Journal of Cancer Therapy
Vol.2 No.4(2011), Article ID:7786,7 pages DOI:10.4236/jct.2011.24062
Study of Celastrol on Akt Signaling Pathway and Its Roles in the Apoptosis of K562 Cells
![]()
1Medical College of WuHan University of Science Technology, Wuhan, China;
2Institute of Hematology, Union Hospital, Tongji Medical College of Huazhong University of Science and Technology, Wuhan, China.
Email: *wxnan@sina.com
Received July 16th, 2011; revised August 20th, 2011; accepted August 31st, 2011.
Keywords: Celastrol, K562 Cells, Akt Signal Transduction Pathway, Apoptosis
ABSTRACT
The purpose Celastrol, the main active compound of the Celastrus genus plants, belonging to Celastraceae, has recently marked antitumour potency on solid tumours of various derivations, Methods: We demonstrate here that Celastrol also present powerful antileukaemic potency through both growth arrest and apoptosis induction in K562 cells, which was accompanied by typical apoptotic morphological and sharp decreased expression of phosphorylation level of Caspase family members and Akt signaling pathway related proteins were determined by western blot before and after celastrol treatment, and further the effect of AKT signaling pathway on celastrol-induced-apoptosis was analyzed. However, in vitro treatment with Celastrol resulted in significantly reduced expression of phophorylation of Akt, Survivin and Bcl-2 significantly in K562 cells. Results: 25 nmol/L WORT (PI3K-Akt inhibitor) can significantly augmented cell apoptosis induced by Celastrol in K562 cells in dose-dependent manner, Moreover, most Caspase3,8,6 were activated in K562 cells during Celastrol treatment, 50 µmol/Lz-VAD-fmk (Caspase inhibitor) can to enhance the apoptosis induced by Celastrol. Discussion: These results suggest that the fact that Akt signaling pathway might act as new targets of Celastrol, correlates well with the sensitivity to Celastrol, as well as the rate of apoptosis induced by Celastrol, Mechanisms that regulate Akt signaling pathway may be provide novel opportunities for drug development.
1. Introduction
Hematologic tumorigenesis mechanisms show that the disorder of Cell autonomous death process-programmed cell death or apoptosis is the pivotal cause of tumorigenesis. In the cell growth, death, and differentiation, the genetic information acts through diverse signal transduction pathways, in which sustained Akt phosphrylation plays an crucial role in leukemic cell malignant transduction. Several agents suppress the growth of leukemic cells by inhibited the Akt signal pathway in vitro [1,2]. Previous studies show Celastrol can suppress diverse tumor cells growth including HL-60 cell, but the mechanism involved is not clear. In recently, cell apoptosis is closely related with several tumors development and progression and chemotherapeutic agents efficacies. High dose chemotherapy and radiotherapy can induce tumor cell apoptosis, but inducing sensitive-specific cell apoptosis is one of other mechanisms that several anticancer drugs works, which has become a novel strategy for anticancer drug development [3]. Celastrol is one monomer of Tripterygium and a triterpenoid pigment with formula C29H38O and molecular weight 450.000. It is reported without suppression the immune response and anti-inflammatory effects [4-5], celastrol exhibited anti-cancer activity in vitro [6-7], but mechanism involved is not fully understood. In this study, K562 cells were used to explore the molecular mechanism of Celastrol—inducedapoptosis, and further investigate the significance of Akt pathway in this process.
2. Materials and Methods
2.1. Materials
K562 cell line is obtained from the China Center for Typical Cuture Collection (Wuhan, China) and maintained in RPMI-1640 (Sigma) medium supplemented with 10% fetal serum (FBS, Gibco-BRI, USA.) at 37˚C in a humidified atmosphere. Celastrol is purchased from the (Sigma). Anti-P-Akt, Akt, Survivin, Bcl-2, Caspases3, 8,6 GAPDH are purchased from Zymed, San Francisco, Calif. MTT was bought from Janssen Chimica Company (New Brunswick, NJ, USA). Hoechst 33258 and dimethyl sulfoxide (DMSO) were purchased from Sigma, USA.
Chemiluminescence (ECL) reagent kits were purchased from Pierce Biotechnology (Rockford, IL, USA). Annexin V-FITC/PI, DNA apoptosis kit is was from Keygen company, China.
2.2. Proliferation Assay
Logarithmically growing K562 cells were seeded in triplicate at a concentration of 5 × 104 cells/ml in culture medium, and then exposed to various concentrations (0.25 - 16.0 μmol/L) and exposure intervals (up to 3 days) of Celastrol. After the incubation period, 10 μl of the MTT labeling reagent (final concentration 0.5 mg/ml) was added to each well. At the end of another 4 h incubation, the medium were removed and 150 μl DMSO was added to each well. After the purple crystals were completely dissolved, the optical density was measured by using a μQuant Microplate Spectrophotometer (BioTeck, Winooski, USA) at a wavelength of 570 nm.
