Journal of Cancer Therapy
Vol.3 No.6(2012), Article ID:25233,8 pages DOI:10.4236/jct.2012.36131
A Single Therapeutic miRNA Rescues the Oncolysis of Androgen Activated Prostate Specific Virus in Androgen Independent Cell Model
![]()
1James Brady Urological Institute, The Johns Hopkins University School of Medicine, Baltimore, USA; 2Department of Urology, Shaikh Zayed Hospital, Lahore, Pakistan; 3Department of Internal Medicine, MedStar Union Memorial Hospital, Baltimore, USA.
Email: *nhoti1@jhmi.edu
Received September 13th, 2012; revised October 15th, 2012; accepted October 26th, 2012
Keywords: Adenovirus; CRAd; miRNA; E1A
ABSTRACT
Prostate specific conditionally replicating adenoviruses (CRAd) are made by placing a tissue specific promoter upstream of one or more of the viral genes required for replication (e.g., E1A, E1B). However, one major problem associated with these vectors is their dependency for androgen receptor (AR), to transactivate the immediate early gene of the virus. This absolute necessity of androgens for prostate specific promoters renders them less useful in patients that have undergone or are currently on total androgen ablation therapy. Therefore, an alternative approach is needed for the existing vectors to be useful in clinical settings. To overcome this problem, we have generated a prostate specific CRAd by introducing a single therapeutic miRNA (miR-34c) that was identified in our library screen to synergize with viral oncolysis. Overexpression of miR-34c from the backbone of virus not only helped in suppressing the proliferation of AR positive cells but also rescued the oncolysis of androgen activated prostate specific virus in androgen independent cell model. To our knowledge this is the first report describing the utility of a single therapeutic miRNA construct to rescue viral oncolysis in advanced androgen resistant AR negative prostate cancer cell line model.
1. Introduction
Metastatic prostate cancers are heterogeneous populations of androgen-dependent and androgen-independent cells [1]. Emerging clinical treatments provide temporary relief with hormone deprivation; however, inevitably the cancer develops androgen independence for which only modest results have been achieved by our current arsenal of chemotherapeutics. This drug resistance is partially due to the unusually slow growth rate of prostate cancers when compared with other cancers. Gene therapy using oncolytic viruses has emerged as an alternative approach to such traditional therapies [2-4]. It infects both quiescent and dividing cells [5], and can be highly tailored to tissues of interest. Although it is still in its infancy, it has shown a safe profile in number of cancer patients and has the potential to be developed into a potent antitumor agent [6-8].
Oncolytic conditionally replicating adenoviruses (CRAds) are made by placing the viral replicative genes (E1A or E1B) in front of a tissue specific promoter to restrict viral replication to the tissue of interest. We have previously developed CRAds, by placing the E1A gene under the control of the Probasin or prostate specific antigen (PSA) promoter and enhancer [4,8,9]. This virus was the first tissue specific conditionally replicating adenoviral vector and was the first to be clinically tested for any organ system [8]. PSA gene regulation, like many genes expressed by the prostate, is controlled by the steroid hormone receptor, androgen receptor (AR) [10,11]. AR is activated by steroid hormones such as testosterone and dihydroxytestosterone resulting in dimerization, nuclear translocation, and transcriptional activation of target genes [12]. Given that adenoviral replication is lytic, such a strategy results in prostate specific oncolysis.
One of the problems associated with CRAds used in prostate cancer gene therapy is their selectivity towards androgen dependent disease. A majority of the prostate specific CRAds utilize tissue specific promoters that have an absolute requirement for androgens to activate viral genes. This absolute necessity of androgens for prostate specific promoters renders them ineffective in patients that have undergone or currently on total androgen ablation therapy or in subset of tumor population that have lost the AR expression. Therefore, an alternative approach is needed for the existing vectors to be useful in these particular clinical settings. To overcome these limitations we have generated a prostate specific CRAd by introducing a therapeutic miRNA-34c that was previously shown to suppress proliferation of prostate cancer cells [13]. miRNAs are small double-stranded 21 - 23 nucleotide non-coding RNA molecules that are known to bind to the mRNA, leading to mRNA cleavage or translational silencing of target protein [14]. Dysregulated expression of these molecules significantly contributes to various aspects of carcinogenesis in many tissues including prostate [15]. Like cellular tumor suppressors and oncogenes; miRNAs are also classified as oncogenic (OncomiRs) or tumor suppressor miRNA based on their function in cancer cells. The miR-34 microRNA precursor family that gives rise to three mature miRNAs (miR-34a, b & c) has been classified into the tumor suppressor class [16]. The positive transcriptional regulation of miR-34c level has been previously reported to be a direct target of p53. In this article, we describe the function of miR-34c that synergizes the oncolysis of androgen dependent prostate specific CRAd in AR negative cells. To our knowledge this is the first report describing prostate specific androgen dependent CRAd that utilizes a single therapeutic miRNA to rescue oncolysis in androgen independent AR negative cells.
