Journal of Cancer Therapy
Vol.2 No.1(2011), Article ID:4518,18 pages DOI:10.4236/jct.2011.21004
Epirubicin-[Anti-HER2/neu] Synthesized with an Epirubicin-(C13-imino)-EMCS Analog: Anti-Neoplastic Activity against Chemotherapeutic-Resistant SKBr-3 Mammary Carcinoma in Combination with Organic Selenium
![]()
1Department of Basic Sciences, College of Veterinary Medicine, Mississippi State University, Mississippi State, USA;
2Department Organic Chemistry, Mississippi State University, Mississippi State, USA;
3College of Osteopathic Medicine, William Cary University, Hattiesburg, USA.
Email: coyne@cvm.msstate.edu
Received October 16th, 2010; revised December 10th, 2010; accepted 17th, 2010.
Keywords: Epirubicin-(C13-Imino)-[Anti-HER2/neu], Chemotherapeutic-Resistant Mammary Carcinoma, HER2/neu, Selenium, Synthesis
ABSTRACT
Purpose: Discover the anti-neoplastic efficacy of epirubicin-(C13-imino)-[anti-HER2/neu] against chemotherapeuticresistant SKBr-3 mammary carcinoma and delineate the capacity of selenium to enhance it’s cytotoxic anti-neoplastic potency. Methods: In molar excess, EMCH was combined with epirubicin to create a covalent epirubicin-(C13-imino)- EMCH-maleimide intermediate with sulfhydryl-reactive properties. Monoclonal immunoglobulin selective for HER2/neu was then thiolated with 2-iminothiolane at the terminal ε-amine group of lysine residues. The sulfhydryl-reactive epirubicin-(C13-imino)-EMCH intermediate was then combined with thiolated anti-HER2/neu monoclonal immunoglobulin. Western-blot analysis was utilized to characterize the molecular weight profiles while binding of epirubicin-(C13-imino)- [anti-HER2/neu] to membrane receptors was determined by cell-ELISA utilizing populations of SKBr-3 mammary carcinoma that highly over-expresses HER2/neu complexes. Anti-neoplastic potency of epirubicin-(C13-imino)-[anti-HER2/ neu] between the epirubicin-equivalent concentrations of 10–12 M and 10–7 M was determined by vitality staining analysis with and without the presence of selenium (5 μM). Results: Epiribucin-(C13-imino)-[anti-HER2/neu] between epirubicin-equivalent concentrations of 10–8 M to 10–7 M consistently evoked higher anti-neoplastic potency than “free” nonconjugated epirubicin which corresponded with previous investigations utilizing epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin-(C3-amide)-[anti-EGFR]. Selenium at 5 mM consistently enhanced the cytotoxic anti-neoplastic potency of epirubicin-(C13-imino)-[anti-HER2/neu] at epirubicin equivalent concentrations (10–12 to 10–7 M). Conclusions: Epirubicin-(C13-imino)-[anti-HER2/neu] is more potent than epirubicin against chemotherapeutic-resistant SKBr-3 mammary carcinoma and selenium enhances epirubicin-(C13-imino)-[anti-HER2/neu] potency. The methodology applied for synthesizing epirubicin-(C13-imino)-[anti-HER2/neu] is relatively time convenient and has low instrumentation requirements.
1. Introduction
Monoclonal immunoglobulin fractions with binding avidity for HER2/neu and EGFR have demonstrated effectiveness in the treatment of mammary carcinoma and other neoplastic disease states that over-express these trophic membrane-associated receptors. Unfortunatelyimmunoglobulin-based therapeutics of this type reportedly have an inability to exert significant cytotoxic activity or completely resolve neoplastic conditions [1-7] unless they are applied in combination with chemotherapy or other forms of anti-cancer treatment [8,9]. Despite general familiarity with the influence of anti-HER2/neu immunoglobulin on cancer cell biology and its application in clinical oncology there is surprisingly little known about covalent anthracycline-[anti-HER2/neu] immunochemotherapeutics and their potential to exert enhanced levels of anti-neoplastic activity against chemotherapeutic-resistant mammary carcinoma [10].
A small collection of organic chemistry reactions can be used to covalently link anthracycline-class chemotherapeutic agents to monoclonal immunoglobulin or other biologically active protein fractions. One common methodology for the synthesis of anthracycline conjugates involves the creation of a covalent amide bond at the single C3 α-monoamine group of the anthracycline carbohydrate moiety [10-13]. Dual combinations of epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin- (C3-amide)-[anti-EGFR] have demonstrated synergistic levels of anti-neoplastic activity against chemotherapeutic-resistant SKBr-3 mammary carcinoma which is complemented by the simultaneous application of competitive P-glyco-protein inhibitors [10].
Generation of an imino covalent bond at the C13-keto group of anthracyclines utilizing reactive hydrazides is an alternative synthesis strategy [14-18]. Chemically reactive anthracycline (C13-imino) intermediates produced in this manner have been applied to synthesize a variety of composite chemotherapeutic preparations through the formation of a secondary covalent bond to a variety of ligands and carrier molecules including HPMA (N-(2-hydroxy-propyl)-methacrylamide) [19,20], polyethylene glycol (PEG) [21], DNA aptamer [22], transferin [23,24], albumin/lactosaminated albumin [14,15]. Similarly, doxorubicin-EMCH derivatives with a sulfhydryl-reactive maleimide moiety (e.g. DOXO-EMCH) have been synthesized which form conjugates with serum albumin following intravenous administration and are subsequently internalized by certain neoplastic cell types [16,25,26].
Immunochemotherapeutics synthesized through the creation of a covalent imino bond at the C13-keto group of anthracyclines have been described [27-29] utilizing only a rather limited spectrum of monocloncal immunoglobulin fractions. Furthermore, in contrast to immunoglobulin-based diagnostic radiopharmaceuticals and radioimmunotherapeutics, there are few descriptions of the molecular design, synthesis and efficacy evaluation of covalent anthracycline immunochemotherapeutics that exert selective anti-neoplastic properties against mammary carcinoma propagated in-vitro in tissue culture [30,31], in-vivo xenografts [32], or natural clinical disease states. Immunochemotherapeutics synthesized as anthracycline (C13-imino)-immunoglobulin with selective “targeted” delivery properties for breast cancer have predominately utilized anti-Lewis Y antigen monoclonal immunoglobulin (e.g. BR96/SGN15) [32-35] in part because it has cross-cancer-efficacy against lung carcinoma [32,35,36], intestinal carcinoma [32-34,37], and ovarian carcinoma [33]. The full anti-neoplastic efficacy of covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutics proportedly increased through enhanced acid-sensitive mediated liberation of the anthracycline within the micro-environment of the phagolysosome/endolysosome (pH 5.0 - 5.5) remains to be delineated. Similarly, although previous investigations have characterized the influence of selenium on the biology of various neoplastic cell type, little is know about the potential for selenium to enhance the cytotoxiic anti-neoplastic potency of epirubicin-(C13-imino)-[anti-HER2/neu] against chemotherapeutic-resistant mammary carcinoma.
2. Materials and Methods
2.1. Synthesis Methodology I: Epirubicin- (C13-keto)-EMCH-Immunoglobulin
Phase-I: Immunoglobulin Thiolation at Lysine ε-Amine Groups-A purified fraction of monoclonal immunoglobulin with binding-avidity specifically for human HER2/neu (ErbB-2, CD 340) was utilized for the semi-synthesis of epirubicin-(C13-imino)-[anti-HER2/neu]. Dessicated antiHER2/neu monoclonal immunoglobulin (1.5 mg) was combined with 2-iminothiolane (2-IT: 6.5 mM final concentration) in PBS (0.1 M, pH 8.0, 250 μl) and incubated at 25˚C for 1.5 hours in combination with simultaneous constant gentle stirring [11,38-40]. Thiolated anti-HER2/ neu monoclonal immunoglobulin was then buffer exchanged into PBS-EDTA (phosphate 0.1, NaCl 0.15 M, EDTA 10 mM, pH 7.3) using micro-scale column chromatography. Moles of reduced sulfhydryl groups covalently introduced into anti-HER2/neu monoclonal immunoglobulin was measured with a 5,5’-dithiobis-(2- nitrobenzoic acid (DTNB reagent) based assay. The average number of thiolated lysine ε-amine groups introduced into anti-HER2/neu fractions (R-SH/IgG) was 3:1 using 2-IT reagent.
