diabetes [112]. For the virus infections that can induce diabetes, Coxsackievirus serotype B (CVB) and hepatitis C virus (HCV) will be discussed [113]. CVB belongs to the species Human enterovirus-B (HEV-B), a member of the family Picornaviridae. There are six serotypes of CVB and a large number of substrains of these small non-enveloped, single-stranded positive-sense RNA viruses. Up to now, the codon usage research of CVB is lacked.
5. CONCLUSION
Blood glucose concentration is very important, disorder of glucose metabolism will lead to a series of serious diseases, especially diabetes. DRPs represent Lys as the most abundant amino acid while GMRPs show Leu-rich and Trp-poor character. Especially, the percentage of Trp can distinguish between type 1and type 2 diabetes mellitus. Amongst the aromatic entities, Tryptophan was found to possess the most amyloidogenic potential. Therefore, targeting aromatic recognition interfaces by tryptophan could be a useful approach for inhibiting the formation of amyloids and a treatment strategy for several human diseases associated with amyloid formation, including Alzheimer’s disease, Parkinson’s disease, diabetes, etc. Moreover, the usage biases of codons in DRPs’genes depend on GC contents to a great extent, in concord with all codons of the highly expressed genes with the terminal of C/G. The deficit of CpG dinucleotides in diabetes related gene coding sequences is largely attributed to the hypermutability of methylated CpGs to UpGs by the mutational pressure. This helps to treat and control diabetes at gene level by site mutant. Additionally, besides a common node insulin receptor, there are some similar node proteins in DRP interaction network (DRPIN) and GMRP interaction network (GMRPIN), such as glucose transporter members GLUT4 and GLUT1, protein tyrosine phosphatases PTPRN and PTP1B, and adipose metabolism signal proteins LIPL and APOA2. Moreover, GMRPIN’s node protein PPARγ could regulate expression of the following genes: ACSL1, adiponectin, ATGL, and DGAT1, as well as the expression of DRPIN’s node proteins GPDM and LIPL. The sharing proteins involve the following signal pathway, such as glucagon receptor, amylin receptor, insulin, PPARγ, angiopoietin, PC-1/ ENPP1, and adiponectin mediated signal pathway. Moreover, most of gene sequences of 15 node proteins in DRPs’ interaction network involved the binding sites of 37 transcription factors divide into four kinds of superclasses, such as basic domain (such as C/EBP), betaScaffold factors (i.e. NF-κB), helix-turn-helix (i.e. HNF), and zinc-coordinating DNA-binding domains (i.g. GATA- 1). The leucine zipper factor family C/EBP distinguishes a nine-nucleotide motif of T-[1]-C-[1]-G/C-[1]-A-Pu-T, while Cys4 zinc finger family GATA-1 binding site displays two reverse symmetry motifs, such as PyTATCA/T and A/T-GATA-Pu. Especially, BAD complex can integrate pathways of glucose metabolism and apoptosis by BH3 domain of BAD directly interacting with GK because of BAD as a substrate for the PKA-PP1 pair, regardless of phosphorylated and dephosphorylated BAD. In conclusion, we try to systematically analyze the basic parameters, interactions, pathways, and networks of proteins related to diabetes and glucose metabolism on mutil-scale and mutil-level, including the amino acid compositions, codon biases, protein-protein interaction network, transcription factor binding sites, and BAD complex structure. This facilitates the discovery of treatment for diabetes mellitus and is helpful for the prediction and evaluation of potential diabetes targets. They are also conducive to the treatment of Alzheimer’s disease.
6. ACKNOWLEDGEMENTS
This work was supported by a grant from Basic Scientific Research Expenses of Central University (020814360012) and National Key Technology R&D Program (2008BAI51B01).