2. Materials and Methods
2.1. Cell Culture & Reagents
DU145 and LNCaP cell line was obtained from American Type Culture Collection (Manassas, VA) and cultured as per the supplier’s protocol. C4-2 a generous gift from Dr. John Issacs Laboratory (Johns Hopkins University) were cultured in RPMI (Cellgro Mediatech, Herndon, VA) supplemented with 10% FBS, and maintained at passages 50 to 60. Virus packaging cell line DPL-S11 described previously [9] was maintained in DMEM (Cellgro Mediatech, Herndon, VA) with 5% FBS and 200 μg/ml G418. The 293HEK cell line (Quantum Biotech) was used for viral titration and was maintained in DMEM with 10% FBS. All media were supplemented with Ciprofloxacin Hydrochloride 5 μg/ml (US Biological, Swampscott, MA) and Gentamicin 50 μg/ml (Quality Biological Inc., Gaithersburg, MD). All cells were maintained at 37˚C in an atmosphere containing 5% CO2. All restriction enzymes used are from NEB (New England Biolabs, MA). Primary mouse monoclonal antibodies for androgen receptor (AR 441) and E1A (M73) were bought from Santa Cruz Biotech (USA). The majority of the chemicals and reagents used in this study were from Sigma-Aldrich unless otherwise specified.
2.2. Generation of Recombinant Adenoviruses
Viruses (Ad5-PSE/PBN-E1A, Ad5-PSE/PBN-E1A-miR- 34c, Ad5-Delta-E1B, Ad5-CMV-miR-34c and Ad5-CMVhTR) were generated using Ad-Easy system (Stratagene, CA) through recombination of linearized shuttle plasmids in DPL-S11 cells. Briefly shuttle plasmids RpsToadPSE/PBN-E1A, pAd-TrackCMV-hTR, or pAdTrack-CMVmiR34c that carries prostate-specific enhancer and rat probasin promoter driving E1A, and CMV driving miR- 34c or hTR (control miRNA) were linearized with pme1 restriction endonucleases. After gel purification the linearized vectors were separately transformed into the electro-competent AdEasier-1 (BJ5183-AD-1) cells for homologous recombination. The desired clones (pAd5- PSE/PBN-E1A or pAd5-TrackCMV-miRNA34c or pAd5- TrackCMV-hTR) after screening were transformed into DH10B cells for large-scale DNA amplification. For viral propagation the recombinant plasmids were linearized with EcoR1 and transfected into adenovirus packaging cell line DPL-S11 to generate recombinant adenoviruses.
Similarly for generating a prostate-specific conditionally replicating adenovirus that carries CMV driving miR-34c after the fiber region, an adenoviral pFEX system [17] was used to recombine a shuttle plasmid RpsToad-PSE/PBN-E1A using homologous recombination between the two LTRs of the adenovirus in BJ5183 bacterial competent cells. After getting the desired recombinant, the fiber-less viral DNA that carried prostate-specific enhancer and rat probasin promoter (pAd5-FexPSE/PBN-E1A) was linearized with Pac1 and transfected into the fiber expressing FBJ cell line (a generous gift of Dr David Johns, Johns Hopkins University) to generate prostate-specific pseudo-typed Ad5-FexPSE/PBN-E1A virus. The Ad5-PSE/PBN-E1A miR-34c that carries miR- 34c under the control of CMV promoter was produced by the transfection and infection experiment using a transient vector RpUC-WT-FIB CMV-miR-34c and a pseudo-typed fiber-less adenovirus Ad5-FexPSE/PBN-E1A in 293Cre57 cells. Once Ad5-PSE/PBN-E1A-miR-34c was made and purified, RCA (replication competent adenovirus) was ruled out using PCR amplification with a set of primers E1A 5’-CGTTCCGGGTCAAAGTTGG 3’ E1A 5’-CCTCCGTGGCAGATAATATGTC-3’ spanning the wild type E1A promoter and E1A gene.
2.3. Western Blot Analyses
Cells were washed with 1× PBS and re-suspended with five volumes of cold lysis buffer (50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 0.5% NP-40) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN). The cell lysate was incubated on ice for 30 min and then centrifuged for 10 min at 4˚C. Equal amounts of proteins were separated by SDS-PAGE, and the resolved proteins were then transferred to a nitrocellulose membrane. After blocking with 5% nonfat milk in TBST overnight at 4˚C, the blot was incubated with primary antibody at 1 h at room temperature. The membrane was then probed with HRP-conjugated secondary antibody for 1 h and developed (ECL-Plus system, Amersham Pharmacia, Piscataway, NJ) using the manufacturer’s protocol.