Phase-II: Synthesis of Epirubicin-(C13-imino)-EMCH Sulfhydryl Reactive Intermediate-The C13-keto group of epirubicin (1.479 × 10–2 mg, 2.55 × 10–5 mMole in methanol) was reacted with the hydrazide group of the heterobifunctional covalent cross-linking reagent, N-ε- maleimi-docaproic acid hydrazide in the presence of trifluoroacetic acid (EMCH: 43.2 μg, 1.275 × 10–4 mmol in methanol) and then incubated at 25˚C for 96 hours in concert with constant gentle stirring [17,24]. Recrystalization of epirubicin-(C13-imino)-EMCH to remove residual unreacted EMCH was performed by the addition of acetonitrile until a slight opalescent appearance developed followed by incubation at –20˚C for 24 hours. Recrystalized epirubicin-EMCH was harvested by centrifugation and rinsed in cold acetonitrile. The resulting recrystalization supernatant was partially evaporated under a stream of nitrogen (N2) gas in order to maximize yield of total epirubicin-(C13-imino)-EMCH product. Prior to Phase II synthesis procedures, residual methanol in aliquots of recrystalized epirubicin-EMCH was removed under a gentle stream of nitrogen gas and then re-dissolved in dimethylsulfoxide (DMSO).
Phase-III: Covalent Incorporation of Epirubicin-(C13- imino)-EMCH into Thiolated Immunoglulin at Lysine ε-Amine Groups-The primary reduced sulfhydryl groups (R-SH) of thiolated lysine ε-amines within HER2/neu monoclonal immunoglobulin contained in PBS-EDTA (phosphate 0.1, NaCl 0.15 M, EDTA 10 mM, pH 7.3) was combined with the sulfhydryl-reactive maleimido group of epirubicin-EMCH and allowed to react while incubating at 25˚C with continual gentle stirring for 2 hours. Residual epirubicin was removed from epirubicin- [anti-HER2/neu] applying micro-scale “desalting” column chromatography after pre-equilibration with PBS (phosphate 0.1, NaCl 0.15 M, pH 7.3).
2.2. Synthesis Methodology II: Epirubicin- (C3-amide)-SMCC-Immunoglobulin
Phase-I: Introduction of Sulfhydryl Groups into Immunoglobulin at Lysine ε-Amine Groups-Monoclonal Immunoglobulins with selective binding avidity against human epidermal growth factor receptor type 1 (EGFR) and HER2/neu (ErbB-2, CD 340) were acquired as desiccated preparations in 1.5 mg amounts. Simultaneous removal of xylose and buffer-exchange into PBS (phosphate 0.1 M, NaCl, 0.15 M, pH 7.3) was performed prior to semi-synthesis procedures using micro-scale “desalting” column chromatography resulting in a final IgG concentration of 13.3 μM (> 2.0 mg/ml in 700 μl).
Individual IgG monoclonal antibodies at a concentration of approximately 2.0 mg/ml in 700 μl of PBS where combined with N-succinimidyl-S-acetylthioacetate (SATA) at a molar ratio of 1:9 (3 μl of a 55 mM SATA formulation in DMSO). Preparations were incubated at 25˚C for 30 minutes and then buffer exchanged into PBS (phosphate 0.1 M, NaCl 0.15 M, pH 7.3) using micro-scale “desalting” column chromatography.
Phase-II: Synthesis of Epirubicin-(C3-amide)-SMCC Sulfhydryl Reactive Intermediate-The α-monoamide group of epirubicin (168.3 μl of 2 mg/ml aqueous stock) was reacted with the N–hydroxysuccinimide (NHS) group of succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC: 5.61 μl of 10 mM stock in DMSO) at a 7.5:1 molar ratio and allowed to mix with constant stirring at 25˚C for 30 minutes.
Phase-III: Covalent Incorporation of Epirubicin-(C3- amide)-SMCC at SATA Modified Immunoglobulin Lysine ε-Amine Groups-Immediately prior to Phase-III synthesis methods, SATA-IgG preparations were deacetylated (activated) with hydroxylamine (0.5 M with EDTA 25 mM in PBS, pH 7.3) at a 10:1 volumetric ratio for 2 hours with continual stirring at 25˚C thereby generating a primary sulfhydryl group. Residual unreacted SATA was removed from MoAb IgG preparations by buffer exchange into PBS-EDTA (phosphate 0.1, NaCl 0.15 M, EDTA 10 mM, pH 7.3) using micro-scale “desalting” column chromatography. Sulhydryl content was subsequently determined using an Ellman’s Reagent based assay system.
The primary sulfhydryl group of deacetylated SATAIgG preparations was subsequently reacted with the maleimido group of SMCC-epirubicin followed by incubation at 25˚C with continual stirring for 30 minutes. Residual epirubicin was removed from covalent epirubicin immunochemotherapeutic preparations by a buffer exchange into PBS (phosphate 0.1, NaCl 0.15 M, EDTA 10 mM, pH 7.3) using micro-scale “desalting” column chromatography.
2.3. Analysis, Characteristics and Properties
General Analysist-Determination of the IgG concentration within covalent epirubicin-[anti-HER2/neu] immunochemotherapeutics was determined by measuring absorbance at 280 nm. Measurement of epirubicin concentrations was established by excitation at 485 nm and measurement of emission at 538 nm using known concentrations of epirubicin to generate a standard reference control curve sufficient to generate a linear equation for determination of epirubicin-equivalent concentrations between 10–9 M and 10–5 M. Determination of the non-conjugated “free” epirubicin concentration contained in covalent epirubicin immunochemotherapeutic preparations was established by chloroform extraction [19,41, 42], with the organic phase collected by pipette, evaporated to dryness under a stream of nitrogen gas, and the resulting residue dissolved in Tris buffered saline (50 mM, pH 7.4) prior to further analysis. Adjusted epirubicin:immunoglobulin molar incorporation indexes were calculated by measuring absorbance at 485 nm and 280 nm respectively and by correcting for absorbance from epirubicin at 280 nm.
Molecular Mass-Dependent Separation of Covalent Epirubicin Immunochemotherapeutics by Non-Reducing SDS-PAGE-Covalent epirubicin-[anti-HER2/neu] immunochemotherapeutics in addition to a anti-HER2/neu immunoglobulin reference control fraction were adjusted to a standardized protein concentration of 60 μg/ml and then combined 50/50 v/v with conventional SDS-PAGE sample preparation buffer (Tris/glycerol/bromphenyl blue/ SDS) formulated without 2-mercaptoethanol or boiling. The epirubicin immunochemotherapeutics, a reference control anti-HER2/neu immunoglobulin fraction (0.9 μg/well) and a mixture of pre-stained molecular weight markers were then developed by non-reducing SDSPAGE (11% acrylamide) performed at 100 V constant voltage at 3˚C for 2.5 hours.
Western-Blot Immunodetection Analyses-Covalent epirubicin-[anti-HER2/neu] immunochemotherapeutics following mass-dependent separation by non-reducing SDSPAGE were equilibrated in tank buffer devoid of methanol. Mass-separated epirubicin-[anti-HER2/neu] immunochemotherapeutics contained in acrylamide SDS-PAGE gels were then laterally transferred onto sheets of nitrocellulose membrane at 20 volts (constant voltage) for 16 hours at 2˚C to 3˚C with the transfer manifold packed in crushed ice.
Nitrocellulose membranes with laterally-transferred epirubicin immunochemotherapeutics were equilibrated in Tris buffered saline (TBS: Tris HCl 0.1 M, NaCl 150 mM, pH 7.5, 40 ml) at 4˚C for 15 minutes followed by incubation in TBS blocking buffer solution (Tris 0.1 M, pH 7.4, 40 ml) containing bovine serum albumin (5%) for 16 hours at 2˚C to 3˚C applied in combination with gentle horizontal agitation. Nitrocellulose membranes were then vigorously rinsed in Tris buffered saline (Tris 0.1 M, pH 7.4, 40 ml, n = 3x).