REFERENCES
- Ahmad, W. (2013) Overlapped metabolic and therapeutic links between alzheimer and diabetes. Molecular Neurobiology, 47, 399-424. doi:10.1007/s12035-012-8352-z
- Cagnin, M., Ozzano, M., Bellio, N., Fiorentino, I., Follo, C. and Isidoro, C. (2012) Dopamine induces apoptosis in APPswe-expressing Neuro2A cells following Pepstatinsensitive proteolysis of APP in acid compartments. Brain Research, 30, 102-107. doi:10.1016/j.brainres.2012.06.025
- Kelliher, M., Fastbom, J., Cowburn, R.F., Bonkale, W., Ohm, T.G., Ravid, R., Sorrentino, V. and O’Neill, C. (1999) Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and β-amyloid pathologies. Neuroscience, 92, 499-513. doi:10.1016/S0306-4522(99)00042-1
- Walsh, D.M., Minogue, A.M., Frigerio, C.S., Fadeeva, J.V., Wasco, W. and Selkoe, D.J. (2007) The APP family of proteins: Similarities and differences. Biochemical Society Transactions, 35, 416-420. doi:10.1042/BST0350416
- Musselman, D.L., Betan, E., Larsen, H. and Phillips, L.S. (2003) Relationship of depression to diabetes types 1 and 2: Epidemiology, biology, and treatment. Biological Psychiatry, 54, 317-329. doi:10.1016/S0006-3223(03)00569-9
- Zinman, B. (1998) Glucose control in type 1 diabetes: From conventional to intensive therapy. Clinical Cornerstone, 1, 29-38. doi:10.1016/S1098-3597(98)90016-3
- Pinero-Pilona, A. and Raskin, P. (2001) Idiopathic type 1 diabetes. Journal of Diabetes and Its Complications, 15, 328-335. doi:10.1016/S1056-8727(01)00172-6
- Gloyn, A.L. (2003) The search for type 2 diabetes genes. Ageing Research Reviews, 2, 111-127. doi:10.1016/S1568-1637(02)00061-2
- Oiknine, R. and Mooradian, A.D. (2003) Drug therapy of diabetes in the elderly. Biomedicine & Pharmacotherapy, 57, 231- 239. doi:10.1016/S0753-3322(03)00052-0
- Wang, X.N., Yang, J., Xu, P.Y., Chen, J., Zhang, D., Sun, Y. and Huang, Z.M. (2011) Construction of drug screening cell model and application to new compounds interfering production and accumulation of beta-amyloid by inhibiting γ-secretase. In: Chang, R.C.-C., Ed., Advanced Understanding of Neurodegenerative Diseases, InTech, Croatia, 169-192.
- Yang, J., Zhu, T.Y., Jiang, Z.X., Chen, C., Wang, Y.L., Zhang, S., Jiang, X.F., Wang, T.T., Wang, L., Xia, W.H., Li, L., Chen, J.J., Wang, J.Y., Wang, W.W. and Zheng, W.J. (2010) Codon usage biases in alzheimer’s disease and other neurodegenerative diseases. Protein & Peptide Letters, 17, 630-645. doi:10.2174/092986610791112666
- Yang, J. and Jiang, X.F. (2010) A novel approach to predict protein-protein interactions related to alzheimer’s disease based on complex network. Protein & Peptide Letters, 17, 356-366. doi:10.2174/092986610790780323
- Riboulet-Chavey, A., Pierron, A., Durand, I., Murdaca, J., Giudicelli, J. and Van Obberghen, E. (2006) Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes, 55, 1289-1299. doi:10.2337/db05-0857
- Karlsson, H.K., Ahlsen, M., Zierath, J.R., Wallberg-Henriksson, H. and Koistinen, H.A. (2006) Insulin signaling and glucose transport in skeletal muscle from first-degree relatives of type 2 diabetic patients. Diabetes, 55, 1283- 1288. doi:10.2337/db05-0853
- Yano, A., Kubota, M., Iguchi, K., Usui, S. and Hirano, K. (2006) Buformin suppresses the expression of glyceraldehyde 3-phosphate dehydrogenase. Bibliographic Information, 29, 1006-1009. doi:10.1248/bpb.29.1006
- Morino, K., Petersen, K.F., Dufour, S., Befroy, D., Frattini, J., Shatzkes, N., Neschen, S., White, M.F., Bilz, S., Sono, S., Pypaert, M. and Shulman, G.I. (2005) Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. The Journal of Clinical Investigation, 115, 3587-3593. doi:10.1172/JCI25151
- Herman, M.A. and Kahn, B.B. (2006) Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. The Journal of Clinical Investigation, 116, 1767-1775. doi:10.1172/JCI29027
- Bouzakr, K., Zachrisson, A., Al-Khalili, L., Zhang, B.B., Koistinen, H.A., Krook, A. and Zierath, J.R. (2006) Sirna-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metabolism, 4, 89-96. doi:10.1016/j.cmet.2006.04.008