2.4. Reporter-Based Viral Replication Assay
We have applied a reporter system to quantify adenoviral replication in real time by linking green fluorescent protein (GFP) expression to the viral major late gene fiber through an Internal Ribosome Entry Site (IRES). In brief, this replication-deficient reporter virus, FFIG expresses GFP in a replication-dependent manner when co-infected with a replicating adenovirus. For reporter experiments, cells were plated into 96-well plates at 1 × 104 per well overnight, and co-infected the next day with replication competent adenoviruses at an MOI of 2 PFU per cell and the reporter virus FFIG at an MOI of 10 - 20 PFU per cell. The GFP fluorescence signals were measured at the indicated time points using a multi-plate fluorometer (FLUOstar Optima Microplate Reader), using an excitation wavelength of 485 ± 20 nm and an emission wavelength of 530 ± 25 nm. Background fluorescence was measured in cells, which were infected with FFIG virus alone. GFP data are plotted as fold GFP induction relative to the mock-infected cells. All samples were performed in either triplicate or quadruplicate with error bars representing the s.e.m.
2.5. MTS Assay
The metabolic viability of the cells was monitored using an MTS assay kit (CellTiter 96) from Promega (Madison, WI). A mixture of MTS and phenazine methosulfate (an electron-coupling reagent; final concentrations, 333 μg·ml−1 and 25 μM) was added, and cells were incubated for 1 hour at 37˚C. Formazan formed from the reduction of MTS was quantified by measurement of absorbance of the medium at 490 nm using a microplate reader. Data obtained from the experiments were normalized to the background signals and plotted as percent cell kill.
2.6. Statistical Analysis
All experiments were done in triplicate or quadruplicate and plotted with s.e.m. All statistical analysis was performed using Excel running on IBM-PC compatible computer on the Windows XP-operating system. Statistical comparisons for paired data were analyzed by the Student’s t-test for the in vitro assays. Statistical significance was defined as P < 0.05.
3. Results
3.1. Library Screen to Identify Therapeutic miRNAs
In order to identify candidate miRNAs that have the potential to suppress the proliferation of prostate cancer cells and an additive effect in combination oncolytic therapy, we performed a miRNA library screen using miRIDIAN microRNA 810 Mimics (Thermo Fisher Scientific, USA). Prostate cancer C4-2 cells plated at the density of 4 × 104 cells were transfected in duplicate with individual miRNAs. Twenty four hours post transfection cells were infected with CN702 (a wild type adenovirus) using 0.2 MOI. MTS assay was performed at 72 h in duplicate to evaluate percent cell kill by each miRNA in the presence of virus. Plates were normalized to mock miRNA tranfected and wild type CN702 infected C4-2 cells. All data were plotted as a waterfall plot to show any cell survival or death when treated with individual miRNA in combination with CN702 virus (Figure 1(a)). Using a cutoff value of at least 20% cell kill above the wild type (CN702) virus and mock transfected miRNA cells, a total of twelve individual miRNAs were identified (Figure 1(b)). Among these twelve therapeutic miRNAs, we specifically choose miR-34c for consequent studies as it has been shown to function downstream in the P53 apoptotic pathway and has also been shown to suppress the proliferative growth of prostate cancer cells. In order to validate that the cell kill was due to miR-34c, we performed the