2.3. Annexin V-FITC Stainin for Evaluation of Apoptosis
According to the method described in Annexin V-FITC detection kit, cells (1 × 106) exposed to various concentrations of Celastrol (0.5 - 2.0 µmol/L) were resuspended in 100 μl of binding buffer after 24 hr culture, afterwords, cells were stained with 5 μl Annexin V-FITC solution and 15 μl propidium iodode solution for 15 min. at room temperature in the dark. Then the samples were diluted with 300 μL of binding buffer and analysed by flow cytometry (FACsirt BD Biosciences).
2.4. Hoechst 33258 Staining
Nuclear fragmenation is visualized by Hoechst 33258 staining of apoptotic nuclei.after 24 hr culture, cels exprosed to 2.0 µmol/L (close IC50) of Celastrol were fixed in fixed in 4% araformaldehyde for 10 min at room temperature before deposition on polylysine-coated coverslips. The adhered cells are permeabilized 0.1% Triton- 100 for 5 min ar room temperature,and then incubated with Hoechst 33258 for 30 min at 37˚C, rinsed with PBS and mounted on slides with glycerol-PBS. Finally, the cells are viewed with an Olympus BH-2 fluorescence microscope (Tokyo, Japan.)
2.5. DNA Fragmentation Assay
DNA is isolated according to the method described in the DNA apoptosis ladder kit. Cells are washed and lysed in 300 µL of digestion buffer after 24 h and or 48 h culture. The genomic DNA is dissolved in TE buffer and 5 µg of DNA samples is subjected to agarose gel (2%) electrophoresis. The gels are visualized and photographed under UV light.
2.6. Western Blot Assay for Protein Expression
After treatment,cells were harvested and lysed in 100 μL of lysis buffer (10 nM This-HCl [pH 7.5], 1 mM EDTA, 1% Triton X-100,150 nM NaCl, 1 mM dithiothreitol, 10% glycerol, 0.2 nM phenylmethylsulfonyl fluoride, and protease inhibitors) by incubation on ice for 30 min, and then the extracts were centrifuged at 29,000 × g for 15 min to remove cell debris after the addition of 5 × loading buffer, protein samples were electrophoresed on a 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membraned. the membranes were then immunoblotted with primary antibodies directed against P-Akt, Akt, Survivin, Bcl-2 Caspases3,8,6 to separately after overnight incubation 4˚, the blots were exposed to the horseradish peroxidase-labeled secondary antibodies at a final dilution of 1:1500 in western washing solution. finally, the blots of the bands was carried out using the Quantity One densitometric analysis software (Bio-Rad, Richmond, CA, USA).
2.7. Statistical Analysis
Student’s t-test and one-way analysis of variance are used to determine statistical significance of differences between values for various experimental and control groups, p values < 0.05 was considered a significant difference.
3. Results
3.1. Celastrol Declined Proliferation of K562 Cells
The cytotoxicity of Celastrol to K562 cells was calculated from the loss of cell viability using MTT assay. As shown in Figure 1, exposure of K562 cells to 0.25 µmol/L Celastrol or 0.5 ower than that for 24 h did not affect their viability. However, at concentrations higher than that, Celastrol significantly inhibited the growth of K562 cells. The degree of growth inhibition depended on both the concentration and the exposure duration to Celastrol. The IC50 value of Celastrol for 36 h was 2.031 ± 0.109 μmol/L.
3.2. Celastrol Induced the Apoptosis of K562 Cells
To address the role of Celastrol in conferring sensitivity to apoptosis, K562 cells are exposed to increasing concentration of Celastrol,and then detected with DNA fragmentation assay and Annexin-V/PI double-labeled cytometry. The degree of apoptotic cell death is quantified
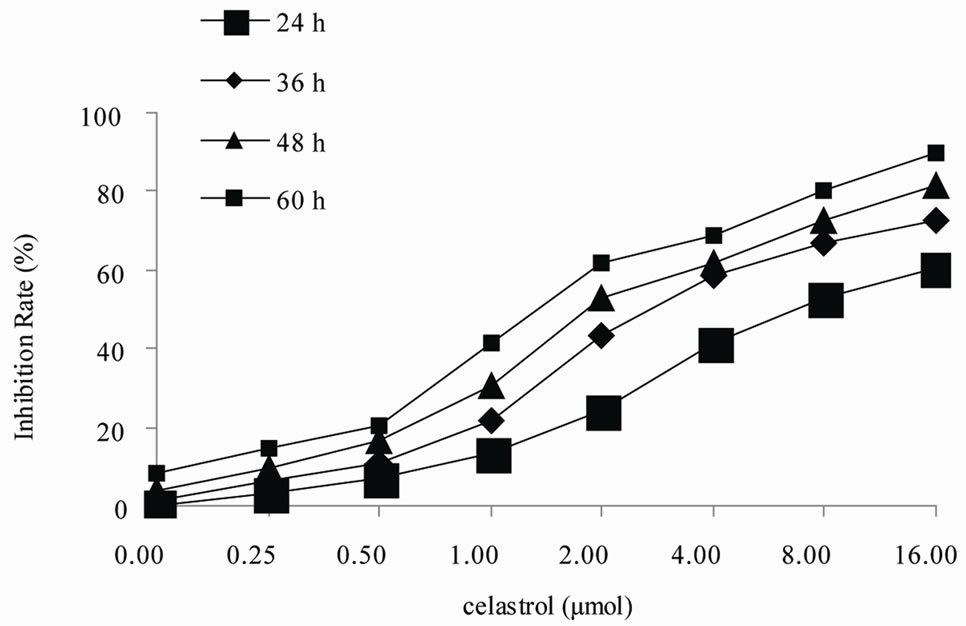
Figure 1. Inhibition of proliferation by Celastrol in K562 cells. Cells are treated in vitro with various concentrations of 0 - 16.0 µmol/L Celastrol for 0 - 60 h,Growth inhibition is determined using a MTT assay and shown as an inhibitory rate. The figures are representative of three independent experiments.
