Journal of Cancer Therapy
Vol.06 No.04(2015), Article ID:55559,11 pages
10.4236/jct.2015.64035
Effect of Carboxyamidotriazole Orotate, a Modulator of Calcium-Dependent Signaling Pathways, on Advanced Solid Tumors
Matthew H. Taylor1, Alan Sandler1,2, Walter J. Urba3, Antonio M. P. Omuro4, Greg S. Gorman5, Rashida A. Karmali6*
1Knight Cancer Institute, Oregon Health and Science University, Portland, OR, USA
2Genentech, San Francisco, CA, USA
3Earle A Chiles Research Institute, Providence Cancer Center, Portland, OR, USA
4Memorial Sloan-Kettering Cancer Center, New York, NY, USA
5Samford University, Birmingham, AL, USA
6Tactical Therapeutics Inc., New York, NY, USA
Email: *rashida@tacticaltherapeutics.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 March 2015; accepted 9 April 2015; published 13 April 2015
ABSTRACT
Pre-clinical studies suggest carboxyamidotriazole orotate (CTO) demonstrates anti-tumor activity through modulation of multiple tyrosine kinase signaling pathways and interactions with the tumor microenvironment. We determined the safety and tolerability, pharmacokinetic profile, maximum tolerated dose, and recommended Phase II dose of CTO monotherapy in patients with advanced solid tumors. In this first-in-human Phase I clinical trial, eligible patients with advanced solid tumors were enrolled to receive a once-daily dose of CTO following a standard 3 + 3 Phase I design (starting at 50 mg/m2/day) with dose escalations of 30% - 100%. Dose limiting toxicity (DLT) was defined in the first cycle of treatment. Measurable disease and response were defined by RECIST version 1.1. Forty-four patients were evaluable for safety. CTO-related grade 3 toxicities included diarrhea (2.5%), fatigue (5.0%), lymphopenia (2.5%) and transient creatine phosphokinase (CPK) elevation (2.5%). There were no grade 4 or 5 toxicities. Steady state plasma levels of CAI (CTO metabolite) were achieved by day 12 with a half life estimate of 55 hr. Although no objective response rates were observed, nine patients with rapidly progressive and treatment-re- fractory tumors achieved stable disease (SD) durable for up to 14 months. The maximum tolerated dose for CTO alone was 427 mg/m2/day. The dose-limiting toxicity was grade 3 fatigue. CTO is orally bioavailable, safe, well tolerated and produces disease stabilization in a broad range of heavily treated refractory tumors. Combination trials of CTO with other antineoplastic agents are ongoing.
Keywords:
CTO, Safety, Ca-Dependent Signaling Pathways

1. Introduction
Carboxyamidotriazole orotate (CTO) is the orotate salt of 5-amino-1-(4-(4-chlorobenzoyl)-3,5-dichlorobenzyl)- 1,2,3-triazole-4-carboxamide (CAI, L651582, NSC 609974), which inhibits non-voltage dependent calcium signaling, resulting in simultaneous modulation of receptor-mediated, calcium-dependent signal transduction pathways, including EGFR, MEK, RAS, HDAC, HSP90, WNT-β catenin and VEGF [1] -[3] . CAI exhibits antiangiogenic, anti-proliferative, and anti-invasive properties with promising efficacy in pre-clinical studies. CAI has been shown to inhibit microvessel growth and diminish expression of endothelial VEGF in an in vitro model system [4] . This result led to investigation of CAI as an inhibitor of angiogenesis and tumor growth.
Previous clinical trials have established that CAI absorption after oral dosing is limited and unpredictable [5] [6] . Its hydrophobic properties result in poor solubility and absorption, which in turn result in limited bioavailability and efficacy in vivo. Several trials have been conducted using bioavailability-enhancing strategies, but these approaches have not been successful [7] - [9] .