colony formation assay independent of CN702 virus. Briefly equal number of C4-2 cells transfected with miR-34c or control hTR plasmid were mixed with DMEM containing 15% fetal bovine serum and 0.5% agar and plated on six-well plates for 14 days. As shown in Figure 1(c), cells that were transfected with miR-34c have approximately 2.5 fold lower number of colonies than that of controls (Figure 1(c)). These data confirmed the previously known anti-proliferative role of miR-34c in prostate cancer [13]. To further investigate whether overexpression of miR-34c in combination with a non-replicative adenovirus Ad5-Delta E1B causes programmed cell death, C4-2 cells transfected with miR-34c (3 μg/well), Ad5-Delta E1B alone, or in combination with Ad5-Delta E1B virus were infected with 1 MOI. Cells were harvested 48 h post infection (PI) and were subjected to western blot analysis for caspase-3 levels. As shown in the Figure 1(d), cells that were both transfected and infected had lower levels of caspase-3 protein levels compare to the miR-34c alone or virus alone. These data support the anti-proliferative role of miR-34c
 (a)
(a)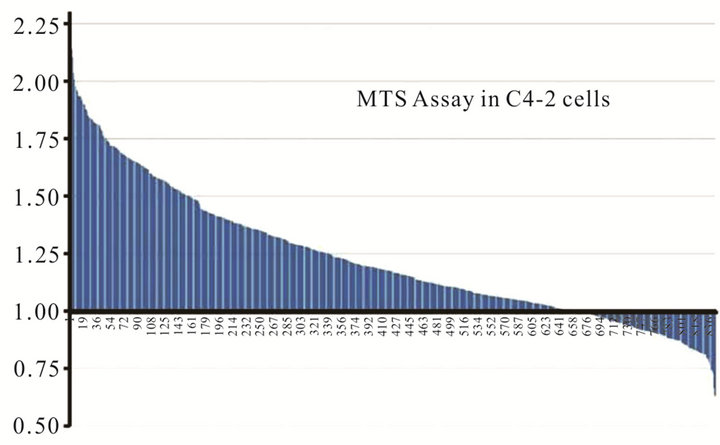 (b)
(b)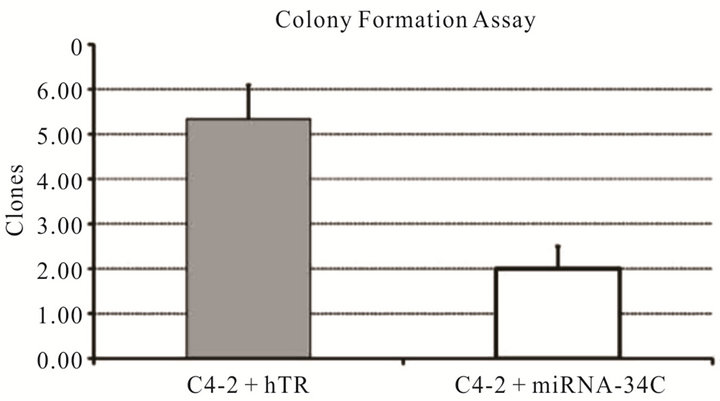 (c)
(c)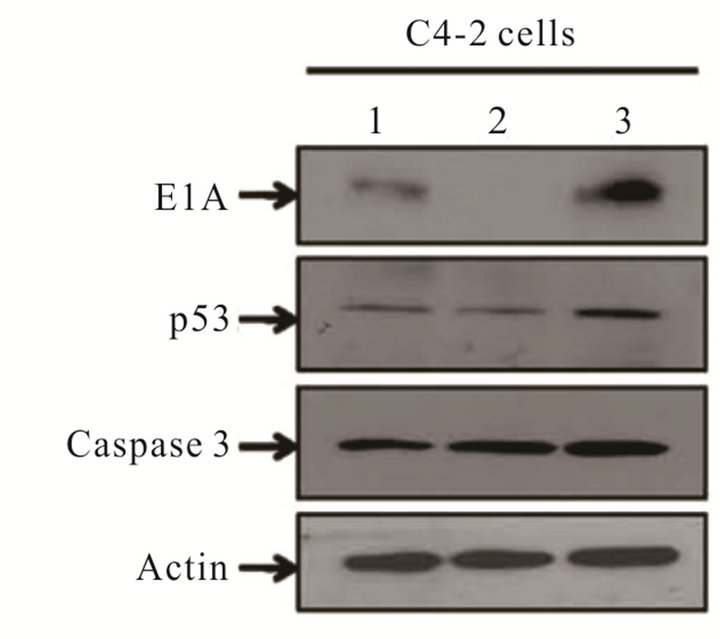 (d)
(d)
Figure 1. Effect of miRNAs in combination with Adenovirus: Waterfall plot chart showing the effect of 810 individual transfected miRNAs in C4-2 cells in combination with wild type CN702 virus at 0.2 MOI 72 h post transfection (a). Candidate miRNAs selected from the MTS assay that have shown at least 20% cell kill above the wild type virus (b). Colony formation assay performed with C4-2 cells transfected with miR-34c and control hTR plasmids. Eight to ten cells counted as a single clone in each treatment were plotted after two weeks to evaluate the function of the contribution of miR-34c in C4-2 cells in the absence of virus (c). Western blot analysis showing the expression of p53 and activated Caspase 3 processing in the presence or absence of non-replicating adenovirus (d).
in C4-2 cells.
3.2. Evaluation of Replication Deficient Ad5-CMV-miR-34c Virus in Androgen Resistant Cell Line
To demonstrate that the therapeutic role of miR-34c was not limited to androgen dependent cells but also affects androgen resistant cells, we constructed a replication incompetent adenovirus that expresses miR-34c and a control hTR virus using CMV promoter. DU145 cells that do not express AR were plated in 96 well plates at the density of 2 × 104 cells. Equal MOI of each non-replicating adenovirus (2 MOI of Ad5CMV-miR-34c or Ad5CMV-hTR) were used to infect cells for five days followed by MTS cell cytotoxicity assay. As shown in Figure 2(a) there was no difference among the two treatments by the end of 24 h, however, by 120 h a significant cell kill was only observed in the Ad5CMVmiR34c infected DU145 cells compared to control Ad5- CMV-hTR infected cells. To further validate these observations, we performed a crystal violet assay using these viruses in DU145 cells. Briefly, 7 × 105 cells plated into 6 well plates were infected with different MOIs (0.5, 5 and 20 MOI) of either Ad5CMV-miR-34c or control Ad5-CMV hTR virus for 10 days. By the end of the experiment wells were stained with crystal violet to evaluate cell cytotoxicity. As shown in Figure 2(b), wells that were infected with miR-34c virus have fewer cells compared to hTR virus (Figure 2(b)). These data further confirm the anti-proliferative role of miR-34c in androgen resistant prostate cancer cell line.