Rinsed BSA-blocked nitrocellulose membranes developed for Western-blot (immunodetection) analyses were incubated with biotinylated goat anti-murine IgG (1:10,000 dilution) at 4˚C for 18 hours applied in combination with gentle horizontal agitation. Nitrocellulose membranes were then vigorously rinsed in TBS (pH 7.4, 4˚C, 50 ml, n = 3) followed by incubation in blocking buffer (Tris 0.1 M, pH 7.4, with BSA 5%, 40 ml). Blocking buffer was decanted from nitrocellulose membrane blots and then rinsed in TBS (pH 7.4, 4˚C, 50 ml, n = 3) before incubation with strepavidin-HRPO (1:100,000 dilution) at 4˚C for 2 hours applied in combination with gentle horizontal agitation. Prior to chemiluminescent development nitrocellulose membranes were vigorously rinsed in Tris buffered saline (Tris 0.1 M, pH 7.4, 40 ml, n = 3). Development of nitrocellulose membranes by chemiluminescent autoradiography following processing with conjugated HRPO-strepavidin required incubation in HRPO chemiluminescent substrate (25˚C; 5 to 10 mins.). Autoradiographic images were acquired by exposing radiographic film (Kodak BioMax XAR) to nitrocellulose membranes sealed in transparent ultraclear re-sealable plastic bags.
2.4. Mammary Carcinoma Tissue Culture Cell Culture
Chemotherapeutic-resistant SKBr-3 human mammary carcinoma populations were utilized as an ex-vivo neoplasia model. Characteristically, SKBr-3 mammary carcinoma uniquely over-expresses epidermal growth factor receptor 1 (EGFR, ErbB-1, HER1), and highly over-expresses epidermal growth factor receptor 2 (EGFR2, HER2/neu, ErbB-2, CD340, p185) at 2.2 × 107/cell and 1 × 106/cell respectively.
Populations of the SKBr-3 mammary carcinoma cell line were propagated in tissue culture flasks (150-cc2) containing McCoy’s 5a Medium Modified supplemented with fetal bovine serum (10% v/v) and penicillin-streptomycin at a temperature of 37˚C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2). Tissue culture media was not supplemented with growth factors, growth hormones or other biological stimulant of any type. Investigations were performed using SKBr-3 mammary carcinoma monolayer populations at a ≥85% level of confluency.
Cell-ELISA IgG Binding Assay-Cell suspensions of SKBr-3 mammary carcinoma were seeded into 96-well microtiter plates in aliquots of 2 × 105 cells per well and allowed to form adherent monolayers over a period of 48 hours (confluency  80%). Growth media within individual wells was removed manually by pipette followed by serially rinsed (n = 3) with PBS and stabilization of adherent SKBr-3 cellular monolayers onto the plastic surface of 96-well plates with paraformaldehyde (4% in PBS for 15 minutes). Stabilized SKBr-3 monolayers were then incubated with epirubicin-(C13-imino)- [anti-HER2/neu] or epirubicin-(C3-amide)-[anti-HER2/ neu] with epirubicin-(C3-amide)-[anti-EGFR] 50/50 immunochemotherapeutic combinations formulated at gradient total IgG concentrations of 0.1, 0.25, 0.5, 1.0, 5.0 and 10 μg/ml (200 μl/well) in tissue culture growth media. Following a 3-hour incubation period at 37˚C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2), monolayer SKBr-3 mammary carcinoma populations were serially rinsed with PBS (n = 3) and then incubated with β-galactosidase conjugated goat anti-mouse IgG (1:500 dilution) at 25˚C for 2 hours. Residual unbound immunoglobulin was then removed by serial rinsing with PBS (n = 3). Final cell-ELISA development required serial rinsing (n = 3) of stabilized SKBr-3 monolayers with PBS followed by incubation with nitrophenyl-β-Dgalactopyranoside substrate (ONPG 100 μl/well formulated fresh at 0.9 mg/ml in PBS at pH 7.2 containing MgCl2 10 mM, and 2-mercaptoethanol 0.1 M). Absorbance within each individual well was then measured at 410 nm (630 nm reference wavelength) after incubation at 37˚C for 15 minutes.
80%). Growth media within individual wells was removed manually by pipette followed by serially rinsed (n = 3) with PBS and stabilization of adherent SKBr-3 cellular monolayers onto the plastic surface of 96-well plates with paraformaldehyde (4% in PBS for 15 minutes). Stabilized SKBr-3 monolayers were then incubated with epirubicin-(C13-imino)- [anti-HER2/neu] or epirubicin-(C3-amide)-[anti-HER2/ neu] with epirubicin-(C3-amide)-[anti-EGFR] 50/50 immunochemotherapeutic combinations formulated at gradient total IgG concentrations of 0.1, 0.25, 0.5, 1.0, 5.0 and 10 μg/ml (200 μl/well) in tissue culture growth media. Following a 3-hour incubation period at 37˚C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2), monolayer SKBr-3 mammary carcinoma populations were serially rinsed with PBS (n = 3) and then incubated with β-galactosidase conjugated goat anti-mouse IgG (1:500 dilution) at 25˚C for 2 hours. Residual unbound immunoglobulin was then removed by serial rinsing with PBS (n = 3). Final cell-ELISA development required serial rinsing (n = 3) of stabilized SKBr-3 monolayers with PBS followed by incubation with nitrophenyl-β-Dgalactopyranoside substrate (ONPG 100 μl/well formulated fresh at 0.9 mg/ml in PBS at pH 7.2 containing MgCl2 10 mM, and 2-mercaptoethanol 0.1 M). Absorbance within each individual well was then measured at 410 nm (630 nm reference wavelength) after incubation at 37˚C for 15 minutes.
Cell Vitality Assay for Measuring Immunochemotherapeutic Cytotoxicity-Individual preparations of epirubicin-(C13-imino)-[anti-HER2/neu] or epirubicin-(C3-amide)- [anti-HER2/neu] in 50/50 combination with epirubicin-(C3-amide)-[EGFR] were formulated in growth media at standardized epirubicin-equivalent concentrations of 10–12, 10–11, 10–10, 10–9, 10–8, and 10–7 M (final concentrations). Each epirubicin-equivalent concentration of epirubicinimmunoglobulin covalent biopharmaceuticals was then transferred in triplicate into 96-well microtiter plates containing SKBr-3 mammary carcinoma monolayers (growth media 200 μl/well total volume).
Potential for selenium to enhance the anti-neoplastic potency of epirubicin-[anti-HER2/neu] against SKBr-3 mammary carcinoma was determined by incubating cell populations with methylseleninate (CH3SeO2H at 5 μM or 0.635 μg/ml) [43-46] at a concentration pre-determined to not promote a markedly detrimental influence on cell vitality. Epirubicin immunochemotherapeutics formulated with or without selenium where then incubated with monolayer populations of SKBr-3 mammary carcinoma for 72-hours at 37˚C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2). Complementary investigations evaluated the influence of increasing selenium concentrations (5 μM to 50 μM) in combination with a fixed epirubicin concentration (10–8 M), in addition to increasing epirubicin concentration (10–12 M to 10–7 M) in combination with a fixed selenium concentration (45 mM) over a 72-hour incubation period with chemotherapeutic-resistant SKBr-3 mammary carcinoma. Lastly, as a point of reference, selenium cytotoxic anti-neoplatic activity was compared to equal micro-molar concentrations of celecoxib in chemotherapeutic-resistant SKBr-3 mammary carcinoma.
The contents of 96-well microtiter plates at 72-hours were removed manually by pipette and SKBr-3 mammary carcinoma monolayers serially rinsed (n = 3) with PBS followed by incubation with MTT vitality stain reagent (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide 5mg/ml) formulated in RPMI-1640 growth media devoid of pH indicator or bovine fetal calf serum. During an incubation period of 3 hours at 37˚C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2) the enzyme mitochondrial succinate dehydrogenase was allowed to convert MTT vitality stain to navy-blue formazone crystals. Contents of the 96-well microtiter plate were then removed, and serially rinsed with PBS (n = 3) followed by dissolving of the resulting blue intracellular formzone crystals with DMSO (300 μl/well). Spectrophotometric absorbance of the navy-blue colored supernatant was then measured at 570 nm using a computer integrated microtiter plate reader.