- Sharma, A., Chavali, S., Mahajan, A., Tabassum, R., Banerjee, V., Tandon, N. and Bharadwaj, D. (2005) Genetic association, post-translational modification, and protein-protein interactions in Type 2 diabetes mellitus. Molecular Cell Proteomics, 4, 1029-1037. doi:10.1074/mcp.M500024-MCP200
- Marzban, L., Trigo-Gonzalez, G. and Verchere, C.B. (2005) Processing of pro-islet amyloid polypeptide in the constitutive and regulated secretory pathways of beta cells. Molecular Endocrinology, 19, 2154-2163. doi:10.1210/me.2004-0407
- Li, L. and Holscher, C. (2007) Common pathological processes in alzheimer disease and type 2 diabetes: A review. Brain Research Reviews, 56, 384-402. doi:10.1016/j.brainresrev.2007.09.001
- Correia, S.C., Santos, R.X., Perry, G., Zhu, X., Moreira, P.I. and Smith, M.A, (2011) Insulin-resistant brain state: The culprit in sporadic alzheimer’s disease? Ageing Research Reviews, 10, 264-273. doi:10.1016/j.arr.2011.01.001
- Casley, C.S., Land, J.M., Sharpe, M.A., Clark, J.B., Duchen, M.R. and Canevari, L. (2002) β-Amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiology of Disease, 10, 258-267. doi:10.1006/nbdi.2002.0516
- Zeng, K.W., Wang, X.M., Ko, H., Kwon, H.C., Cha, J.W. and Yang, H.O. (2011) Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid β-protein via the PI3K/Akt/Bad/BclXL-regulated mitochondrial apoptotic pathway. European Journal of Pharmacology, 672, 45-55. doi:10.1016/j.ejphar.2011.09.177
- Danial, N.N., Gramm, C.F., Scorrano, L., Zhang, C-Y., Krauss, S., Ranger, A.M., Datta, S.R., Greenberg, M.E., Licklider, L.J., Lowell, B.B., Gygi, S.P. and Korsmeyer, S.J. (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature, 424, 952-956. doi:10.1038/nature01825
- Yang, J., Dong, X.C. and Leng, Y. (2006) Conformation biases of amino acids based on tripeptide microenvironment from PDB database. Journal of Theoretical Biology, 240, 374-384. doi:10.1016/j.jtbi.2005.09.025
- Zhan, C.Y., Yang, J., Dong, X.C. and Wang, Y.L. (2007) Molecular modeling of purinergic receptor P2Y12 and interaction with its antagonists. Journal of Molecular Graphics and Modelling, 26, 20-31. doi:10.1016/j.jmgm.2006.09.006
- Wright, F. (1990) The ‘effective number of codons’ used in a gene. Gene, 87, 23-29. doi:10.1016/0378-1119(90)90491-9
- Sharp, P.M., Tuohy, T. and Mosurski, K. (1986) Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Research, 14, 5125-5143. doi:10.1093/nar/14.13.5125
- Ikemura, T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the E. coli translational system. Journal of Molecular Biology, 151, 389-409. doi:10.1016/0022-2836(81)90003-6
- McInerney, J.O. (1998) GCUA: General codon usage analysis. Bioinformatics, 14, 372-373. doi:10.1093/bioinformatics/14.4.372
- Yang, J., Dong, X.C. and Leng, Y. (2006) Application of FTTP to alpha-helix or beta-strand motifs. Journal of Theoretical Biology, 242, 199-219. doi:10.1016/j.jtbi.2006.02.014
- Sandberg, R., Bränden, C-I., Ernberg, I. and Cöster, J. (2003) Quantifying the species-specificity in genomic signatures, synonymous codon choice, amino acid usage and G+C content. Gene, 311, 35-42. doi:10.1016/S0378-1119(03)00581-X
- Sharp, P.M. and Li, W.H. (1986) Codon usage in regulatory genes in Escherichia coli does not reflect selection for “rare” codons. Nucleic Acids Research, 14, 7737- 7749. doi:10.1093/nar/14.19.7737
- Ghosh, T.C., Gupta, S.K. and Majumdar, S. (2000) Studies on codon usage in Entamoeba histolytica. International Journal for Parasitology, 30, 715-722. doi:10.1016/S0020-7519(00)00042-4
- Zhou, T., Sun, X. and Lu, Z.H. (2006) Synonymous codon usage in environmental chlamydia UWE25 reflects an evolutional divergence from pathogenic chlamydiae. Gene, 368, 117-125. doi:10.1016/j.gene.2005.10.035
- Greenacre, M. (1984) Theory and applications of correspondence analysis. Academic Press, London.