3.3. miR-34c Rescues the Oncolytic Activity of Androgen Dependent CRAd in Androgen Resistant Model
Although tissue specificity that is achieved through AR dependent promoters reduces toxicity by restricting replication to prostate tissue this poses a problem for those
 (a)
(a)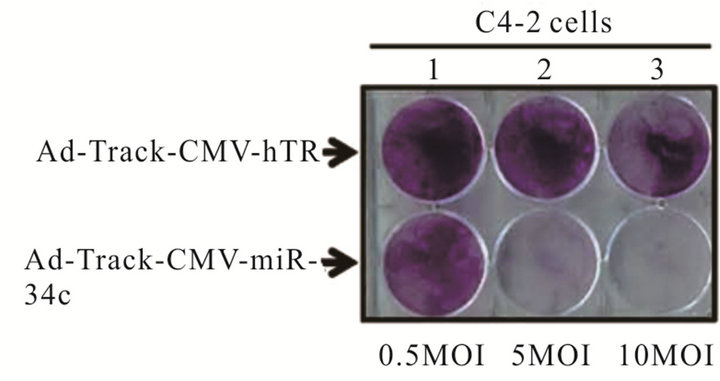 (b)
(b)
Figure 2. Anti-proliferative role of miR-34c in non replicating Adenovirus: MTS assay performed in C4-2 cells infected with non-replicating adenovirus carrying miR-34C or control hTR virus at 24 and 120 h (a). Crystal violet assay for C4-2 cells infected either Ad-Track-CMV-miR 34c or control Ad-Track CMV hTR with different MOIs (0.5 MOI, 5 MOI and 10 MOI) for 5 days (b). Experiments were performed in triplicates and results were plotted as ±SE of mean.
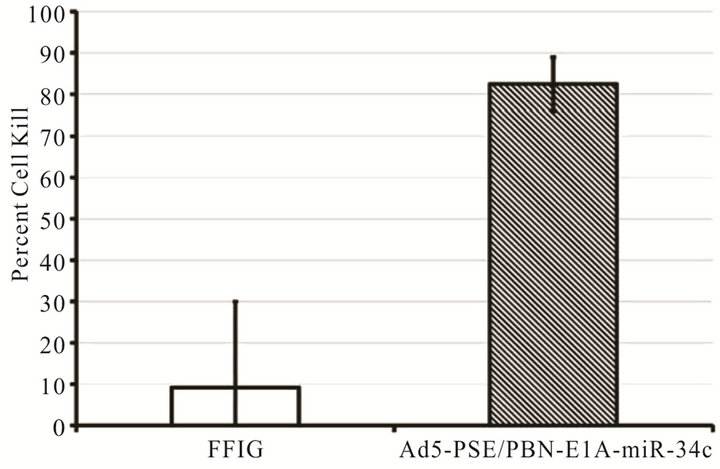
patients on androgen ablation therapy or in tumors that have loss AR expression. Thus an alternative method is needed to rescue the use of prostate specific CRAds that are dependent on androgen for their replication. Furthermore, using tissue specific promoters and enhancer (PSA or PBN and PSE) in the viral backbone utilize much of the genomic space that can be accommodated within the viral capsid. To overcome these problems, we investigated if a single therapeutic miRNA (miR-34c) that has shown therapeutic effect in our in-vitro cell line models will rescue the viral oncolysis in AR negative cells without compromising replication potential in androgen dependent cells. A prostate specific CRAd (Ad5- PSE/PBN-E1A-miR34c) that expresses miR-34c from CMV promoter was engineered using the pFEX system. To demonstrate that miR-34c expressing CRAd retains its replication potency we performed the FFIG reporter assay system, which places a GFP gene under the control of the major late promoter (MLP), allowing us the ability to measure viral replication in real time in a non-invasive high throughput assay. C4-2 cells plated at a density of 5 × 104 cells per well were infected with MOI of 2 with Ad5-PSE/PBN-E1A-miR34c in the presence of a reporter virus FFIG (MOI 5) in complete medium supplemented with 5 nM of R1881. As shown in Figure 3(a), the replication kinetics, measured by GFP signal, was observed as early as 36 h PI, suggesting that Ad5-PSE/ PBN-E1A-miR34C retains it replicative potency. Similarly, to assess the oncolytic potential of Ad5-PSE/PBNE1A-miR34c, MTS assay was performed by 96 h PI in C4-2 cells, as shown in Figure 3(b) cells that were infected with Ad5-PSE/PBN-E1A-miR-34c had more than 80 percent cell death compared to the FFIG reporter virus.
To further evaluate whether the addition of miRNA (miR-34c) in androgen dependent prostate specific CRAd can rescue the oncolysis of virus in AR negative DU145 cells, we took LNCaP (AR positive) and DU145 cells (AR negative) and stably expressed GFP or RFP with the help of lentiviral vectors. Stably selected LNCaP-GFP or DU145-RFP cells mixed in 3:1 ratio (LNCaP and DU145) were infected with 0.5 MOI of Ad5-PSE/PBN-E1A or Ad5-PSE/PBN-E1A-miR-34c or a non replicating control Ad5-PSE/PBN-Luc virus. At the end of 96 h viral oncolysis was measured using florescent microscopy. As shown in the Figure 3(c), the prostate specific CRAd that does not express miR-34c was only able to kill the green AR positive LNCaP-GFP cells but was unable to cause any significant toxicity in the DU145-RFP cells when compared to the control Ad5-PSE/PBN-Luc virus. However, Ad5-PSE/PBN-E1A-miR34c that was engineered to express the miR-34c was able to cause significant oncolysis of both cell types providing evidence of its therapeutic potential in a heterogeneous androgen dependent and independent in-vitro prostate cancer cell model.