3. Results
In Phase I of the synthesis method, the C13-keto group of epirubicin was reacted with the hydrazide group of N-ε-maleimidocaproic acid hydrazide (EMCH) to create an imino bond resulting in the formation of a covalent epirubicin-(C13-imino)-EMCH intermediate (Figure 1). In Phase II of the synthesis method, the thiol-reactive maleimide group of the epirubicin-(C13-imino)-EMCH intermediate was covalently linked to sulfhydryl groups introduced at the ε-amine of lysine amino acid residues within HER2/neu immunoglobulin fractions following thiolation with 2-iminothiolane (2-IT) reagent (Figure 1). The anthracycline molar incorporation index for epirubicin-(C13-imino)-[anti-HER2/neu] was 40% while epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin-(C3-amide)- [anti-EGFR] were produced at a level of 27.5% and 40.7% respectively utilizing essentially identical conditions [10]. The percent of non-covalently bound anthracycline contained in covalent epirubicin immunochemotherapeutics following the application of micro-scale desalting/buffer exchange column chromatography was consistently < 4.0% where residual non-covalently bound anthracycline generally can not be removed by performing serial/repeated separations by column chromatography) [23]. Higher epirubicin molar incorporation indexes are possible to achieve with modifications in the synthesis methodology but the harsher conditions required for such purposes are accompanied by substantial reductions in the final yield of covalent immunochemotherapeutic [47], and declines in antigen-immunoglobulin binding affinity (e.g. cell-ELISA analysis).
Covalent epirubicin-[anti-HER2/neu] immunochemotherapeutics mass-separated by SDS-PAGE and developed by Western-blot immunodetection analysis in combination with chemiluminescent autoradiography detected a condensed 150-kDa band between a 5.0-kDa to 450-kDa molecular weight range (Figure 2) The molecular weight of 150-kDa detected for epiribucin-[antiHER2/neu] immunochemotherapeutics directly corresponded with the molecular weight/mass detected for the anti-HER2/neu immunoglobulin reference control fraction and the known molecular weight for immunoglobulin (Figure 2). Analogous results have been reported for similar covalent immunochemotherapeutics [10,14,48].
Cell-ELISA Binding AnalysisUtilizing standardized total immunoglobulin-equivalent concentrations formulated at 0.1, 0.25, 0.5, 1.0, 5.0, and 10.0 μg/ml the evaluation
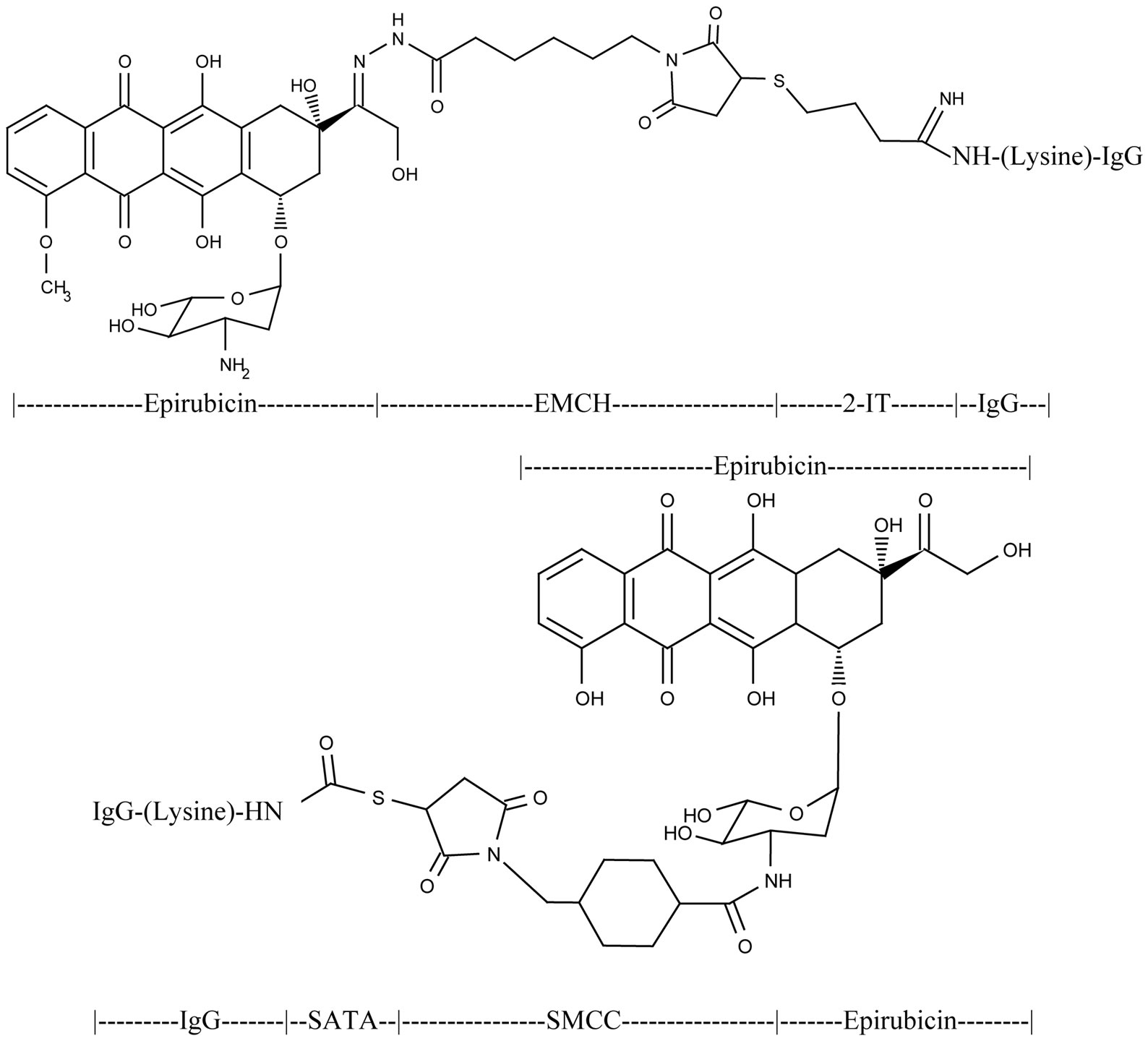
Figure 1. Top Panel-Covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic created using a 2-phase synthesis scheme. Phase-I: a maleimodocaproyl-hydrazone is created at the C13-keto group of the anthracycline through the carbonyl-reactive hydrazide group of N-ε-maleimidocaproic acid hydrazide (EMCH). Phase-II: the sulfhydryl-reactive maleimide group of the epirubicin-(C13-imino) maleimodocaproyl-hydrazone intermediate is covalently linked at/to thiolated lysine α-amine groups residing within anti-HER2/neu monoclonal immunoglobulin fractions. Bottom PanelCovalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic created using as 2-phase synthesis scheme. Phase I: an amide bond is created at the C3 α-monoamino group of the anthracycline through the amine-reactive N-hydroxysuccinimide (NHS ester) group of succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (SMCC). Phase-II: the sulfhydryl-reactive maleimide group of the epirubicin-(C3-amide) reactive intermediate becomes covalently linked at/to thiolated lysine ε-amine groups residing within anti-HER2/neu monoclonal immunoglobulin fractions.
of epirubicin-(C13-imino)-[anti-HER2/neu] by cell-ELISA produced profiles that revealed proportional increases in antibody bound to the external membrane of SKBr-3 mammary carcinoma populations in a manner that validated retained selective binding at HER2/neu receptor sites (Figure 3). Differences in total membrane-bound immunoglobulin detected between anti-HER2/neu compared to anti-EGFR reflect the greater membrane expression densities for HER2/neu (2.2 × 107/cell) compared to EGFR (1 × 106/cell) in populations of SKBr-3 mammary carcinoma (Figure 3) [10].
Cytotoxicity Analysis-The anti-neoplastic potency of

Figure 2. Western-blot autoradiography of covalent epirubicin-(C13-imino)-[anti-HER2/neu] and epirubicin-(C3-amide)-[antiHER2/neu] immunochemotherapeutics. Legend: Lane-1-murine anti-human EGFR immunoglobulin; Lane-2-murine antihuman HER2/neu immunoglobulin; Lane-3-covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic; Lane-4-covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic; and Lane-5-covalent epirubicin-(C3-amide)- [anti-EGFR] immunochemotherapeutic. Immunoglobulin preparations were mass-separated by SDS-PAGE and transferred laterally onto sheets of nitrocellulose membrane to facilitate detection with biotinylated goat anti-mouse IgG. Subsequent analysis entailed incubation of membranes with conjugated strepavidin-HRPO in combination with the use of an HRPO chemiluminescent substrate to facilitate the acquisition of autoradiography images.