- Musto, H., Romero, H., Zavala, A., Jannari, K. and Bernardi, G. (1999) Synonymous codon choices in the extremely GC-poor genome of Plasmodium falciparum: Compositional constraints and translational selection. Journal of Molecular Evolution, 49, 27-35. doi:10.1007/PL00006531
- Karlin, S. and Burge, C. (1995) Dinucleotide relative abundance extremes: A genomic signature. Trends Genet, 11, 283-290. doi:10.1016/S0168-9525(00)89076-9
- Alfonso, V. and Florencio, P. (2002) Computational methods for the prediction of protein interactions. Current Opinion in Structural Biology, 12, 368-373. doi:10.1016/S0959-440X(02)00333-0
- Eckmann, J.P. and Moses, E. (2002) Curvature of colinks uncovers hidden thematic layers in the world wide web. Proceedings of the National Academy of Sciences of the United State of Amereica, 99, 5825-5829. doi:10.1073/pnas.032093399
- Albert, R., Jeong, H. and Barabási, A-L. (2000) Error and attack tolerance of complex networks. Letters to Nature, 406, 378-382. doi:10.1038/35019019
- Ma, J., Zhou, T., Gu, W., Sun, X. and Lu, Z. (2002) Cluster analysis of the codon use frequency of MHC genes from different species. Biosystems, 65, 199-207. doi:10.1016/S0303-2647(02)00016-3
- Yang, J. (2010) Molecular modeling of human BAD, a pro-apoptotic Bcl-2 family member, integrating glycolysis and apoptosis. Protein & Peptide Letters, 17, 206-220. doi:10.2174/092986610790226003
- Yang, J., Li, J.H., Wang, J. and Zhang, C-Y. (2010) Molecular modeling of BAD complex resided in a mitochondrion integrating glycolysis and apoptosis. Journal of Theoretical Biology, 266, 231-241. doi:10.1016/j.jtbi.2010.06.009
- Oxenkrug, G.F. (2010) Metabolic syndrome, age-associated neuroendocrine disorders, and dysregulation of tryptophan-kynurenine metabolism. Annals of the New York Academy of Sciences, 1199, 1-14. doi:10.1111/j.1749-6632.2009.05356.x
- Grosjean, H. and Fiers, W. (1982) Preferential codon usage in prokaryotic genes: The optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene, 18, 199-209. doi:10.1016/0378-1119(82)90157-3
- Gupta, S.K. and Ghosh, T.C. (2001) Expressivity is the main factor in dictating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene, 273, 63-70. doi:10.1016/S0378-1119(01)00576-5
- Grocock, R.J. and Sharp, P.M. (2002) Synonymous codon usage in Pseudomonas aeruginosa PA01. Gene, 289, 131-139. doi:10.1016/S0378-1119(02)00503-6
- Romero, H., Zavala, A. and Musto, H. (2000) Codon usage in Chlamydia trachomatis is the result of strandspecific mutational biases and a complex pattern of selective forces. Nucleic Acids Research, 28, 2084-2090. doi:10.1093/nar/28.10.2084
- Tao, P., Dai, L., Luo, M., Tang, F., Tien, P. and Pan, Z. (2009) Analysis of synonymous codon usage in classical swine fever virus. Virus Genes, 38, 104-12. doi:10.1007/s11262-008-0296-z
- Akimoto, Y., Miura, Y., Toda, T., Wolfert, M.A., Wells, L., Boons, G.J., Hart, G.W., Endo, T. and Kawakami, H. (2011) Morphological changes in diabetic kidney are associated with increased O-GlcNAcylation of cytoskeletal proteins including α-actinin 4. Clinical Proteomics, 8, 15. doi:10.1186/1559-0275-8-15
- Ohtsubo, K., Chen, M.Z., Olefsky, J.M. and Marth, J.D. (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nature Medicine, 17, 1067-1075. doi:10.1038/nm.2414