4. Discussion
The development of effective therapy for solid tumors remains a challenge to cancer researchers. At least in prostate cancer, it has been realized that no matter how effective the induction therapy works, the disease will recur in an aggressive form almost in all prostate patients. This debilitating phenotype is due to the complexity of prostate cancer with multiple redundant, interacting signaling pathways that help cancerous cells to acquire resistance to treatments. Thus, new agents, with unique mechanisms of action capable of confronting this complexity, are needed that can be used alone or in combination with other drugs for therapeutic efficacies.
Oncolytic virotherapy is an emerging treatment modality that utilizes the lytic property of viruses to lyse and destroys cancer cells. We, and other researchers, have
 (a)
(a)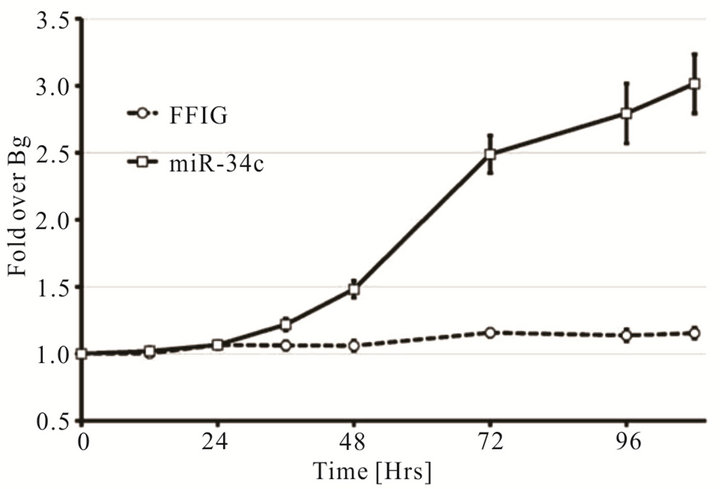 (b)
(b)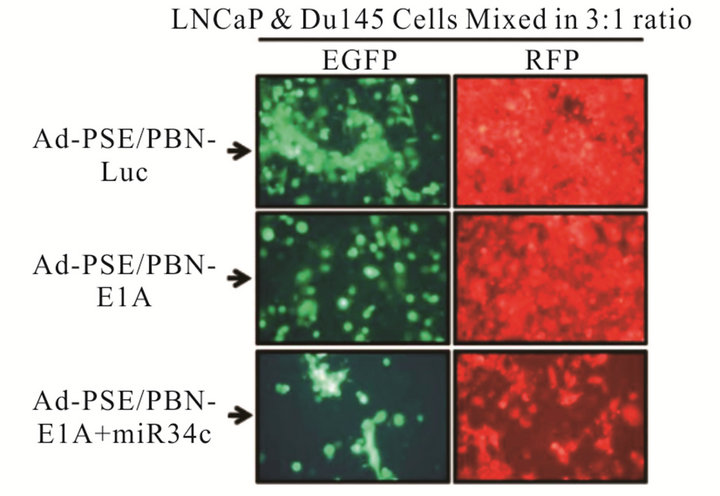 (c)
(c)
Figure 3. A single therapeutic miRNA rescues the oncolysis of Androgen dependent CRAd in Androgen independent model: The Prostate specific CRAd virus expressing miR-34c after the fiber region was assessed for kinetics and degree of viral replication by measuring green fluorescent protein (GFP) expression from the major late promoter in the reporter virus FFIG. As the MLP is only active in late viral replication, this activity is surrogate for viral replication. GFP expression was measured and plotted as fold induction of GFP over the FFIG background control (a). MTS assay of the prostate specific Ad-PSE/PBN-E1A-miR34c and control FFIG virus at 96h PI showed oncolysis of LNCaP cells (b). LNCaP and DU145 cells stably infected with Lentivirus to express GFP or RFP were mixed in 3:1 ratio and infected with either Ad5-PSE/PBN-E1A or Ad5-PSE/PBN-E1A-miR-34C or control Ad5-PSE/PBN-Luc (0.5 MOI) for 96h. Only Prostate specific virus that expresses miR-34c (Ad5-PSE/PBN-E1A-miR34c) was able to kill AR negative DU145-RFP cells compared to the Ad5-PSE/PBN-E1A or control Ad5-PSE/PBN-Luc virus. Data shown represent mean ± SE of the quadruplicate experiment.
previously developed several oncolytic prostate specific viruses by placing the adenoviral genes responsible for controlling replication (E1A, E1B or E4) [4,8,18,19]. Although this strategy has shown clinical efficacy in early clinical trials, the potency of these viruses has been inadequate for a single modality therapy. A common theme among these viruses is the AR activation of their promoters to drive the immediate early genes. This strategy however, made them less efficacious in cellular milieus that have lost or having lower levels of activated AR receptors thus an alternative approach was needed to make them useful in patient population that has undergone or on total ablation therapy with heterogeneous tumor cell population in terms of AR expression.