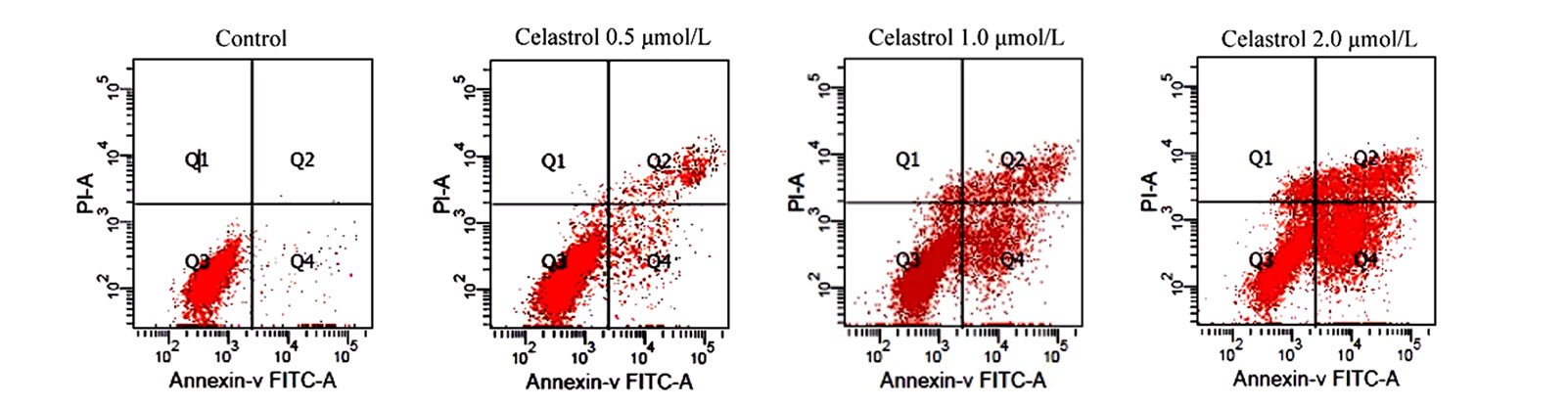
as the percentage of the Annexin-V FITC positive cells. There are little binding of Annexin-V FITC in untreated K562 cells, However, after treatment with Celastrol of 2.0 μmol/L for 24 h, the percentage of apoptotic cells was increased from 2.24% to 39.23% (P < 0.01, Figure 2(a)). as shown in Hoechst 33258 staining figures, apoptotic bodies containing nuclear fragments are found in Celastrol-treated cells, and the chromatin becomes condensed, marginalized, the nuclear envelope appear lytic and the cytoplasm becomes shrinkaged. While the normal K562 cells present intact plasma membrane and order chromatin folding (Figure 2(c)).
3.3. Celastrol Induces K562 Cell Apoptosis through Channel of CASPASE Activation
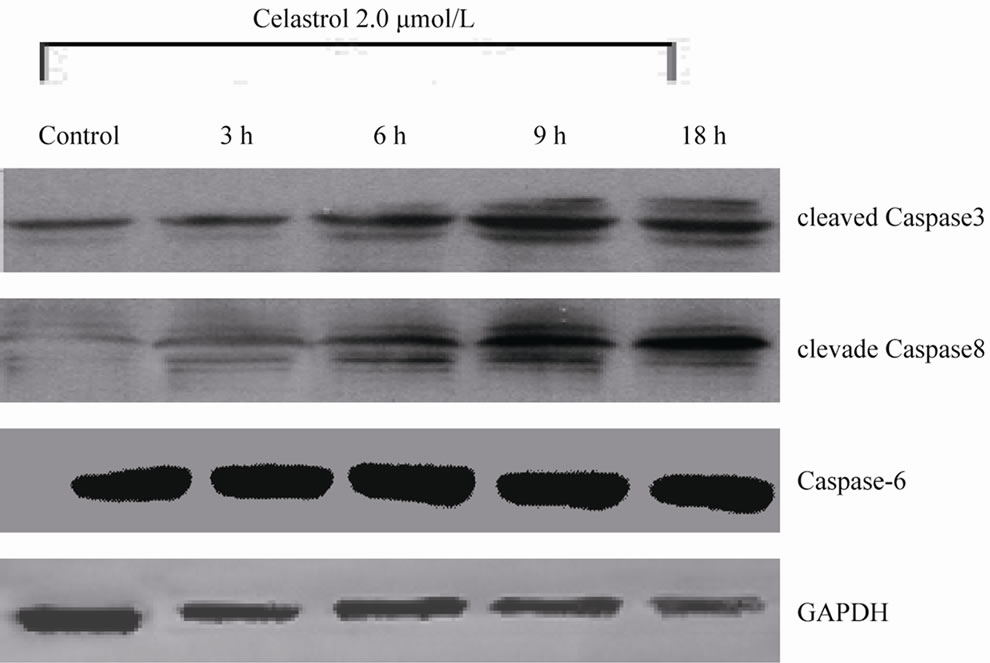
Among the Caspase members, the cleavage of Caspase3, 8 is implicated in the process of Celastrol-induced K562 cell apoptosis (Figure 3(a)). In K562 cells, Celastrol induces rapid and significant activation of Caspase3 but has no effect on the activation of Caspase 6. 50 μmol/L Capase inhibitor z-VAD-fmk significantly block Celastrol-induced K562 cell apoptosis. (Figure 3(b))
3.4. Celastrol Inhibit Akt Activity of K562 Cells