- Rogers, C., Moukdar, F., McGee, M.A., Davis, B., Buehrer, B.M., Daniel, K.W., Collins, S., Barakat, H. and Robidoux, J. (2012) EGF receptor (ERBB1) abundance in adipose tissue is reduced in insulin-resistant and type 2 diabetic women. The Journal of Clinical Endocrinology & Metabolism, 97, E329-E340. doi:10.1210/jc.2011-1033
- Monteiro, L.Z. and Foss-Freitas, M.C., Júnior Montenegro, R.M. and Foss, M.C. (2012) Body fat distribution in women with familial partial lipodystrophy caused by mutation in the lamin A/C gene. Indian Journal of Endocrinology Metabolism, 16, 136-138. doi:10.4103/2230-8210.91209
- Kitagawa, M., Takasawa, S., Kikuchi, N., Itoh, T., Teraoka, H., Yamamoto, H. and Okamoto, H. (1991) Rig encodes ribosomal protein S15. The primary structure of mammalian ribosomal protein S15. FEBS Letters, 283, 210-214. doi:10.1016/0014-5793(91)80590-Y
- Ma, X.W., Chang, Z.W., Qin, M.Z., Sun, Y., Huang, H.L., He, Y. (2009) Decreased expression of complement regulatory proteins, CD55 and CD59, on peripheral blood leucocytes in patients with type 2 diabetes and macrovascular diseases. Chinese Medical Journal (English Edition), 122, 2123-2128.
- Lachaud, A.A., Auclair-Vincent, S., Massip, L., AudetWalsh, E., Lebel, M. and Anderson, A. (2010) Werner’s syndrome helicase participates in transcription of phenolbarbital-inducible CYP2B genes in rat and mouse liver. Biochemical Pharmacology, 79, 463-470. doi:10.1016/j.bcp.2009.09.002
- Ravichandran, L.V., Chen, H., Li, Y., Quon, M.J. (2001) Phosphorylation of PTP1B at Ser50 by Akt impairs its ability to dephosphorylate the insulin receptor. Molecular Endocrinology, 15, 1768-1780. doi:10.1210/me.15.10.1768
- Delcommenne, M., Tan, C., Gray, V., Rue, L., Woodgett, J., Dedhar, S. (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proceedings of the National Academy of Sciences of the United State of Amereica, 95, 11211-11216. doi:10.1073/pnas.95.19.11211
- McGettrick, A.J., Feener, E.P., Kahn. C.R. (2005) Human insulin receptor substrate-1 (IRS-1) polymorphism G972R causes IRS-1 to associate with the insulin recaptor and inhibit receptor autophosphorylation. The Journal of Biological Chemistry, 280, 6441-6446. doi:10.1074/jbc.M412300200
- Marini, M.A., Frontoni, S., Mineo, D., Bracaglia, D., Cardellini, M., De Nicolais, P., Baroni, A., D’Alfonso, R., Perna, M., Lauro, D., Federici, M., Gambardella, S., Lauro, R. and Sesti, G. (2003) The Arg972 variant in insulin receptor substrate-1 is associated with an atherogenic profile in offspring of type 2 diabetic patients. The Journal of Clinical Endocrinology & Metabolism, 88, 3368- 3371. doi:10.1210/jc.2002-021716
- Danial, N.N. and Korsmeyer, S.J. (2004) Cell death: Critical control points. Cell, 116, 205-219. doi:10.1016/S0092-8674(04)00046-7
- Zong, W.X., Lindsten, T., Ross, A.J., MacGregor, G.R. and Thompson, C.B. (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Development, 15, 1481-1486. doi:10.1101/gad.897601
- Yang, J., Liu, C.Q. (2000) Molecular modeling on human CCR5 receptors and complex with CD4 antigens and HIV-1 envelope glycoprotein gp120. Acta Pharmacologica Sinica, 21, 29-34.
- Yang, J., Zhan, C.Y., Dong, X.C., Yang, K. and Wang, F.X. (2004) Interaction of human fibrinogen receptor (GPIIb-IIIa) with decorsin. Acta Pharmacologica Sinica, 25, 1096-1104.
- Chen, T., Yang, J., Wang, Y.L., Zhan, C.Y., Zang, Y.H. and Qin, J.C. (2005) Design of recombinant stem cell factor-macrophage colony stimulating factor fusion proteins and their biological activity in vitro. Journal of Computer-Aided Molecular Design, 19, 319-328.