In this study, we selected a therapeutic miRNA (miR- 34c) from the library pool of 810 miRNA mimics that demonstrated an additive effect in MTS assay in combination with virus. Based on these data we developed a prostate-specific CRAd using the Cre-recombinase based pFex system where the therapeutic miR-34c was placed downstream of the fiber gene under the control of CMV promoter. The CRAd was rendered prostate specific by placing the E1A gene under the control of the PSA enhancer fused to the rat probasin promoter. Using the FFIG reporter assay we were able to show in real time that the addition of the hairpin loop structure did not compromise replication and oncolytic potential of Ad5- PSE/PBN-E1A-miR-34c in C4-2 cells.
The downregulated expression of miR-34c has been previously reported in a number of different malignancies, including prostate cancer. Similarly, overexpression of miR-34c has been shown to suppress the ability of colon carcinoma cells (HT-29) and A549 lung carcinoma cells to migrate and invade upon HGF stimulation [13]. Our data also support the anti-proliferative role of miR-34c as over-expression of miR-34c significantly reduced the number of C4-2 clones in the colony formation assay independent of adenovirus (Figure 1(c)) and in the crystal violet assay when expressed from a replication deficient adenovirus in DU145 cells (Figure 2(b)). Based on the anti-proliferative role of miR-34c in both AR positive and negative prostate cell lines, we constructed a prostate specific CRAd that expresses miR-34c. One advantage of overexpressing miR-34c, which is the downstream transcription target of p53, in replication competent virus might be that the E1B protein of adenovirus during its replicative cycle targets and degrades p53 which cripples the intrinsic apoptotic pathway. However, molecules like miR-34c that affects downstream in the p53 apoptotic pathway will have an additive therapeutic advantage in combinatory oncolytic therapy. Furthermore, shorter sequences like therapeutic miRNAs have an advantage over larger genes in terms of viral packaging. Using tissue specific prostate enhancer and promoters utilizes much of the adenoviral genomic space that can be accommodated inside the viral capsid. By engineering prostate specific CRAd with a single therapeutic miR-34c we were able to demonstrate that viral oncolysis can be rescued in the AR negative androgen resistant DU145 cells. Provided further in vivo validation, these results have significant clinical implication and will form the basis of future attempts to develop novel therapies, especially in patients that have androgen independent disease.
5. Acknowledgements
This study was supported by grant awards from Flight Attendant Medical Research Institute (FAMRI) and Wendy Will Case Cancer Fund to N. Höti. We would like to thank Wasim H. Chowdhury for data analysis in a portion of this work and Dr. Ronald Rodriguez for allowing his lab resources to accomplish this work.
REFERENCES
- N. Craft, C. Chhor, C. Tran, A. Belldegrun, J. DeKernion, O. N. Witte, J. Said, R. E. Reiter and C. L. Sawyers, “Evidence for Clonal Outgrowth of Androgen-Independent Prostate Cancer Cells from Androgen-Dependent Tumors through a Two-Step Process,” Cancer Research, Vol. 59, No. 19, 1999, pp. 5030-5036.
- T. Hakkarainen, M. Rajecki, M. Sarparanta, M. Tenhunen, A. J. Airaksinen, R. A. Desmond, K. Kairemo and A. Hemminki, “Targeted Radiotherapy for Prostate Cancer with an Oncolytic Adenovirus Coding for Human Sodium Iodide Symporter,” Clinical Cancer Research, Vol. 15, No. 17, 2009, pp. 5396-5403. doi:10.1158/1078-0432.CCR-08-2571
- M. S. Ferguson, N. R. Lemoine and Y. Wang, “Systemic Delivery of Oncolytic Viruses: Hopes and Hurdles,” Advances in Virology, Vol. 2012, 2012, Article ID: 805629.