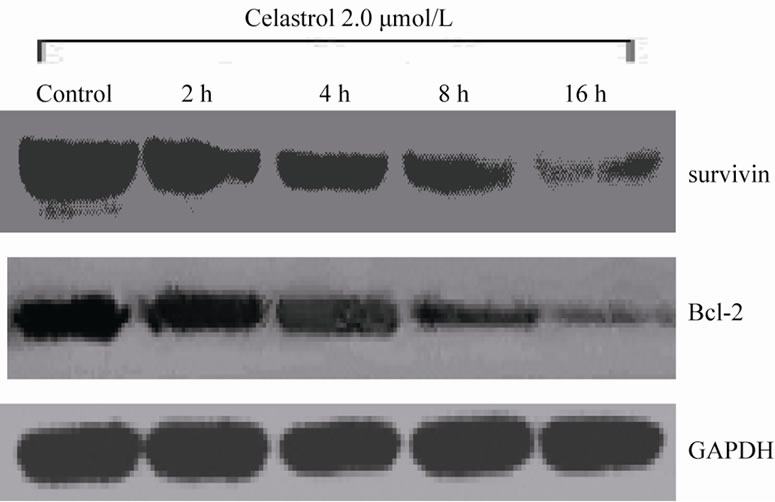
The Akt phosphorylation level of K562 cell can be significantly inhibited with 2.0 μmol/L Celastrol for 4 h and the intensity of inhibiton is increased with time elongated. However, the total level of Akt expression has no evident change by Celastrol (Figure 4(a)). Simultaneously, we examined the protein levels of downstream molecules of Akt including survivin and Bcl-2 and we found that the activity of survivin and Bcl-2 is rapidly decreased in K562 cells treated by Celastrol (Figure 4(b)).
3.5. PI3K Inhibitor Significantly Enhances Celastrol-Induced K562 Cellapoptosis
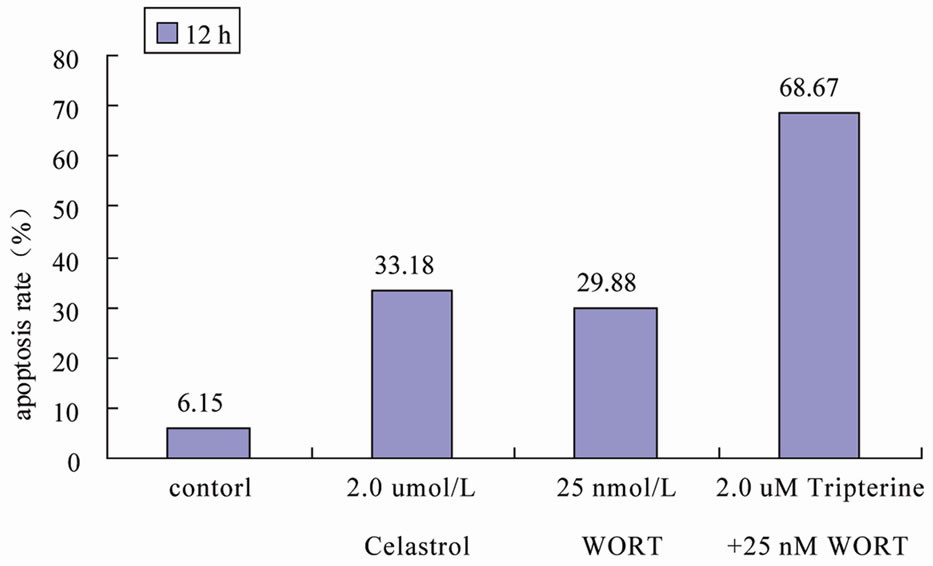
We used signaling pathway inhibitor WORT to treat K562 cells for 1 hour, and then affected K562 cells with 2.0 μmol/L Celastrol for 6 h. The result shows that WORT can significantly improve the apoptotic proportion of Celastrol-treated K562 cells (Figure 5(a)). Meanwhile, we investigated the activation level of Caspase3 intracellularly and found that the expression level of activated Caspase3 in cells treated with both WORT and Celastrol is further increased in contrast to cells only treated with Celastrol (Figure 5(b)).