- Strausberg, R.L., Feingold, E.A., Grouse, L.H., Derge, J.G., Klausner, R.D., Collins, F.S, Wagner, L., Shenmen, C,M., Schuler, G.D., Altschul, S.F., Zeeberg, B., Buetow, K.H., Schaefer, C.F., Bhat, N.K., Hopkins, R.F., Jordan, H., Moore,T., Max, S.I., Wang, J. and Marra, M.A. (2002) Generation and initial analysis of more than 15,000 fulllength human and mouse cDNA sequences. Proceedings of the National Academy of Sciences of the United State of Amereica, 99, 16899-16903. doi:10.1073/pnas.242603899
- Zheng, J., Knighton, D.R., Xuong, N.H., Taylor, S.S., Sowadski, J.M. and Ten Eyck, L.F. (1993) Crystal structures of the myristylated catalytic subunit of cAMP-dependent protein kinase reveal open and closed conformations. Protein Science, 2, 1559-1573. doi:10.1002/pro.5560021003
- Yang, J. (2010) Molecular modeling of human BAD, a pro-apoptotic Bcl-2 family member, integrating glycolysis and apoptosis. Protein & Peptide Letters, 17, 206-220. doi:10.2174/092986610790226003
- Terrak, M., Kerff, F., Langsetmo, K., Tao, T., Dominguez, R. (2004) Structural basis of protein phosphatase 1 regulation. Letters to Nature, 429, 780-784. doi:10.1038/nature02582
- Newlon, M.G., Roy, M., Morikis, D., Carr, D.W., Westphal, R., Scott, J.D. and Jennings, P.A. (2001) A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. The EMBO Journal, 20, 1651-1662. doi:10.1093/emboj/20.7.1651
- Yang, J. (2010) Molecular modeling of human hepatocyte PKA (cAMP-dependent protein kinase type-II) and its structure analysis. Protein & Peptide Letters, 17, 646-659. doi:10.2174/092986610791112792
- Yang, J. and Jiang, X.F. (2010) A novel approach to predict protein-protein interactions related to alzheimer’s disease based on complex network. Protein & Peptide Letters, 17, 356-366. doi:10.2174/092986610790780323
- Li, X.C., Carretero, O.A., Shao, Y. and Zhuo, J.L. (2006) Glucagon receptor-mediated extracellular signal-regulated kinase 1/2 phosphorylation in rat mesangial cells: Role of protein kinase a and phospholipase C. Hypertension, 47, 580-585. doi:10.1161/01.HYP.0000197946.81754.0a
- Fu, W., Ruangkittisakul, A., Mactavish, D., Shi, J.Y., Ballanyi, K. and Jhamandas, J.H. (2012) Amyloid beta (Aβ) peptide directly activates Amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. The Journal of Biological Chemistry, 287, 18820-18830. doi:10.1074/jbc.M111.331181
- Boller, S., Joblin, B.A., Xu, L., Item, F., Trüb, T., Boschetti, N., Spinas, G.A. and Niessen, M. (2012) From signal transduction to signal interpretation: An alternative model for the molecular function of insulin receptor substrates. Archives of Physiology Biochemistry, 118, 148- 155. doi:10.3109/13813455.2012.671333
- Lee, D.H., Huang, H., Choi, K., Mantzoros, C. and Kim, Y.B. (2012) Selective PPARγ modulator INT131 normalizes insulin signaling defects and improves bone mass in diet-induced obese mice. Endocrinology and Metabolism: American Journal of Physiology, 302, E552-E560. doi:10.1152/ajpendo.00569.2011
- Chen, J.X., Tuo, Q., Liao, D.F. and Zeng, H. (2012) Inhibition of protein tyrosine phosphatase improves angiogenesis via enhancing Ang-1/Tie-2 signaling in diabetes. Experimental Diabetes Research, 2012, 836759.