CTO, the orotate salt of CAI synthesized by a novel process which uses different ingredients to synthesize intermediates, has increased bioavailability, faster clearance, and minimal toxicity compared to CAI [10] [11] . These characteristics suggest that lower drug doses may achieve desired plasma concentrations [12] . In in vivo animal studies, the time to peak plasma concentration (Cmax) is much shorter for CTO (12.0 ± 2.5, ng-hr/ml) compared to CAI (14.5 ± 1.5 ng-hr/ml), and AUC for CTO (158,354 ± 10,233 ng-hr/ml) is significantly higher than for CAI (84,234 ± 9756 ng-hr/ml). CTO has demonstrated activity in the tumor microenvironment, inhibiting exosome-induced activation of multiple tyrosine kinase signaling pathways including Akt, ERK, and Bcr-Abl in a chronic myeloid leukemia xenograft model [2] . CAI has been shown to inhibit tumor cell proliferation and angiogenesis in vitro and in vivo [13] - [18] .
The antitumor activity of CTO alone or in combination with temozolomide, 5 fluorouracil, or paclitaxel has been demonstrated in xenograft models. Preclinical studies with xenograft mouse models of human glioblastoma [19] , colon cancer [19] , melanoma [20] , and ovarian cancer [21] were conducted with CTO. Statistically significant decreases in tumor growth were observed in all xenograft models. Plasma CAI levels of 10,530 ± 2703 ng/mL (SD) were recorded after CTO was orally administered, and CTO was demonstrated to cross the blood brain barrier in the glioblastoma xenograft model [19] [20] .
We conducted a Phase I trial to evaluate the safety, tolerability, and efficacy of CTO in patients with advanced, treatment-refractory solid tumors. Secondary endpoints of the trial included pharmacokinetic studies, overall response rate, and determination of the maximum tolerated dose (MTD) of CTO in patients in a broad range of refractory solid tumors.
2. Materials and Methods
2.1. Patient Population
Key Eligibility Criteria
Patients were required to have advanced or metastatic treatment-refractory and histologically or cytologically confirmed solid tumors. Measurable disease and response rate were defined by RECIST. Patients had either received prior anticancer therapy, or were not eligible for any established conventional therapy. Patients were required to have adequate bone marrow, renal, and hepatic function. Female patients of childbearing potential were required to have a negative serum or urine pregnancy test at the time of pre-treatment screening. Recovery from all adverse effects (excluding alopecia) of prior therapies to ≤ grade 1 prior to study entry was required. Patients were ≥ 18 years old with a life expectancy of at least 12 weeks, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 - 2. Prior therapy must have been completed 4 weeks prior to study entry.
The protocol was approved by the appropriate Institutional Review Board/Independent Ethics Committee (IRB/IEC) of participating sites. The investigator obtained written informed consent from each patient, or their authorized representative, who participated in the study.
2.2. Drug Supply and Administration
CTO was provided in 25 and 100 mg capsules (Johnson Matthey Inc, Xcellience LLC, Pharmatek Labs, Inc.). Patients received CTO orally after a two-hour fast and remained fasting for one more hour.
An initial cohort of three patients with advanced or metastatic solid tumors received CTO at a starting dose of 50 mg/m2/day orally. Patients in subsequent cohorts received dose escalations of 30% to 100% depending on the pharmacokinetics of the previous cohort. The criteria for dose escalation specified a 50% increase in dose when a peak plasma concentration of >1500 and ≤3300 ng/mL was achieved in any of the patients treated, and when no CTO-related dose-limiting toxicity (DLT) was observed. CTO was administered on Cycle 1, Day 1 and Days 8-35 (28 doses) to ensure complete clearance of CAI after the first dose. Subsequent cycles comprised 28 days. The patients could receive up to 12 cycles unless unacceptable toxicity, disease progression, or inter current illness required discontinuation. Patients could continue treatment beyond 12 cycles if clinical benefit continued, as long as toxicity remained acceptable. All patients received their designated dose throughout therapy; there was no intra-patient dose escalation.
Safety data included vital signs, laboratory parameters, and adverse events. The severity of adverse events and laboratory abnormalities was reported according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Hematological and blood chemical parameters were assessed at regular intervals. ECG was performed at Screening and Cycle 2, Day 1 to evaluate for QTc prolongation.