4. Conclusions
Dysregulation of cell proliferation, differentiation, and apoptosis is responsible for the tumor development and progression. Previous studies demonstrated celastrol suppressed many tumor growth in vitro. Our study shown celastrol inhibited the K562 cell proliferation through apoptosis in a doseand time-dependent manner. Results form Hoechst 33258 staining assay found nuclei pyknosis, chromatin condensation, DNA fragmentation, and apoptosis body in K562 cells after celastrol treatment. DNA ladder assay also confirmed celastrol initiated endogenous endonuclease activity to induce DNA fragmentation. Apoptotic cell proportion is positive correlation with the celsatrol dose, which is consistent with the literature [8]. The present study also found activation of caspase is involved in the apoptosis process induced by celastrol in K562 cells, and caspase inhibitors block it, which confirmed caspase is responsible for the celastrolinduced-apoptosis consistent to the literature [9].
 (a)
(a)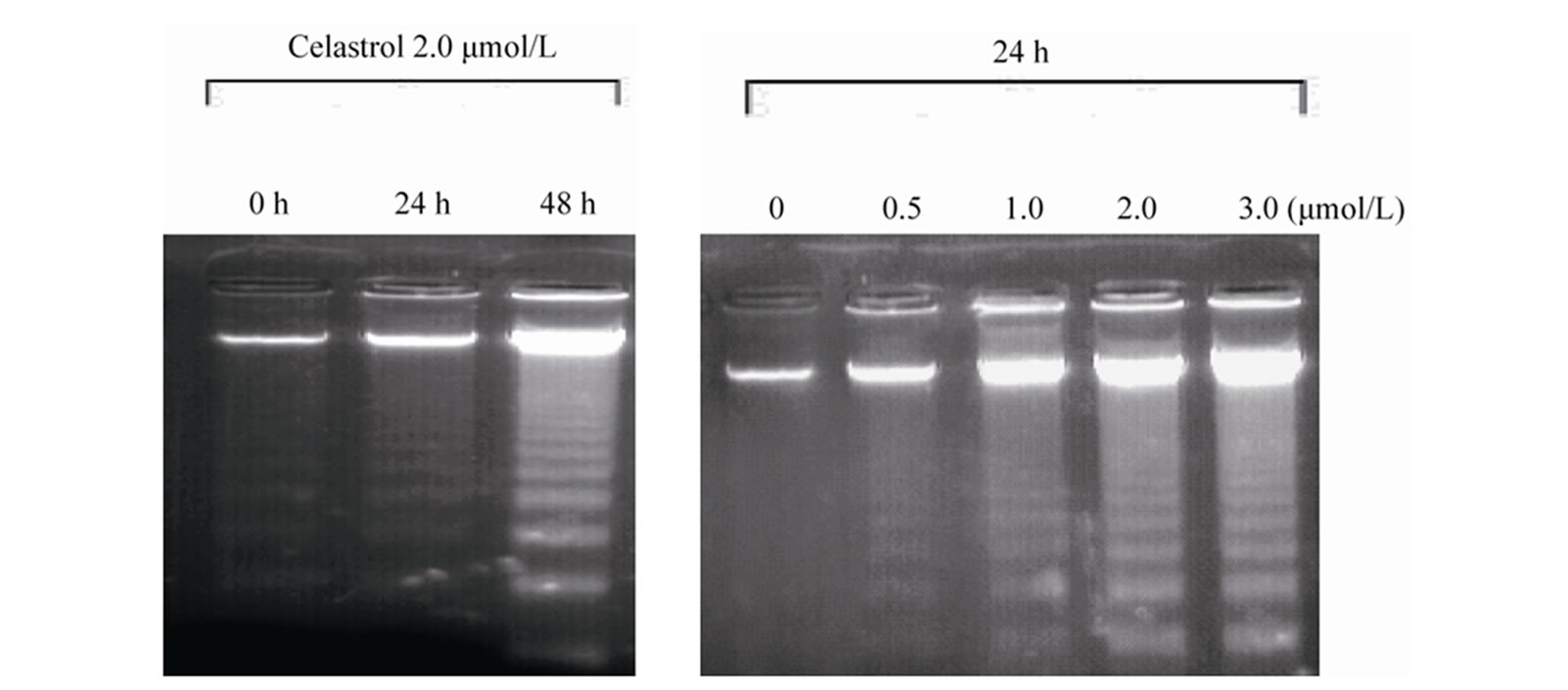 (b)
(b)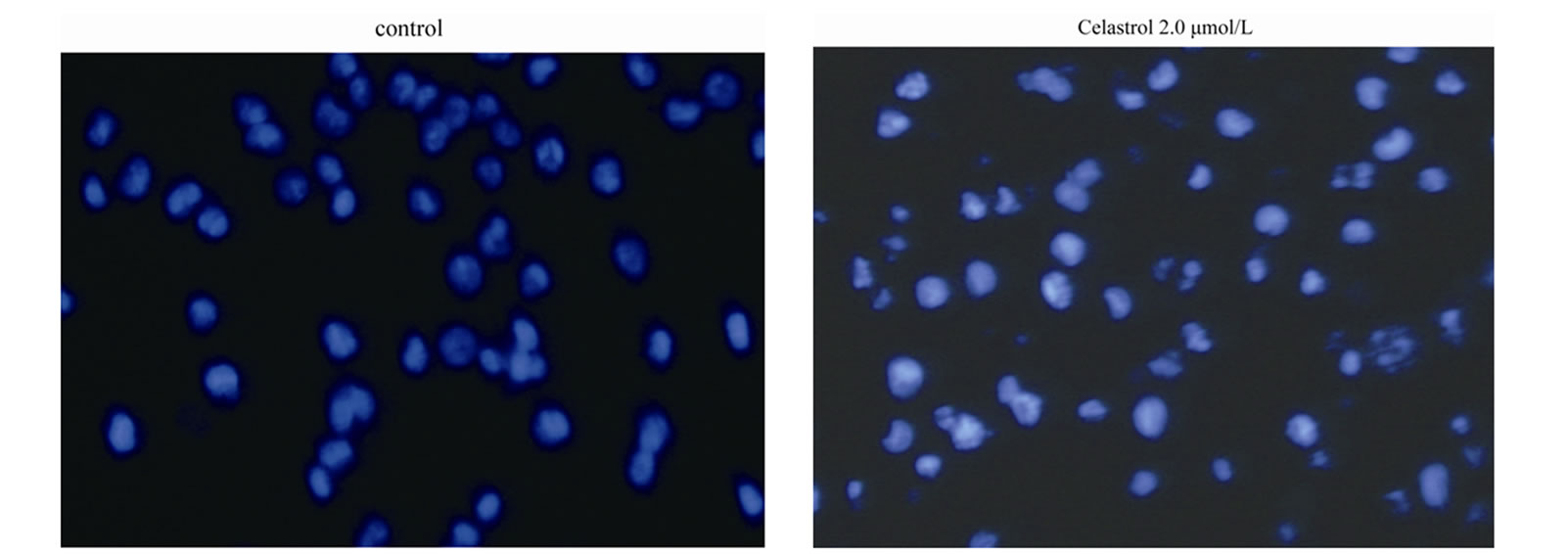 (c)
(c)