- Zhou, H.H., Chin, C.N., Wu, M., Ni, W., Quan, S., Liu, F., Dallas-Yang, Q., Ellsworth, K., Ho, T., Zhang, A., Natasha, T., Li, J., Chapman, K., Strohl, W., Li, C., Wang, I.M., Berger J, An, Z., Zhang, B.B. and Jiang, G. (2009) Suppression of PC-1/ENPP-1 expression improves insulin sensitivity in vitro and in vivo. European Journal of Pharmacology, 616, 346-352. doi:10.1016/j.ejphar.2009.06.057
- Karki, S., Chakrabarti, P., Huang, G., Wang, H., Farmer, S.R. and Kandror, K.V. (2011) The multi-level action of fatty acids on adiponectin production by fat cells. PLoS One, 6, e28146. doi:10.1371/journal.pone.0028146
- Morris, D.L., Cho, K.W. and Rui, L. (2010) Critical role of the src homology 2 (SH2) domain of neuronal SH2B1 in the regulation of body weight and glucose homeostasis in mice. Endocrinology, 151, 3643-3651. doi:10.1210/en.2010-0254
- Samuel, V.T. and Shulman, G.I. (2012) Mechanisms for insulin resistance: Common threads and missing links. Cell, 148, 852-871. doi:10.1016/j.cell.2012.02.017
- Tahrani, A.A., Bailey, C.J., Del Prato, S. and Barnett, A.H. (2011) Management of type 2 diabetes: New and future developments in treatment. Lancet, 378, 182-197. doi:10.1016/S0140-6736(11)60207-9
- Morris, J.K. and Burns, J.M. (2012) Insulin: An emerging treatment for alzheimer’s disease dementia? Current Neurology and Neuroscience Reports, 12, 520-527. doi:10.1007/s11910-012-0297-0
- Danial, N.N. (2009) BAD: Undertaker by night, candyman by day. Oncogene, 27, S53-S70. doi:10.1038/onc.2009.44
- Gramma, P., Tripathy, D., Sanchez, A., Yin, X. and Luo, J. (2011) Brain microvasculature and hypoxia-related proteins in alzheimer’s disease. International Journal of Clinical and Experimental Pathology, 4, 616-627.
- Mandrekar-Colucci, S. and Landreth, G.E. (2011) Nuclear receptors as therapeutic targets for alzheimer’s disease. Expert Opinion on Therapeutic Targets, 15, 1085- 1097. doi:10.1517/14728222.2011.594043
- Nicolakakis, N. and Hamel, E. (2011) Neurovascular function in alzheimer’s disease patients and experimental models. Journal of Cerebral Blood Flow & Metabolism, 31, 1354-1370. doi:10.1038/jcbfm.2011.43
- Duarte, A.I., Moreira, P.I. and Oliveira, C.R. (2012) Insulin in central nervous system: More than just a peripheral hormone. Journal of Aging Research, 2012, 384017.
- Berglund, E.D., Kang, L., Lee-Young, R.S., Hasenour ,C.M., Lustig, D.G., Lynes, S.E., Donahue, E.P., Swift, L.L., Charron, M.J. and Wasserman, D.H. (2010) Glucagon and lipid interactions in the regulation of hepatic AMPK signaling and expression of PPARα and FGF21 transcripts in vivo. Endocrinology and Metabolism: American Journal of Physiology, 299, E607- E614. doi:10.1152/ajpendo.00263.2010
- Zraika, S., Aston-Mourney, K., Marek, P., Hull, R.L., Green, P.S., Udayasankar, J., Subramanian, S.L., Raleigh, D.P. and Kahn, S.E. (2010) Neprilysin impedes islet amyloid formation by inhibition of fibril formation rather than peptide degradation. The Journal of Biological Chemistry, 285, 18177-18183. doi:10.1074/jbc.M109.082032
- Zraika, S., Hull, R.L., Udayasankar, J., Aston-Mourney, K., Subramanian, S.L., Kisilevsky, R., Szarek, W.A. and Kahn, S.E. (2009) Oxidative stress is induced by islet amyloid formation and timedependently mediates amyloid-induced beta cell apoptosis. Diabetologia, 52, 626- 635. doi:10.1007/s00125-008-1255-x
- Yao, M., Nguyen, T.V. and Pike, C.J. (2005) Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. The Journal of Neuroscience, 25, 1149-1158. doi:10.1523/JNEUROSCI.4736-04.2005
- Kitamura, Y., Shimohama, S., Kamoshima, W., Ota, T., Matsuoka, Y., Nomura, Y., Smith, M.A., Perry, G., Whitehouse, P.J. and Taniguchi, T. (1998) Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in alzheimer’s disease. Brain Research, 780, 260-269. doi:10.1016/S0006-8993(97)01202-X
- Fang, X., Gao, G., Xue, H., Zhang, X. and Wang, H. (2012) Exposure of perfluorononanoic acid suppresses the hepatic insulin signal pathway and increases serum glucose in rats. Toxicology, 294, 109-115. doi:10.1016/j.tox.2012.02.008
- Yang, J., Li, J.H., Wang, J. and Zhang, C.Y. (2010) Molecular modeling of BAD complex resided in a mitochondrion integrating glycolysis and apoptosis. Journal of Theoretical Biology, 266, 231-241. doi:10.1016/j.jtbi.2010.06.009
- Fountoulakis, M., Cairns, N. and Lubec, G. (1999) Increased levels of 14-3-3 gamma and epsilon proteins in brain of patients with alzheimer’s disease and down syndrome. Journal of Neural Transmission. Supplementa, 57, 323-325.