Dose-limiting toxicity (DLT) was defined as any of the following adverse events occurring in the first cycle of treatment and considered to be possibly related to study treatment: nausea/vomiting ≥ Grade 3 despite maximal antiemetic therapy, diarrhea ≥ Grade 3 despite maximal antidiarrheal therapy, or any other Grade 3 or 4 non- hematologic toxicity. Hematologic DLTs were defined as Grade 4 neutropenia lasting more than 5 days, Grade 3 neutropenia associated with fever or sepsis, and Grade 4 thrombocytopenia.
Doses were escalated in cohorts of three patients per dose level if no DLTs were observed in the first cycle. If a DLT was observed in one of three patients in a cohort, three more patients were enrolled at the same dose level. The maximum tolerated dose (MTD) was defined as one dose level below the dose at which a DLT was observed in at least two patients. If two of six patients in the cohort had DLTs in Cycle 1, that dose exceeded the MTD, and dose escalation ceased.
2.3. Pharmacokinetics
To determine blood levels of CAI derived from CTO, blood samples were drawn throughout participation in the trial. During Cycle 1, blood samples were drawn pre-dose, 0.5, 1, 2, 4, 8, 24, 32, and 48 hours post first dose and every 24 hours thereafter through Cycle 1 Day 7. Blood samples were drawn pre-dose during Cycle 1 on Days 8, 15, 22, and 29. On Day 36 (i.e., Cycle 2 Day 1), blood samples were drawn pre-dose and after dosing for a period of 8 hours (0.5, 1, 2, 4, and 8 hours). Cycle 1 was 36 days to ensure complete clearance of CAI. In subsequent cycles, blood samples were collected prior to CTO administration on Days 15 and 29 (i.e., Day 1 of the next cycle).
Urine was collected on Cycle 1 Day 1 and Cycle 2 Day 1 in two 12-hour collections, 12 and 12 - 24 hours. All samples were assayed by a validated LC-tandem mass spectrometric method.
2.4. Tumor Response Assessment
Tumor measurements by CT scan were done serially after every two cycles of treatment. Assessment was performed by the treating physician and response determined according to RECIST. The calculated response recorded from the start of the treatment until disease progression/recurrence was categorized as complete response (CR, −100%), partial response (PR, −100% to −30%), stable disease (SD, −30% to +20%) and progressive disease (PD, an increase of 20% or more or new lesions).
2.5. Tumor Genotype
For this exploratory part of the study, archived tissue samples were utilized for mutation screening by Next- Generation Sequencing (NGS). These samples were used to determine tumor genotype. Genomic typing was carried out by Knight Diagnostic Laboratories, Oregon Health & Science University.
3. Results
3.1. Patient Characteristics
Forty-four patients with various advanced solid tumors received CTO doses ranging from 50 mg/m2/day to 555 mg/m2/day (Table 1). All patients were deemed evaluable for safety assessment. There were no treatment-re- lated deaths.
3.2. Toxicity and MTD
Thirty-three (82.5%) patients had AEs that were considered related to treatment, including a total of five (12.5%) developing a grade 3, and none developing grades 4 or 5 AEs. The most common AEs were fatigue, nausea, vomiting, dizziness, and anorexia (Table 2). Treatment-related grade 3 AEs included diarrhea, fatigue, lymphopenia, and transient blood creatine phosphokinase elevation (CPK). One patient receiving 219 mg/m2/day discontinued treatment due to the treatment-related DLT of fatigue.
One patient receiving 555 mg/m2/day had Grade 3 fatigue. One patient receiving 555 mg/m2/day developed a transient grade 3 CPK elevation, which subsequently resolved. As two of the six patients at the 555 mg/m2/day dose level experienced a DLT, the previous lower dose level, 427 mg/m2/day was determined to be the MTD. There were no grades 4 or 5 treatment-related AEs, with no treatment-related deaths.

Table 1. Patient demographics.
Table 2. CTO-related adverse events reported by > 10% of patients overall.
3.3. Pharmacokinetics
3.3.1. Single-Dose Pharmacokinetics
PK data are available for 44 patients. Single-dose administration of 50, 75, 112.5, 146, 219, 285, 427, and 555 mg/m2/day yielded concentration profiles of plasma CTO levels, measured as CAI, through 168 hours after the initial single oral dose.