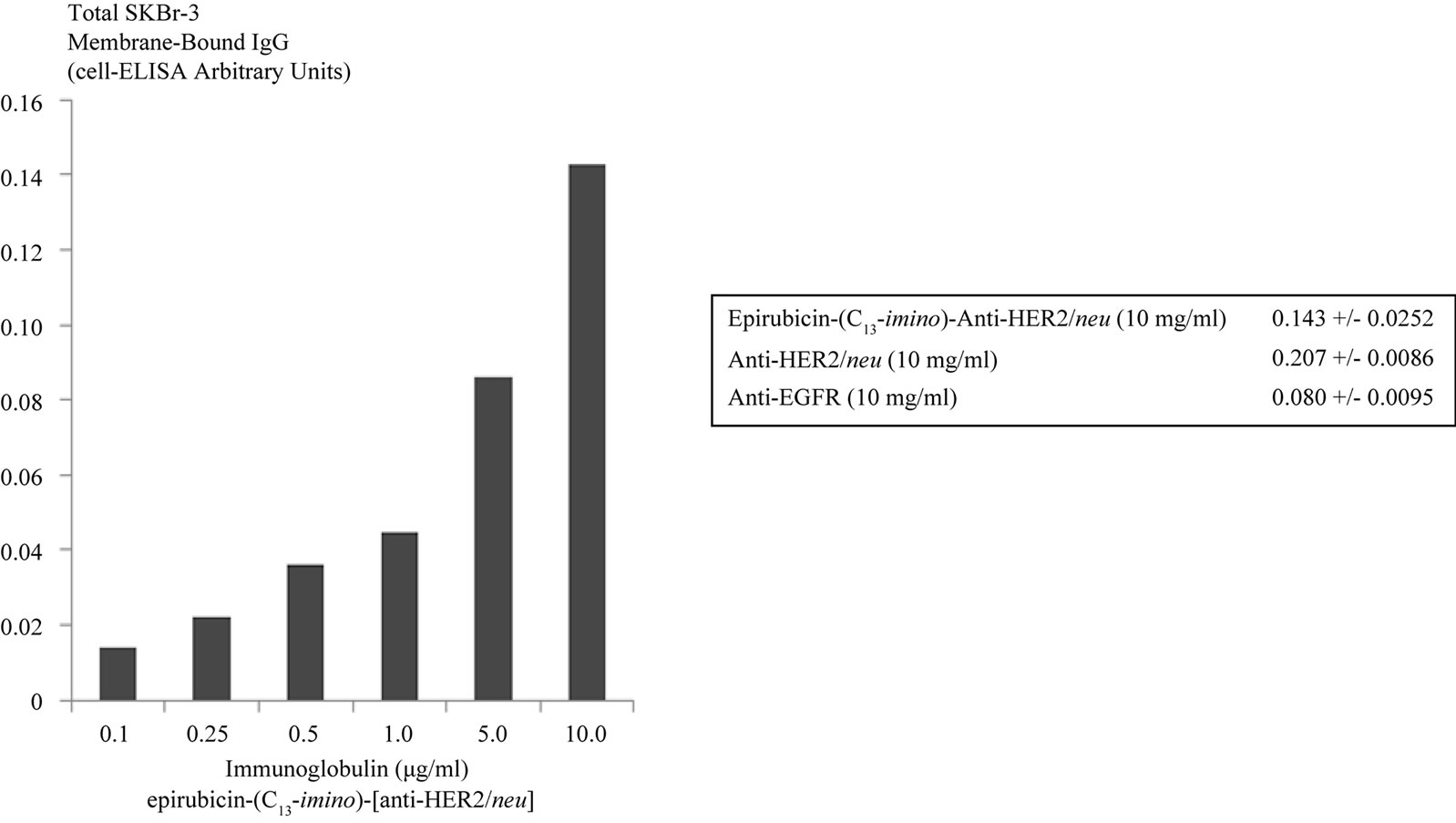
Figure 3. Immunoglobulin cell binding analysis for populations of chemotherapeutic resistant SKBr-3 mammary carcinoma. Legend: covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic incubated with monolayer populations of SKBr-3 mammary carcinoma over a 4-hour period total immunoglobulin bound on the exterior surface membrane was measured by cell-ELISA.
epirubicin-(C13-imino)-[anti-HER2/neu] against chemotherapeutic-resistant SKBr-3 mammary carcinoma was greater than chemotherapeutic-equivalent concentrations of epirubicin with differences at 10–8 M and especially 10–7 M being the most significant (Figure 4). Compared to a synergistic 50/50 combination of epirubicin-(C3- amide)-[anti-HER2/neu] with epirubicin-(C3-amide)-[antiEGFR] [10], essentially equivalent levels of anti-neoplastic activity were detected for epirubicin-(C13-imino)- [anti-HER2/neu] when formulated at epirubicin-equivalent concentrations between 10–12 M and 10–7 M (Figure 5).
Incubation of SKBr-3 mammary carcinoma populations with methylseleninate [49] (5 μM) produced cell vitality/proliferation levels of 89.5% following a 72-hour incubation period. Dual simultaneous exposure of chemotherapeutic-resistant SKBr-3 mammary carcinoma to selenium (5 μM) in combination with epirubicin-(C13- imino)-[anti-HER2/neu] consistently created lower levels of survivability than epirubicin-(C13-imino)-[anti-HER2/ neu] when compared at epirubicin-equivalent concentrations between 10–12 M to 10–7 M (Figure 6). Gradient increases in selenium concentration (5 μM to 50 μM) at a fixed epirubicin concentration (10–8 M) produced additive increases in cytotoxic anti-neoplastic potency against chemotherapeutic-resistant SKBr-3 mammary carcinoma populations (Figure 7). Similarly, gradient increases in

Figure 4. Influence of covalent bonding anti-HER2/neu monoclonal immunoglobulin on the cytotoxic anti-neoplastic potency of epirubicin against chemotherapeutic-resistant SKBr-3 mammary carcinoma. Legend: (u) covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic, and (n) epirubicin chemotherapeutic. Monolayer populations of SKBr-3 mammary carcinoma were incubated with epirubicin immunochemotherapeutic for 72-hours and cytotoxicity measured as a function of cell MTT vitality relative to matched negative reference controls.
epirubicin concentration (10–12 M to 10–7 M) in combination with a fixed selenium concentration (45 mM) additively increased total cytotoxic anti-neoplastic potency exerted against chemotherapeutic-resistant SKBr-3 mammary carcinoma (Figure 8). When utilized as a combination, epirubicin and selenium exerted progressive increases in cytotoxic anti-neoplastic potency where a detectable threshold effect was observed individually with either epirubicin or selenium (Figures 7 and 8). Compared to celecoxib, micro-molar equivalent concentrations of selenium exerted higher cytotoxic anti-neoplastic potency against chemotherapeutic resistant SKBr-3 mammary carcinoma either alone or in combination with epirubicin chemotherapeutic (Figure 9).
Individual fractions of anti-HER2/neu and anti-EGFR monoclonal immunoglobulin alone did not exert any detectable cytotoxic anti-neoplastic activity against SKBr-3 mammary carcinoma at 72-hours (Figure 7) which is in accord with previous investigations [10,48,50-53].
4. Discussion
One proposed attribute of covalent anthracycline immunochemotherapeutics synthesized utilizing a reactive hydrazide is that it creates a covalent but “acid-sensitive”
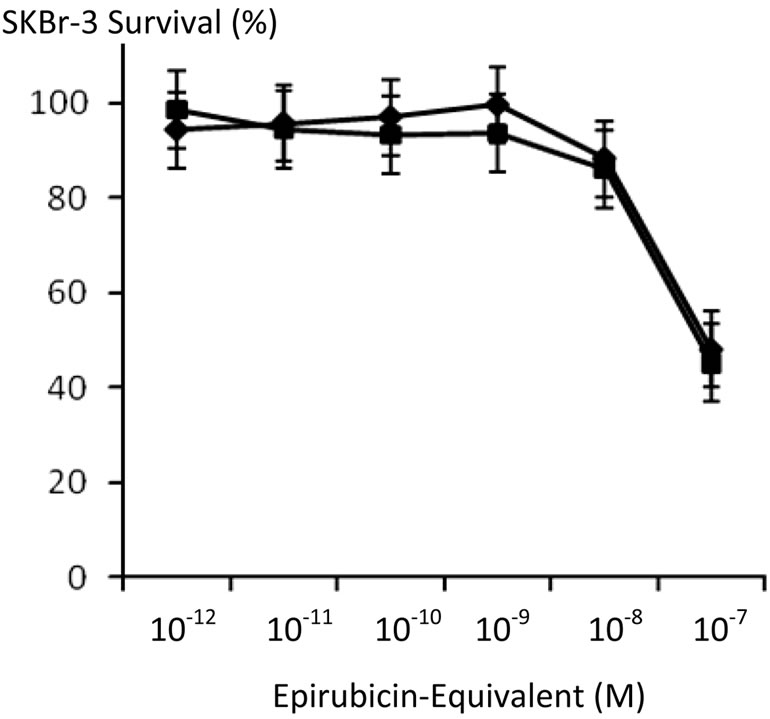
Figure 5. Relative anti-neoplastic potency of covalent epirubicin-(C13-imino) and epirubicin-(C3-amide) immunochemotherapeutics against chemotherapeutic-resistant SKBr-3 mammary carcinoma. Legend: (u) covalent epirubicin- (C13-imino)-[anti-HER2/neu] epirubicin immunochemotherapeutic, and (n) covalent epirubicin-(C3-amide)-[antiHER2/neu] with epirubicin-(C3-amide)-[anti-EGFR] formulated as a 50/50 immunochemotherapeutic combination. Previous investigations revealed that a 50/50 immunochemotherapeutic combination of covalent epirubicin-(C3- amide)-[anti-HER2/neu] with epirubicin-(C3-amide)-[antiEGFR] was more potent than either covalent epirubicin- (C3-amide) immunochemotherapeutic alone [10]. Monolayers of SKBr-3 mammary carcinoma were incubated with covalent epirubicin-immunochemotherapeutic for 72-hours and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
imino bond at the C13-keto position of doxorubicin or epirubicin and the formation of a thiol-reactive anthracycline-(C13-imino)-EMCH intermediate [14,15,20,21,54].