- Chong, Z.Z. and Maiese, K. (2004) Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histology and Histopathology, 19, 495-504.
- Shang, Y.C., Chong, Z.Z., Wang, S. and Maiese, K. (2012) Prevention of β-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3- K/mTOR pathway, bad, and Bcl-xL. Aging (Albany NY), 4, 187-201.
- Scapoli, C., Bartolomei, E., De Lorenzi, S., Carrieri, A., Salvatorell, G., Rodriguez-Larralde, A., Barrai, I. (2009) Codon and aminoacid usage patterns in mycobacteria. Journal of Molecular Microbiology and Biotechnology, 17, 53-60. doi:10.1159/000195674
- Higgs, P.G. and Ran, W. (2008) Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Molecular Biology Evolution, 25, 2279-2291. doi:10.1093/molbev/msn173
- Zhang, Q., Zhao, S., Chen, H., Liu, X., Zhang, L. and Li, F. (2009) Analysis of the codon use frequency of AMPK family genes from different species. Molecular Biology Reports, 36, 513-519. doi:10.1007/s11033-007-9208-x
- Zhou, M., Tong, C.F. and Shi, J.S. (2007) A preliminary analysis of synonymous codon usage in poplar species. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao, 3, 285-293.
- Lobry, J.R. and Gautier, C. (1994) Hydrophobicity, expressiveity and aromaticity are the major trends of aminoacid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Research, 22, 3174-3180. doi:10.1093/nar/22.15.3174
- D’Onofrio, G., Ghosh, T.C. and Bernardi, G. (2002) The base composition of the genes is correlated with the secondary structures of the encoded proteins. Gene, 300, 179-187. doi:10.1016/S0378-1119(02)01045-4
- Sueoka, N. (1992) Directional mutation pressure, selective constraints, and genetic equilibria. Journal of Molecular Evolution, 34, 95-114. doi:10.1007/BF00182387
- Fan, S. and Zhang, X. (2009) CpG island methylation pattern in different human tissues and its correlation with gene expression. Biochemical and Biophysical Research Communications, 383, 421-425.
- Gaffney, D.J. and Keightley, P.D. (2008) Effect of the assignment of ancestral CpG state on the estimation of nucleotide substitution rates in mammals. BMC Evolutionary Biology, 8, 265. doi:10.1186/1471-2148-8-265
- Zeeberg, B. (2012) Shannon information theoretic computation of synonymous codon usage biases in coding regions of human and mouse genomes. Genome Research, 12, 944-955. doi:10.1101/gr.213402
- Al-Hello, H., Ylipaasto, P., Smura, T., Rieder, E., Hovi, T. and Roivainen, M. (2009) Amino acids of coxsackie B5 virus are critical for infection of the murine insulinoma cell line,MIN-6. Journal of Medical Virology, 81, 296-304. doi:10.1002/jmv.21391
- Koike, K. and Moriya, K. (2005) Metabolic aspects of hepatitis C viral infection: Steatohepatitis resembling but distinct from NASH. Journal of Gastroenterology, 40, 329-336. doi:10.1007/s00535-005-1586-z
- Hindersson, M., Orn, A., Harris and R.A., Frisk, G. (2004) Strains of Coxsackie virus B4 differed in their ability to induce acute pancreatitis and the responses were negatively correlated to glucose tolerance. Archives of Virology, 149, 1985-2000.
NOTES
*Corresponding author.