Pharmacokinetic parameters (maximum concentration Cmax, time to reach maximum concentration Tmax, area under the curve AUC, and half-life t1/2,) for a single dose of CTO in all eight cohorts with doses ranging from 50 to 555 mg/m2 are shown in Table 3.
In general, CAI exposure exhibited a stepwise increase with increasing dose of CTO. Median time-to-peak concentration (Tmax) ranged between 8 - 36 h post-dose (Figure 1). Using concentration data out to 168 h, average t1/2 of CAI across all CTO doses was 55 ± 28 h. After single-dose administration, significant fall-off was observed in the Cmax of CAI with increasing dose. There was a non-statistically significant fall-off in partial AUC with increasing dose (Figure 2).
The AUC and average Cmax values for Cohorts 1-4 are from 0 - 48 hours, and for Cohorts 5-8, 168 hours. The average Cmax and AUC values for Cohorts 1-3 show a dose dependent increase followed by a small but not statistically significant decrease in average Cmax and a plateau in the AUC for Cohort 4 (146 mg/m2/day). A dose dependent increase in both AUC and average Cmax was observed between Cohorts 5 (219 mg/m2/day) and 6 (285 mg/m2/day); however, the differences are not statistically significant. A plateau was observed in the average Cmax beginning with Cohort 6 (285 mg/m2/day) and continuing through Cohort 8 (555 mg/m2/day) while the AUC values continued to show a dose-dependent increase. The average Tmax values did not show any statistically significant dose dependent correlation for Cohorts 1-4 (0 - 48 hours) and varied from 16 hours in Cohort 3 (112.5 mg/m2/day) to 36 hours in Cohort 1 (50 mg/m2/day). For Cohorts 5-8 (0 - 168 hours), the differences were not statistically different within this group or between Cohorts 1-4 and 5-8.
3.3.2. Multiple-Dose Pharmacokinetics
Consistent with half-life estimates for CAI, which predict achievement of steady state in approximately 12 days, the majority of patients reach steady state levels of CAI 12 days after the start of daily dosing with CTO. Maximum observed mean plasma concentrations after 36 days of daily administration ranged from 431 ng/mL at 50 mg/m2 to 3070 ng/mL at 555 mg/m2. Like the single-dose PK data, in general, CAI exposure exhibited a stepwise increase with increasing dose of CTO following multiple doses (Table 4).
As observed after single-dose administration, after multiple doses, fall-off was observed in the Cmax and partial AUC of CAI with increasing dose (Figure 3).
3.4. Clinical Activity
No objective tumor responses were observed at the evaluated dose levels. Nine patients with rapidly progressive and treatment-refractory tumors achieved stable disease that was durable between 3 and 14 months. While none
Table 3. Mean (CV%) PK parameters for CAI following a single-dose of CTO in patients with advanced or metastatic solid tumors.
aMedian value for Tmax; mean ± SD for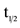 ; bestimates of
; bestimates of
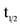 were generally estimated with fewer subjects than group N; cAUClast = 48 h; dAUClast = 168 h.
were generally estimated with fewer subjects than group N; cAUClast = 48 h; dAUClast = 168 h.
Table 4. Mean (CV%) PK parameters for CAI following multiple-dose administration of CTO.
aMedian value for Tmax.
 The CAI plasma concentrations following single-dose administration of CTO is shown on a log-linear scale. While only partial CAI concentration profiles were obtained with CTO doses <219 mg/m2, complete profiles were obtained with CTO doses ≥219 mg/m2 because the PK sampling was taken out to 168 hours. CAI was absorbed slowly at each dose level, with the median time-to-peak concentration (Tmax) ranging between 8 and 36 hours post-dose. Thereafter, plasma concentrations of CAI declined log-linearly over time. Using concentration data taken out to 168 hours, the average half-life (t1⁄2) of CAI across all CTO doses was 55 ± 28 hours.
The CAI plasma concentrations following single-dose administration of CTO is shown on a log-linear scale. While only partial CAI concentration profiles were obtained with CTO doses <219 mg/m2, complete profiles were obtained with CTO doses ≥219 mg/m2 because the PK sampling was taken out to 168 hours. CAI was absorbed slowly at each dose level, with the median time-to-peak concentration (Tmax) ranging between 8 and 36 hours post-dose. Thereafter, plasma concentrations of CAI declined log-linearly over time. Using concentration data taken out to 168 hours, the average half-life (t1⁄2) of CAI across all CTO doses was 55 ± 28 hours.