Figure 2. Induction of apoptosis by celastrol in K562 cells. Celastrol induces a dose-dependent apoptosis in K562 cells as determined by Annexin V-FITC/PI double-labeled cytometry (a), DNA fragmentation assa (b), and Hoechst 33258 staining assay (c).
PTEN depletion resulted in PI3K-Akt sustained activation plays a important role in maligment transformation of K562 cells. Multiple anti-cancer agents act through attenuated PI3K and Akt levels [10,11]. The present study found celastrol could significantly suppress the phosphrylation level of Akt, as well as the downstream molecules survivin and Bcl-2. 25 nmol/L WORT, a PI3K/Akt inhibitor, can greatly enhanced celsatrol induced apoptosis. Furthermore, 25 nmol/L WORT alone can strongly suppress the phosphorylation of Akt,. Bcl-2 is one of the well-known antiapoptotic oncogenes that can block multiple general apoptosis approaches in sev-
 (a)
(a)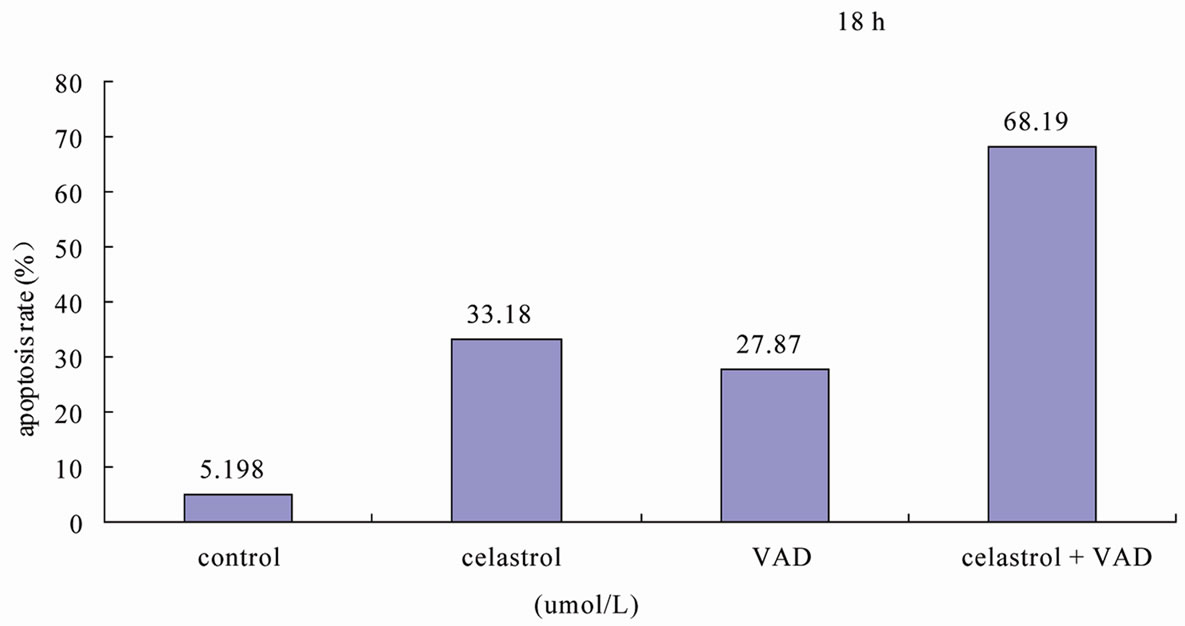 (b)
(b)
Figure 3. Celastrol induces K562 cell apoptosis depending on the pathway of Caspase activation. (a) The process of K562 cell apoptosis induced by Celastrol accompanies with Caspase activation. (b) Caspase inhibitor z-VAD-fmk to enhanced the Celastrol-induced K562cell apoptosis.