The maleimide moiety of anthracycline-(C13-imino)- EMCH intermediates is subsequently reacted with reduced sulfhydryls (R-SH) groups. Hydrazides utilized in related synthesis strategies have included 4-[N-maleimidomethyl] cyclohexane-1 carboxyl-hydrazide [52,55], 6- maleimidocaproyl-hydrazide (3,3’-N-[ε-maleimidocaproic acid] hydrazide) [14-16,26], and other analogous heterobifunctionally reactive reagents [15]. The sulfhydrylreactive maleimide groups incorporated into anthracyline intermediates then form a covalent bond with either cysteine amino acid residues [14,15,18] or thiolated ε-amine groups of lysine amino acid residues [19,27,52,56,57] within biological protein fractions.
The acid-sensitive properties of anthracycline-(C13- imino) immunochemotherapeutics generated through the

Figure 6. Influence of selenium on the potency of covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic against chemotherapeutic-resistant SKBr-3 mammary carcinoma. Legend: (u) covalent epirubicin-(C13- imino)-[anti-HER2/neu] immunochemotherapeutic, and (n) covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic with selenium (5 mM). Monolayers of SKBr-3 mammary carcinoma were incubated for 72-hours and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
formation of a covalent imino (hydrazone) bond is vulnerabile to pH-dependent hydrolysis. Due to this property, the epirubiin moiety is reportedly more extensively liberated within the acidic micro-environment of the phagolysosome (pH 5.0 - 5.5) [28,29,58] following internalization by mechanisms of receptor-mediated endocytosis [59].
Cell Binding-Total membrane bound IgG profiles in SKBr-3 mammary carcinoma populations for epirubicin-(C13-imino)-[anti-HER2/neu] proportionally increased with elevations in total immunoglobulin (Figure 3). Seemingly modest alterations in synthesis chemistry and elevations in chemotherapeutic molar incorporation index can profoundly influence immunoglobulin binding properties [53]. Given this perspective, the relatively mild conditions employed during the course of synthesis procedures and relatively modest 40% molar incorporation index collectively contributed to the high biological integrity of epirubicin-(C13-imino)-[anti-HER2/neu] based on results from SDS-PAGE/immunodetection and cellELISA analyses. Relatively higher anthracycline immunoglobulin molar incorporation indexes can be attained by implementing harsher synthesis conditions. Such modifications often result in only modest declines in
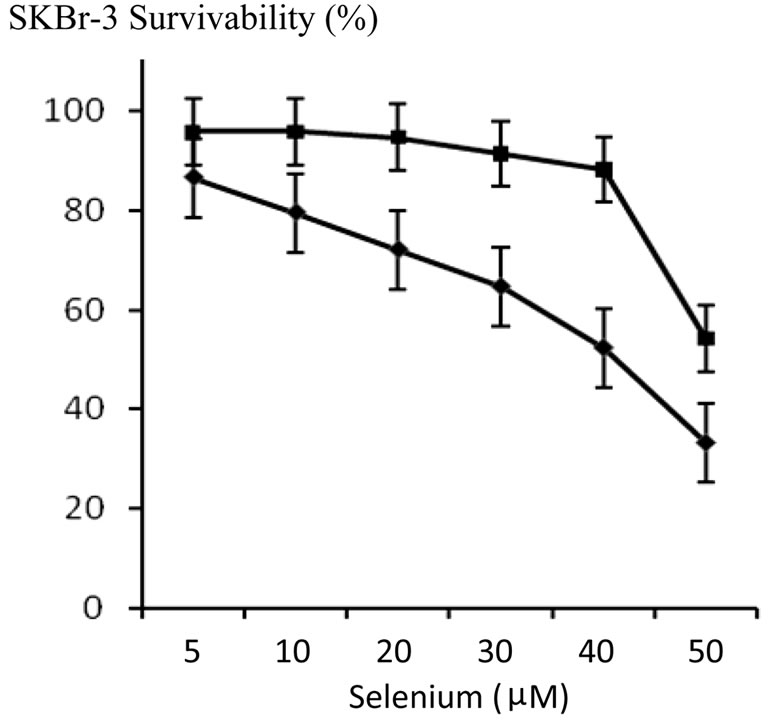
Figure 7. Influence of increasing selenium concentrations on the cytotoxic anti-neoplastic potency of epirubicin (10–8 M) measured as a function of chemotherapeutic-resistant SKBr-3 mammary carcinoma cell vitality. Legend: (n) selenium (methylseleninate); and (u) selenium (methylseleninate) in combination with epirubicin (10–8 M). SKBr-3 monolayers were incubated with immunochemotherapeutics over a 72-hour period and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
immunoreactivity (e.g. 86% for a 73:1 ratio) but disproportionate declines in anti-neoplastic activity down to potency levels substantially lower than those measured for non-conjugated “free” anthracycline [60]. Internalization of covalent epirubicin-[anti-HER2/neu] and epirubicin-[anti-EGFR] immunochemotherapeutics following binding to HER2/neu and EGFR receptors over-expressed by SKBr-3 mammary carcinoma occurs predominately by mechanisms of receptor-mediated endocytosis analogous to previous descriptions for similar covalent anthracycline immunochemotherapeutics [59]. Although specific data for SKBr-3 HER2/neu and EGFR receptor complexes is limited [10], other neoplastic cell lines like metastatic multiple myeloma are known to internalize and metabolize approximately 8 × 106 molecules of anti-CD74 monoclonal antibody per day [61].