Figure 1. Single dose pharmacokinetics of CAI.

 A representation of the single-dose exposure relationships for CTO, in which a quadratic equation is used to visualize the non-linear PK relationship.
A representation of the single-dose exposure relationships for CTO, in which a quadratic equation is used to visualize the non-linear PK relationship.
Figure 2. CTO single-dose proportionality relationship.
of the patients achieved a partial response per RECIST criteria, some patients experienced a reduction in tumor mass on CT scan of up to −30% (SD). Four of these patients for whom archived tissues were available for mutation screening had four different genomic mutations consistent with multiple signaling pathways. The genotypes of available archived tissues screened by NGS included mutations in phosphatidyl inositol-3 kinase (PIK3CA, squamous non small cell lung cancer [NSCLC]), proto-oncogene B-Raf (BRAFV600E, colorectal cancer), epidermal growth factor receptor (EGFR mutation, NSCLC adenocarcinoma), and both PIK3CA and neuroblastoma RAS viral oncogene homolog ([NRAS], tonsillar squamous cell carcinoma). At cycle 4 of treatment (285 mg/m2/day), the patient with the BRAFV600E mutation demonstrated a 15% decrease in the longest diameter of the tumor. This patient had received four prior therapies, and the maximum concentration of plasma CAI at 285 mg/m2/day was 679 ng/mL. The tumor histologies and genotypes for which CTO-mediated SD were observed are subtypes for which slowing of tumor progression is unusual in the absence of therapeutic intervention.
For patients exhibiting SD, Figure 4 lists prior therapies administered and depicts their duration of disease

 A representation of the multiple dose exposure relationships for CTO, in which a quadratic equation is used to visualize the non-linear PK relationship.
A representation of the multiple dose exposure relationships for CTO, in which a quadratic equation is used to visualize the non-linear PK relationship.
Figure 3. CTO multiple dose proportionality relationship.
stability on CTO. One patient with renal cell carcinoma achieved stable disease. Time to progression was 12 months at 75 mg/m2/day CTO, and the maximum plasma CAI was 365 ng/ml. A patient with small cell lung cancer achieved stable disease with time to progression of 4 months at 146 mg/m2/day CTO, and the maximum plasma CAI was 2015 ng/ml. Two patients with NSCLC treated with 219 mg/m2/day of CTO had stable disease with time to progression of 11 and 14 months. A patient with tonsillar squamous cell carcinoma achieved stable disease with time to progression of 8 months at 284 mg/m2/day CTO, and the maximum plasma CAI was 2196 ng/mL. Lastly, a patient with ovarian cancer achieved stable disease with time to progression of 5 months at 427 mg/m2/day CTO, and the maximum plasma CAI was 4249 ng/mL.
4. Discussion
In this dose-escalation, first-in-human Phase I study in advanced solid tumors, CTO was well tolerated with doses ranging to 555 mg/m2/day. Treatment-related and dose-limiting AEs included diarrhea, fatigue, lymphopenia, and transient CPK elevation at a dose of 555 mg/m2/day. The MTD of CTO was 427 mg/m2/day (329 mg/m2/day CAI); the recommended Phase II dose is 427 mg/m2/day for single agent CTO, and the starting dose

Figure 4. Patients demonstrating stable disease. Left: Prior therapies administered, number indicates therapies within class; Center: Diagnosis and CTO dose; Right: Time on study. Text within bar indicates genetic mutation. Archived tumor tissue was unavailable for mutation screening for tumors without the genetic mutation information.
is 219 mg/m2/day (169 mg/m2/day CAI) in a dose-escalation trial when given in combination with chemotherapy. In this regard, of significant importance is the fact that MTDs reported for previous trials of CAI occurr at 300, 150 and 75 mg/m2/day for different formulations including micronized and PEG-500 gel cap formulations [5] [6] [8] . In fact, Berlin et al. questioned the MTD of 300 mg/m2/day CAI set by the NCI, and concluded that doses higher than 150 mg/m2/day micronized CAI could result in neurotoxicities, including reversible vision loss [8] .