eral cells, and survivin, a strongest apoptotic factor at present [12], directly acts on the caspase cascade final proteins caspase-3 and -7 and indirectly p21 to inhibit caspases [13], and subsequently blocks apoptosis. Ikehara et al. [12] regarded survivin was responsible for the invasion, migration, therapy and prognosis. Our study demonstrated survivin and Bcl-2 were downregulated at the same time in celastrol induced apoptosis, whereas caspase-3,8 activities were greatly enhanced. Accordingly, celsatrol induced apoptosis in K562 cells might involve in mitochondrial pathway, whenas Bcl-2 acts againstion imbalance resulted in mitochondria rapture to affect the release of Cytc C from mitochondria so as to block caspase cascade to suppress apoptosis [14]. Therefore, during the process of celastrol induced apoptosis, downregulation of survivin directly initiated caspase to facilitated apoptosis and decrease of Bcl-2 expression level enhanced the sensitivity of tumor cell to apoptosis, and affected on the caspase upstream to activate Cytc C release from mitochondria and subsequently enable caspase to induce apoptosis. Consequently, downregulation of survivin and Bcl-2 simultaneous played a synergistically role in celastrol induced K562 cell apoptosis.
In short, the present study demonstrated celsatrol can significantly inhibite the proliferation, and induce apoptosis in K562 cells through caspase activation, accomplished by decrease of Akt phosphorylation. Though activity reduce of Akt is not the only reason for celastrol
 (a)
(a)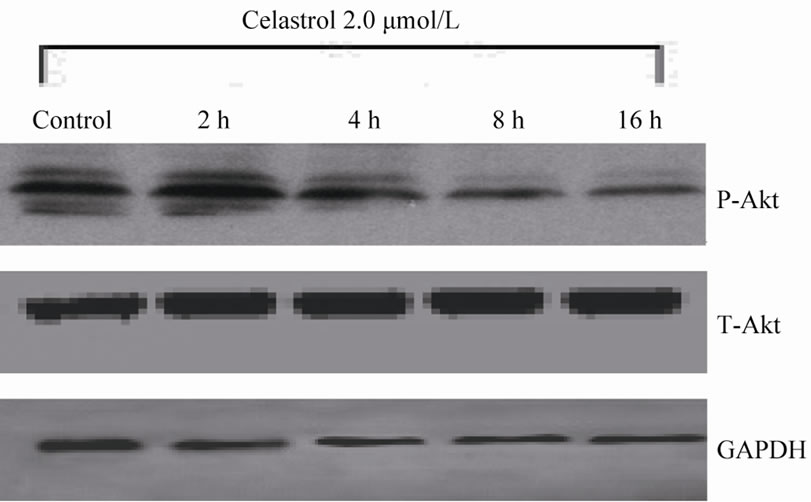 (b)
(b)
Figure 4. The effect of Celastrol on Akt signaling pathway in K562 cells. (a) Celastrol inhibits the Akt phosphorylation level in K562 cells, (b) Celastrol inhibits protein level of Survivin and Bcl-2 in K562 cells.
 (a)
(a)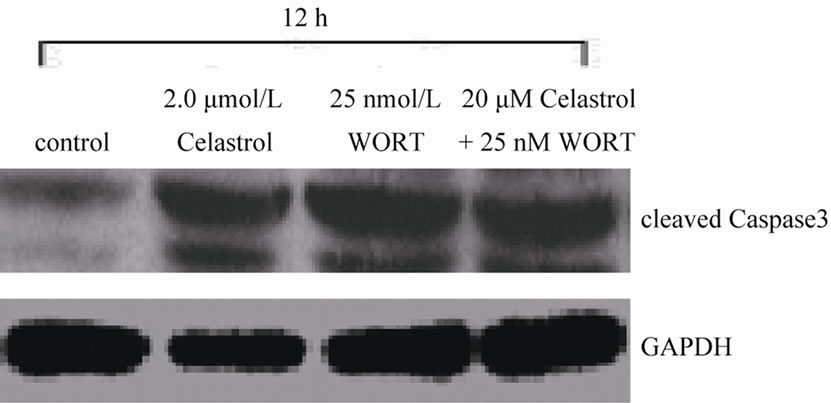 (b)
(b)
Figure 5. WORT’s effect on Celastrol-induced K562 cell apoptosis. (a) 25 nmol/L WORT can observably increased the K562 cell apoptotic proportion induced by Celastrol. (b) 25 nmol/L WORT can further promote Caspase3 cleavage activation induced by Celastrol.
induced apoptosis, it involved in the PI3K/Akt inhibitor and celastrol induced K562 cell apoptosis.