Cytotoxicity-Covalent bonding of epirubicin to monoclonal immunoglobulin fractions enhanced the anti-neoplastic potency of epirubicin-(C13-imino)-[anti-HER2/neu] against chemotherapeutic-resistant SKBr-3 mammary carcinoma relative to epirubicin formulated at chemotherapeutic-equivalent concentrations (Figure 4). Similar properties have been identified for epirubicin-(C3-amide)- [anti-HER2/neu] [10] and epirubicin-(C3-amide)-[anti-
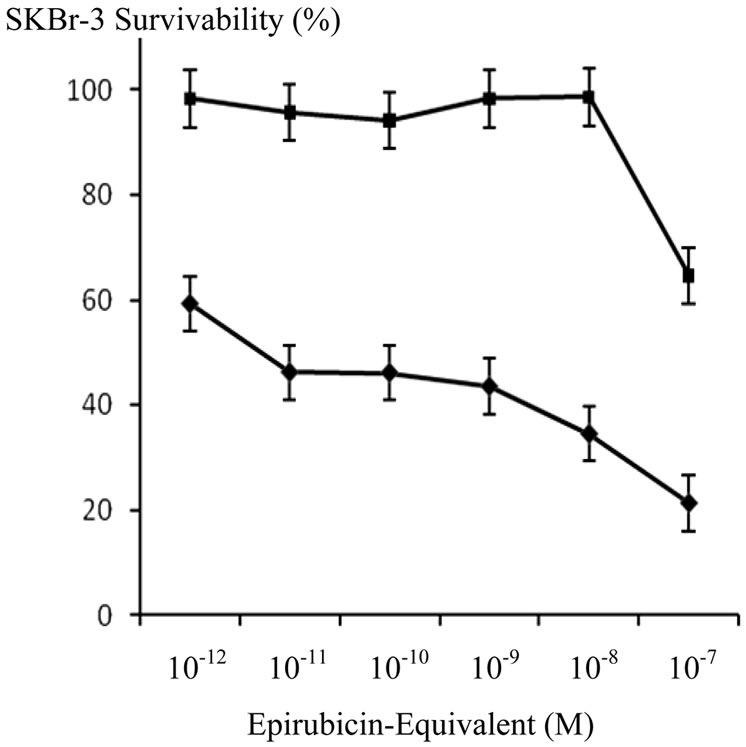
Figure 8. Influence of increasing epirubicin concentrations on the cytotoxic potency of selenium (45 μM) measured as declines in the cellular vitality of chemotherapeutic-resistant SKBr-3 mammary carcinoma. Legend: (u) epirubicin chemotherapeutic in combination with selenium (methylseleninate 45 μM); and (n) epirubicin chemotherapeutic alone. SKBr-3 monolayers were incubated with immunochemotherapeutics over a 72-hour period and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
EGFR] [10] in addition to other analogous covalent immunochemotherapeutic preparations designed to selectively “target” the delivery of anthracyclines on the exterior surface membrane of neoplastic cell types [28,50,51, 53,62,63]. The mechanisms that account for the greater cytotoxic anti-neoplastic activity of epirubicin-(C13-imino)- [anti-HER2/neu], epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin-(C3-amide)-[anti-EGFR] relative to molar-equivalent epirubicin concentrations can collectively be attributed to selective membrane deposition and continual receptor-mediated endocytosis that in turn facilitate progressive epirubicin intracellular internalization and cytosol accumulation [53,59,64,65]. Receptor-mediated endocytosis at membrane HER2/neu complexes can result in intracellular chemotherapeutic levels that approach and exceed 8.5 [65] to > 100 x fold greater [64] concentrations than is possible by simple anthracycline passive diffusion. In this scenario, > 50% of selectively “targeted” anthracycline at 24 hours is retained intracellularly [65] and becomes primarily associated with either membrane structures or is distributed throughout the cytosol environment [59,66]. Conversely, “free” non-conjugated anthracycline following simple passive diffusion across intact lipid bilayer membranes is primarily detected within complexes associated with nuclear DNA less than 30 minutes after initial exposure [59] while anthracycline liberated from covalent anthracycline immunochemotherapeutics reportedly distributes to, and

Figure 9. Relative cytotoxic anti-neoplastic potency of selenium and celecoxib measured as a function of chemotherapeutic-resistant SKBr-3 mammary carcinoma survivability with and without epirubicin (10–7 M). Legend: (n) selenium (methylseleninate) 30 μM, 40 μM, and 50 μM with and without the presence of epirubicin at 10–7 M fixed concentration; (n) celecoxib at 30 μM, 40 μM, and 50 μM with and without the presence of epirubicin at 10–7 M fixed concentration. Monolayers of SKBr-3 mammary carcinoma were incubated with immunochemotherapeutics over a 72-hour period and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
accumulates within the nucleus, mitochondria and golgi compartments [23].
Epirubicin-(C13-imino)-[anti-HER2/neu] produced essentially equivalent levels of cytotoxic anti-neoplastic activity against chemotherapeutic-resistant SKBr-3 mammary carcinoma populations as did epirubicin-(C3-amide)- [anti-HER2/neu] applied synergistically in a 50/50 combination with epirubicin-(C3-amide)-[anti-EGFR] (Figure 5) [10]. Such properties imply that epirubicin-(C13- imino)-[anti-HER2/neu] is slightly more potent than either epirubicin-(C3-amide)-[anti-HER2/neu] or epirubicin-(C3-amide)-[anti-EGFR] when compared at epirubicin-equivalent concentrations [10]. In part these differences in cytotoxic anti-neoplastic activity could be attributed to the acid-sensitive anthracycline imino (hydrazone) bond structure within epirubicin-(C13-keto)-[antiHER2/neu] in contrast to epirubicin-(C3-amide)-[antiHER2/ neu] or epirubicin-(C3-amide)-[anti-EGFR] (Figure 1). Despite the proposed attributes of acid-labile anthracycline-aconityl conjugates, some reagents and methodologies have been found to yield preparations that liberate only 45% of the total covalently bound chemotherapeutic in an acid-labile manner [67].
Several modifications in analytical methodology could have been implemented to substantially increase the level of anti-neoplastic activity exerted by epirubicin-(C13- imino)-[anti-HER2/neu] and epirubicin-(C3-amide)-[antiHER2/neu]. Cytotoxic anti-neoplastic potency could have alternatively been assessed with a breast cancer cell type that was not chemotherapeutic-resistant similar to populations utilized to evaluate majority of the covalent biochemotherapeutics reported in the literature to date. Rare exceptions in this regard have been the evaluation of anti-chondroitin sulfate proteoglycan 9.2.27 surface marker daunorubicin conjugates (against chemotherapeutic-resistant M21 metastatic melanoma) [50,53,68]; anthracycline conjugates of epidermal growth factor (EGF) or EGF fragment (evaluated for their anti-neoplastic potency against chemotherapeutic-resistant MCF-7AdrR mammary carcinoma) [69]; and epirubicin-[anti-HER2/ neu] or epirubicin-[anti-EGFR] potency (assessed against chemotherapeutic-resistant SKBr-3 mammary carcinoma) [10]. Similarly, the cytotoxic anti-neoplastic potency of covalent epirubicin-[anti-HER2/neu] or epirubicin-[antiEGFR] immunochemotherapeutics would likely have been higher if it had been accessed in-vivo since many anti-cancer immunochemotherapeutics produce much greater cytotoxic activity in-vivo compared to their in-vitro level of potency [62,70]. Enhanced levels of cytotoxic anti-neoplastic activity of covalent epirubicin immunochemotherapeutics can presumably be attained at least in part by several host mechanisms that can not be effectively assessed in-vitro in a tissue culture environment. Relevant examples in this regard include, 1) potentially additive or synergistic anti-neoplastic activity achieved through dual chemotherapeutic/anti-trophic receptor immunoglobulin and dual anti-trophic receptor/anti-trophic receptor immunoglobulin combinations (Figure 10) [10]; 2) antibody/antigen/complement complex induced cytolysis; and 3) antibody-dependent cell-mediated immune responses. Lastly, analytical methodologies could have been modified to detect cytotoxic anti-neoplastic potency in chemotherapeutic-resistant SKBr-3 mammary carcinoma in a more sensitive manner than is possible with MTT vitality/proliferation stain assay systems. Instead, declines in viability could have been measured by employing either the [H3]-thymidine cell proliferation assay, or ATP-based assay since they reportedly are ≥ 10-fold more sensitive than MTT vitality stain assays [71,72]. Despite this consideration, measurement of cell vitality utilizing MTT reagent based assays have, and continue to be extensively employed for the routine assessment of chemotherapeutic anti-neoplastic activity [73-76]. However, it is important to note that the creation of lethal injury detected with MTT reagent based assays usually
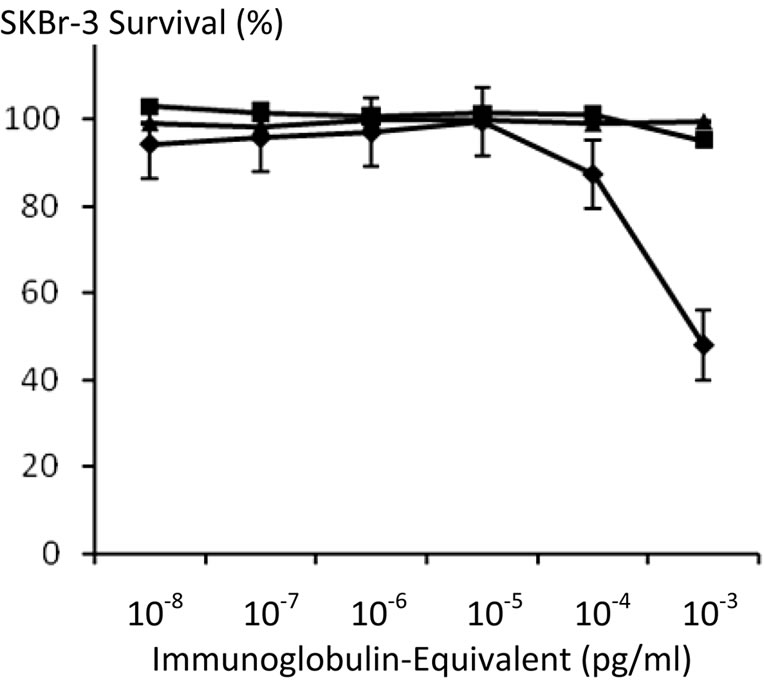
Figure 10. Relative anti-neoplastic potency of covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic compared to monoclonal immunoglobulin antiHER2/neu and anti-EGFR fractions against chemotherapeutic-resistant SKBr-3 mammary carcinoma. Legend: (u) covalent epirubicin-(C13-imino)-[anti-HER2/neu] immunochemotherapeutic; (n) anti-HER2/neu monoclonal antibody; and (p) anti-EGFR monoclonal antibody. Monolayers of SKBr-3 mammary carcinoma were incubated with immunochemotherapeutics over a 72-hour period and cytotoxicity measured as a function of MTT cell vitality relative to matched negative reference controls.