In the present study, CTO is administered orally and the MTD of 427 mg/m2/day (329 mg/m2/day CAI) is determined based on Grade 3 fatigue, a feature of CAI administration reported previously [6] .
In our study, we were able to use much higher doses of CTO and achieve plasma concentrations of CAI that were previously reported to be associated with dose limiting neurotoxicity and gastrointestinal toxicity in clinical studies of CAI [5] .
CTO exhibited linear pharmacokinetics over the dose range studied with some exceptions, which could be due to inter-patient variability and disease state. Steady-state plasma levels of CAI were reached by 12 days after the start of daily dosing of CTO and remained consistent as previously described for CAI pharmacokinetics [6] . We also measured steady-state levels of CAI after 36 days, and the Cmax ranged from 636 ng/ml at 50 mg/m2/day to 4870 ng/ml at 555 mg/m2/day. These levels are well within the range of CAI concentrations from 800 ng/ml to 8000 ng/ml previously shown to be active in vitro and in vivo [6] .
Although there was a dose-related increase in the steady state levels of CAI, we found that the steady state level at 555 mg/m2/day (Cohort 8) was closer to that observed in Cohort 5 (219 mg/m2/day) than Cohort 7 (427 mg/m2/day). In Cohorts 1-3 and 5-6, we observed dose-dependent increases in Cmax and AUC and, in Cohort 4, a small decrease in Cmax and a plateau in AUC. In Cohorts 6-8, we observed a plateau in Cmax, whereas AUC showed a dose-dependent increase.
The unexpected low steady state level for Cohort 8 is ascribed to inter-patient variability. The large standard deviations in Cmax and AUC leading to non-statistically significant differences are likely due to the various disease states of the patients, contributing to inter-patient variability. The plateauing in the values of these two parameters suggests the potential for saturation of exposure, or limitations on absorption, also due to inter-patient variability.
With respect to inter-patient variability, CTO can be considered as a drug with a moderate-to-high degree of PK variability. Multiple dose linearity performed using steady state PK parameters produced results similar to single-dose linearity. There was a fall-off in AUC with increasing dose; additionally, CTO doses in excess of approximately 200 - 300 mg/m2 resulted in an observable fall-off in CAI bioavailability.
Multiple dosing of CTO achieved substantially higher CAI concentrations than did a single dose, indicating significant drug accumulation over time. This accumulation over time is consistent with a drug that is administered more frequently (every 24 h) than its t1/2 (55 h).
The absence of dose-limiting neurotoxicity and gastrointestinal toxicity even at 555 mg/m2/day (427 mg/m2/day CAI) is noteworthy, as this dose is double the dose at which CAI was associated with neurotoxicity based on previous clinical studies. The association of neurotoxicities with median concentration of CAI previously observed was 2070 ng/mL (400 - 3710) for Grade I episodes and 2710 ng/mL (1160 - 4840) for Grade II/III episodes [7] . In the present study, CAI levels as high as 6524 ng/mL were achieved without any evidence of neurotoxicity. We believe that this higher threshold is due to a safer toxicity profile of CTO compared with CAI, and is achieved because of changes made in the method of CAI synthesis. These changes ensured that ingredients used in the synthesis of different intermediates had a safe toxicity profile [10] [11] . Specifically, the original method to synthesize CAI used sodium azide as an ingredient to synthesize intermediates [10] . The new method substituted sodium azide, a highly toxic agent, with diphenylphosphorylazide [11] , and the CAI produced was reacted with orotic acid to produce CTO, demonstrating a safe toxicity profile both in animals and in patients. Because some of the dose-limiting neurotoxicity and gastrointestinal toxicity observed in clinical studies of CAI manufactured by the old method can be caused by sodium azide, the inference to draw is that the serious toxicities observed with CAI are related to sodium azide impurities.
In this Phase I study, nine patients with aggressive treatment-refractory cancers achieved stable disease on CTO. The plasma concentrations of CAI in these patients ranged from 365 ng/mL to 4249 ng/mL (0.8 µM to 6.7 µM), suggesting a wide range of effective CAI plasma levels. In contrast, previous studies reported target CAI plasma levels to be 2000 and 5000 ng/ml CAI (4.7 and 11.8 µM) [7] .