5. Acknowledgements
This work was supported by grant from the Education Foundation of Hubei province of China (no: B20101107). The authors would like to thank the Department of Immunology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China, for offering relevant experimental facilities and technical support.
REFERENCES
- C. Yu, M. Rahmani and J. Almenara, “Induction of Apoptosis in Human Leukemia Cells by the Tyrosine Kinase Inhibitor Adaphostin Proceeds through a RAF-1/MEK/ ERK and Akt-Dependent Process,” Oncogene, Vol. 23, No. 7, 2004, pp. 1364-1376. doi:10.1038/sj.onc.1207248
- M. Al-Rasheed and P. S. Manogaran, “Curcumin Induces Apoptosis via Inhibition of PI3K-Kinase/Akt Pathway in Acute T Cell Leukemia,” Apoptosis, Vol. 11, No. 2, 2006, pp. 245-254.
- A. Salminen, M. Lehtonen and T. Paimela, “Celastrol: Molecular Targets of Thunder God Vine,” Biochemical and Biophysical Research Communications, Vol. 394, No. 3, 2010, pp. 439-442. doi:10.1016/j.bbrc.2010.03.050
- D. H. Kim, E. K. Shin and Y. H. Kim, “Suppression of Inflammatory Responses by Celastrol, a Quinone Methide Triterpenoid Isolated from Celastrus Regelii,” European Journal of Clinical Investigation, Vol. 39, No. 9, 2009, pp. 819-827.
- Y. Kim, K. Kim and H. Lee, “Celastrol Binds to ERK and Inhibits FcepsilonRI Signaling to Exert an Anti-Effect,” European Journal of Pharmacology, Vol. 612, No. 1-3, 2009, pp. 131-142. doi:10.1016/j.ejphar.2009.03.071
- G. Sethi, K. S. Ahn and M. K. Pandey, “Celastrol, a Novel Triterpene, Potentiates TNF-Pinduced Apoptosis and Suoppresses Invasion of Tumor Cells by Inhibiting NF-KappaB Activation,” Blood, Vol. 109, No. 7, 2007, pp. 2727-2735.
- D. H. Zhang, A. Marconi and L. M. Xu, “Tripterine Inhibits Expression of Adhesion Molecules Activated Endothelial Cells,” Journal of Leukocyte Biology, 2006, Vol. 80, No. 2, pp. 309-319. doi:10.1189/jlb.1005611
- Y. H. Xu and J. Yan, “Effect and Possible Mechanism of Tripterine on Inducing Apoptosis of Human Acute Myelocytic Leukemia HL-60 Cells,” Journal of Zhejiang University (Science Edition), Vol. 35, No. 3, 2008, pp. 311- 314.
- S. Mukherjee Chakraborty, U. Ghosh and N. P. Bhattacharyya, “Curcumin-Induced Apoptosis in Human Leukemia Cell HL-60 Is Associated with Inhibition of Telomerase Activity,” Molecular and Cellular Biochemistry, Vol. 297, No. 1-2, 2007, pp. 31-39. doi:10.1007/s11010-006-9319-z
- H. Yang, D. Chen and Q. C. Cui, “Celastrol, a Triterpene Extracted from the Chinese Thunder of God Vine, Is a Potent Proteasomc Inhibitor and Suppresses Human Prostate Cancer Growth in Nude Mice,” Cancer Research, Vol. 66, No. 9, 2006, pp. 4758-4765.
- A. Petronelli, G. Pannitteri and U. Testa, “Triterpenoids as New Promising Anticancer Drugs,” Anticancer Drugs, Vol. 20, No. 10, 2009, pp. 880-892. doi:10.1097/CAD.0b013e328330fd90
- J. Wesierska-Gadek, J. Bednarek and Z. M. Kiliańska, “New Face of Antiapoptotic Proteins. II. Survivin,” Postepy Biochemii, Vol. 53, No. 32, 2007, pp. 239-253.
- A.Nassar, D. Lawson and G. Cotsonis, “Survivin and Caspase-3 Expression in Breast Cancer: Correlation with Prognostic Parameters, Proliferation, Angiogenesis, and Outcome,” Applied Immunohistochemistry & Molecular Morphology, Vol. 16, No. 2, 2008, pp. 113-120. doi:10.1097/PAI.0b013e318032ea73
- R. Zhao, G. A. Follows and P. A. Beer, “Inhibition of the Bcl-xL Deamidation Pathway in Myeloproliferative Disorders,” The New England Journal of Medicine, Vol. 359, No. 26, 2008, pp. 2778-2789. doi:10.1056/NEJMoa0804953