- N. Hoti, W. H. Chowdhury, S. Mustafa, J. Ribas, M. Castanares, T. Johnson, M. Liu, S. E. Lupold and R. Rodriguez, “Armoring CRAds with p21/Waf-1 shRNAs: The Next Generation of Oncolytic Adenoviruses,” Cancer Gene Therapy, Vol. 17, No. 8, 2010, pp. 585-597. doi:10.1038/cgt.2010.15
- R. D. Gerard and D. Collen, “Adenovirus Gene Therapy for Hypercholesterolemia, Thrombosis and Restenosis,” Cardiovascular Research, Vol. 35, No. 3, 1997, pp. 451- 458. doi:10.1016/S0008-6363(97)00134-X
- M. Cristofanilli, S. Krishnamurthy, L. Guerra, K. Broglio, B. Arun, D. J. Booser, K. Menander, J. Van Wart Hood, V. Valero and G. N. Hortobagyi, “A Nonreplicating Adenoviral Vector That Contains the Wild-Type p53 Transgene Combined with Chemotherapy for Primary Breast Cancer: Safety, Efficacy, and Biologic Activity of a Novel Gene-Therapy Approach,” Cancer, Vol. 107, No. 5, 2006, pp. 935-944. doi:10.1002/cncr.22080
- E. J. Small, M. A. Carducci, J. M. Burke, R. Rodriguez, L. Fong, L. van Ummersen, D. C. Yu, J. Aimi, D. Ando, P. Working, D. Kirn and G. Wilding, “A Phase I Trial of Intravenous CG7870, a Replication-Selective, ProstateSpecific Antigen-Targeted Oncolytic Adenovirus, for the Treatment of Hormone-Refractory, Metastatic Prostate Cancer,” Molecular Therapy, Vol. 14, No. 1, 2006, pp. 107-117. doi:10.1016/j.ymthe.2006.02.011
- T. L. DeWeese, H. van der Poel, S. Li, B. Mikhak, R. Drew, M. Goemann, U. Hamper, R. DeJong, N. Detorie, R. Rodriguez, T. Haulk, A. M. DeMarzo, S. Piantadosi, D. C. Yu, Y. Chen, D. R. Henderson, M. A. Carducci, W. G. Nelson and J. W. Simons, “A Phase I Trial of CV706, a Replication-Competent, PSA Selective Oncolytic Adenovirus, for the Treatment of Locally Recurrent Prostate Cancer Following Radiation Therapy,” Cancer Research, Vol. 61, No. 20, 2001, pp. 7464-7472.
- N. Hoti, Y. Li, C. L. Chen, W. H. Chowdhury, D. C. Johns, Q. Xia, A. Kabul, J. T. Hsieh, M. Berg, G. Ketner, S. E. Lupold and R. Rodriguez, “Androgen Receptor Attenuation of Ad5 Replication: Implications for the Development of Conditionally Replication Competent Adenoviruses,” Molecular Therapy, Vol. 15, No. 8, 2007, pp. 1495-1503. doi:10.1038/sj.mt.6300223
- J. Lindzey, M. V. Kumar, M. Grossman, C. Young and D. J. Tindall, “Molecular Mechanisms of Androgen Action,” Vitamins & Hormones, Vol. 49, 1994, pp. 383-432. doi:10.1016/j.bbr.2011.03.031.
- E. P. Gelmann, “Molecular Biology of the Androgen Receptor,” Journal of Clinical Oncology, Vol. 20, No. 13, 2002, pp. 3001-3015. doi:10.1200/JCO.2002.10.018
- Z. Culig, H. Klocker, G. Bartsch and A. Hobisch, “Androgen Receptors in Prostate Cancer,” Endocrine-Related Cancer, Vol. 9, No. 3, 2002, pp. 155-170. doi:10.1677/erc.0.0090155
- Z. Hagman, O. Larne, A. Edsjo, A. Bjartell, R. A. Ehrnstrom, D. Ulmert, H. Lilja and Y. Ceder, “miR-34c Is Downregulated in Prostate Cancer and Exerts Tumor Suppressive Functions,” International Journal of Cancer, Vol. 127, No. 12, 2010, pp. 2768-2776. doi:10.1002/ijc.25269
- Y. K. Kim and V. N. Kim, “Processing of Intronic microRNAs,” EMBO Journal, Vol. 26, No. 3, 2007, pp. 775-783. doi:10.1038/sj.emboj.7601512
- M. Maugeri-Sacca, V. Coppola, D. Bonci and R. De Maria, “MicroRNAs and Prostate Cancer: From Preclinical Research to Translational Oncology,” Cancer Journal, Vol. 18, No. 3, pp. 253-261. doi:10.1097/PPO.0b013e318258b5b6
- H. Hermeking, “The miR-34 Family in Cancer and Apoptosis,” Cell Death & Differentiation, Vol. 17, No. 2, pp. 193-199. doi:10.1038/cdd.2009.56
- S. E. Lupold, T. A. Kudrolli, W. H. Chowdhury, P. Wu and R. Rodriguez, “A Novel Method for Generating and Screening Peptides and Libraries Displayed on Adenovirus Fiber,” Nucleic Acids Research, Vol. 35, No. 20, 2007, p. e138. doi:10.1093/nar/gkm914
- M. Satoh, H. Wang, S. Ishidoya, H. Abe, T. Moriya, H. Hamada and Y. Arai, “Oncolytic Virotherapy for Prostate Cancer by E1A, E1B Mutant Adenovirus,” Urology, Vol. 70, No. 6, 2007, pp. 1243-1248. doi:10.1016/j.urology.2007.09.031
- D. C. Yu, Y. Chen, M. Seng and J. Dilley, “The Addition of Adenovirus Type 5 Region E3 Enables Calydon Virus 787 to Eliminate Distant Prostate Tumor Xenografts,” Cancer Research, Vol. 59, No. 17, 1999, pp. 4200-4203.
NOTES
*Corresponding author.