Single-agent CTO (at doses of 75 - 555 mg/m2/day) showed SD lasting 3-14 months by RECIST. In six heavily treated tumor-burdened patients with rapidly progressive disease, the time to tumor progression was 6+ months on CTO (Figure 4). CTO was well tolerated, and a clinical benefit was observed across tumors within a wide range of oncogenic mutations that were unlikely to demonstrate slowing of tumor progression in the absence of therapeutic intervention. The clinical evaluation of CTO is continuing in combination with other cytotoxic agents and radiation therapy in patients with malignant glioma and glioblastoma.
5. Conclusion
CTO is well tolerated and demonstrates modest clinical activity (SD up to 14 months) in several patients with heavily pre-treated refractory malignancies. Given the mechanism of action of CTO observed in both tumor cells and the tumor microenvironment (inhibiting calcium-mediated signaling transduction pathways), CTO may synergize with other currently available cytotoxic, targeted or immunotherapeutic agents. Clinical trials evaluating combinations of CTO with other antineoplastic agents are currently underway.
Funding
This study was supported by Tactical Therapeutics, Inc.
Acknowledgements
We thank J. Abbott, who provided medical writing services on behalf of Tactical Therapeutics.
References
- Cole, K. and Kohn, E. (1994) Calcium-Mediated Signal Transduction: Biology, Biochemistry, and Therapy. Cancer and Metastasis Reviews, 13, 31-44. http://dx.doi.org/10.1007/BF00690417
- Corrado, C., Flugy, A.M., Taverna, S., Raimondo, S., Guggino, G., Karmali, R., et al. (2012) Carboxyamidotriazole- Orotate Inhibits the Growth of Imatinib-Resistant Chronic Myeloid Leukemia Cells and Modulates Exosomes-Stimu- lated Angiogenesis. PLoS ONE, 7, e42310. http://dx.doi.org/10.1371/journal.pone.0042310
- Karmali, R.A. and Miele, G. (2013) Pharmacodynamic Transcriptional Markers of Carboxyamidotriazole Orotate (CTO) Exposure in Anagen Hair. Molecular Cancer Therapeutics, 12, A233. http://dx.doi.org/10.1158/1535-7163.TARG-13-A233
- Bauer, K.S., Cude, K.J., Dixon, S.C., Kruger, E.A. and Figg, W.D. (2000) Carboxyamido-Triazole Inhibits Angiogenesis by Blocking the Calcium-Mediated Nitric Oxide Synthase-Vascular Endothelial Growth Factor Pathway. The Journal of Pharmacology and Experimental Therapeutics, 292, 31-37.
- Berlin, J., Tutsch, K.D., Hutson, P., Cleary, J., Rago, R.P., Arzoomanian, R.Z., et al. (1997) Phase I Clinical and Pharmacokinetic Study of Oral Carboxyamidotrazole, a Signal Transduction Inhibitor. Journal of Clinical Oncology, 15, 781-789.
- Kohn, E.C., Figg, W.D., Sarosy, G.A., Bauer, K.S., Davis, P.A., Soltis, M.J., et al. (1997) Phase I Trial of Micronized Formulation Carboxyamidotriazole in Patients with Refractory Solid Tumors: Pharmacokinetics, Clinical Outcome, and Comparison of Formulations. Journal of Clinical Oncology, 15, 1985-1993.
- Bauer, K.S., Figg, W.D., Hamilton, J.M., Jones, E.C., Premkumar, A., Steinberg, S.M., et al. (1999) A Pharmacokinetically Guided Phase II Study of Carboxyamido-Triazole in Androgen-Independent Prostate Cancer. Clinical Cancer Research, 5, 2324-2329.
- Berlin, J., Tutsch, K.D., Arzoomanian, R.Z., Alberti, D., Binger, K., Feierabend, C., et al. (2002) Phase I and Pharmacokinetic Study of a Micronized Formulation of Carboxyamidotriazole, a Calcium Signal Transduction Inhibitor: Toxicity, Bioavailability and the Effect of Food. Clinical Cancer Research, 8, 86-94.
- Desai, A., Innocenti, F., Janisch, L., DeMario, M., Shepard, D., Ramirez, J., et al. (2004) A Phase I Trial of Pharmacokinetic Modulation of Carboxyamidotriazole (CAI) with Ketoconazole in Patients with Advanced Cancer. Cancer Chemotherapy and Pharmacology, 54, 377-384. http://dx.doi.org/10.1007/s00280-004-0841-y
- Bochis, R.J., Chabala, J.C. and Fisher H. (1986) 5 Amino or Substituted Amino 1, 2, 3-Triazoles. US Patent 4,590,201.
- Karmali, R.A. (2013) Compositions and Processes for Preparing 5-Amino or Substituted Amino 1, 2, 3-Triazoles and Tri-Azole Orotate Formulations.US Patent 8,377,973.
- Grover, G.J., Kelly, J., Moore, G., Jacoby, H., Karmali, R.A. and Gorman, G.S. (2007) Comparative Pharmacokinetics profile of Carboxyamidotriazole and Carboxyamidotriazole-Orotate. Cancer Therapy, 5, 437-442.
- Kohn, E.C. and Liotta, L.A. (1990) L651582: A Novel Antiproliferative and Antimetastasis Agent. Journal of the National Cancer Institute, 82, 54-60. http://dx.doi.org/10.1093/jnci/82.1.54
- Felder, C.C., Ma, A.L., Liotta, L.A. and Kohn, E.C. (1991) The Antiproliferative and Antimetastatic Compound L651582 Inhibits Muscarinic Acetylcholine Receptor-Stimulated Calcium Influx and Arachidonic Acid Release. The Journal of Pharmacology and Experimental Therapeutics, 257, 967-971.
- Kohn, E.C., Sandeen, M.A. and Liotta, L.A. (1992) In Vivo Efficacy of a Novel Inhibitor of Selected Signal Transduction Pathways Including Calcium, Arachidonate, and Inositol Phosphates. Cancer Research, 52, 3208-3212.
- Kohn, E.C., Felder, C.C., Jacobs, W., Holmes, K.A., Day, A., Freer, R., et al. (1994) Structure-Function Analysis of Signal and Growth Inhibition by Carboxyamido-Triazole, CAI. Cancer Research, 54, 935-942.
- Kohn, E.C., Alessandro, R., Spoonster, J., Wersto, R.P. and Liotta, L.A. (1995) Angiogenesis: Role of Calcium-Me- diated Signal Transduction. Proceedings of the National Academy of Sciences of the United States of America, 92, 1307-1311. http://dx.doi.org/10.1073/pnas.92.5.1307
- Lambert, P.A., Somers, K.D., Kohn, E.C. and Perry, R.R. (1997) Antiproliferative and Antiinvasive Effects of Carbo- xyamidotriazole on Breast Cancer Cell Lines. Surgery, 122, 372-379. http://dx.doi.org/10.1016/S0039-6060(97)90029-5
- Karmali, R.A., Maxuitenko, Y., Gorman, G. and Page, J.G. (2011) Carboxyamidotriazole Orotate and Cytotoxic Chemotherapy Have a Synergistic Effect on Tumor Inhibition in Glioblastoma and Colon Xenograft Mouse Models. Cancer Therapy, 8, 71-80.
- Karmali, R.A., Maxuitenko, Y.Y., Gorman, G.S. and Qu, Z.C. (2012) Combinatorial Treatment with Carboxyamidotriazole-Orotate and Temozolomide in SC-Implanted Human LOX IMVI Melanoma Xenografts. Journal of Solid Tumors, 2, 13-28. http://dx.doi.org/10.5430/jst.v2n5p13
- Karmali, R.A., Maxuitenko, Y. and Gorman, G. (2013) Treatment with Paclitaxel Orotate and Carboxyamidotriazole Orotate in SC-Implanted OVCAR-5 Human Ovarian Tumor Xenografts. Journal of Cancer Therapy, 4, 857-871. http://dx.doi.org/10.4236/jct.2013.44098
NOTES

*Corresponding author.