occurs at higher chemotherapeutic doses than those required to produce alteration in transcription/translation, or [H3]-thymidine and ATP-based assay systems.
Selenium in simultaneous combination with epirubicin- (C13-imino)-[anti-HER2/neu] was analyzed to determine if this element enhances the cytotoxic anti-neoplastic potency of covalent anthracyline immunochemotherapeutics against chemotherapeutic-resistant SKBr-3 mammary carcinoma (Figure 6). Consistently, selenium (methylseleninate 5 mM fixed concentration) additively enhanced [77] the cytotoxic anti-neoplastic potency of epirubicin-(C13-imino)-[anti-HER2/neu] formulated at anthracycline-equivalent concentrations between 10-12 M to 10-7 M, but this effect was not statistically significant (Figure 6). Alterations were most pronounced for epirubicin-(C13-imino)-[anti-HER2/neu] at epirubicinequivalent concentrations < 10–7 M where vitality/proliferation profiles implied that the cytotoxic anti-neoplastic activity detected at 10–7 M was almost entirely evoked through liberation of the anthracycline moiety. When the epirubin equivalent concentration was fixed at 10–8 M, gradient increases in selenium concentration from 5 mM to 50 mM produced significantly higher levels of cytotoxic activity that observed for epirubicin alone (Figure 7). Since the results for both epirubicin-(C13-imino)-[antiHER2/neu] and epirubicin at 10–8 M with and without the presence of selenium at 5 mM were not significant different, it is fully anticipated that compared to epirubicin, highly analogous results would have been detected for epirubicin-(C13-imino)-[anti-HER2/neu] formulated at epirubicin equivalent concentrations of 10–8 M in combination with or without selenium at final concentrations of 5 mM and 50 mM. In the interpretation of this latter consideration, it is important to keep in perspective that the cytotoxic activity of epirubicin-(C13-imino)-[antiHER2/neu] is greater than epirubicin at epirubicinequivalent concentrations (Figure 4) Selenium therefore can potentially be utilized in combination with lower epirubicin-equivalent concentrations of epirubicin-(C13- imino)-[anti-HER2/neu] or epirubicin to attain additive increases in cytotoxic anti-neoplastic potency (Figures 6, 7 and 8). Interestingly, selenium exerts greater cytotoxic anti-neoplastic activity compared to celecoxib [78-81] when analyzed at micro-molar equivalent concentrations (Figure 9).
Serum selenium concentrations provide prognostic implications because they can serve as a positive indicator for predicting effective dosage levels, optimum delivery routes, treatment response, and long-term survival [82]. The biology of cancer cells is modified by selenium based on it’s capacity to 1) induce apoptosis in doxorubicin-resistant lung small-cell carcinoma (selenite 10 μM) [46]; 2) promote severe ER stress (leukemia cell types) [83]; 3) reduce vitality of multidrug-resistant leukemia (selenitetriglycerides 10 μg/ml to 40 μg/ml) [84]; and 4) influence Fas signaling in MCF-7 mamary carcinoma in the presence of doxorubicin (methylseleninate 5 μM) [43]. Relatively few investigations have previously described the comparative cytotoxic anti-neoplastic activity of multiple selenium analogs.
Selenium does sensitize breast cancer populations (MCF-7) to the cytotoxic anti-neoplastic activity of doxorubicin (methylseleninate 2.5 μM and 5 μM) [44] and it increases the vulnerability of B-cell lymphoma 2.5-fold to the cytotoxic anti-neoplastic activity of doxorubicin, etoposide, 4-hydroxyperoxycyclophosphamide, melphalan, and 1-β-D-arabinofuranosylcytosine (methylseleninate 10 - 100 μM) [45]. Furthermore, selenium can additively and synergistically enhance the anti-neoplastic potency of the anthracyclines [44,85,86], irinotecan [87], and taxol [86]. In these scenarios, synergism achieved with selenium in combination with conventional chemotherapeutics can result in transformation towards a simple additive level of enhancement with extended incubation periods (ex-vivo), or prolonged duration of treatment (in-vivo) [77].
Enhancements in anthracycline anti-neoplastic potency in the presence of selenium coincides with increases in apoptosis [44,86] detected as increases in DNA fragmentation, declines in DNA synthesis, elevations in “round-cell” formation, membrane “blebbing” phenomenon and decreased N-(methylamino)-isobutyric acid uptake (MeAIB: non-metabolizable amino acid analog) [86]. Each of these alterations is selenium dose-dependent whereby an optimal effect occurs at “free” elemental selenium concentrations between 4 ng/ml and 40 ng/ml over a 72-hour period [86].
Several biochemical mechanisms modified by selenium likely contribute to it’s enhancement of anthracycline potency against different neoplastic cell types. Selenium (selenite) alone causes cell death through activation of the pro-apoptotic transcription factor GADD153 and in leukemia cells high concentrations promote p53 activation [83]. Independently, selenium (selenite) mediates anti-neoplastic activity through p53 activation and increased oxidative stress which collectively precipitate mitochondrial dysfunction and caspase activation (leukemia cell types) [83]. Selenium promotes apoptosis by also increasing Fas-associated death domain (FADD) expression and enhanced caspase-8 recruitment to Fas and FADD (MCF7 breast cancer) [43]. Progressive increases in selenium concentrations (1 μM to 10 μM) induce dose-dependent increases in the amount and activity of thioredoxin reductase in non-resistant neoplastic cells, but it precipitates declines in thioredoxin reductase in doxorubicin-resistant neoplastic cells (small cell carcinoma) [46]. Functionally, thioredoxin reductase biochemically reduces thioredoxin responsible for mediating the final step of the electron-transfer pathway for nucleoside diphosphate reduction which is essential in cancer cells for growth and survival.
Selenium induces a number of effects within neoplastic cells that are known to increase anthracycline antineoplastic activity and potency. High selenium concentrations elevate oxidative stress [83] in part by reducing catalase enzyme activity (↓ H2O2 → H2O + O2) [85] that in turn complements doxorubicin-induced declines in peroxidase activity (↓ ROOR’ + 2 e– + 2H+ → ROH + R’OH) [85]. Cytotoxic synergy produced by selenium and anthracyclines combinations is further achieved through an elevated plane of apoptosis as a result of mitochondrial caspase-9 activation (MCF7 breast cancer cells) [43]. Such alterations correlate with the detection of selenium-induced DNA strand breaks/fragmentation (leukemia) [84] that occur at a 2.5-fold greater level in the presence of selenium/doxorubicin combinations (B-cell lymphoma) [45]. Selenium-mediated DNA fragmentation and strand breaks (e.g. selenium-induced apoptosis) develop secondary to increases in the formation or molecular stability of topoisomerase II-DNA complexes [88]. Such a property is likely complemented by the greater capacity of anthracyclines to create oxygen “free” radical species in neoplastic cell types that have characteristically high [Fe+2/Fe+3] cytosol concentrations, in addition to anthracycline binding to and inhibition of topoisomerase II.
The collective effect of selenium with anthracyclines on Akt is presently considered to be one of, if not the most important molecular mechanism responsible for the complementary anti-neoplastic activity exerted by this dual combination (MCF-7 breast cancer) [44]. Selenium (selenite) at high concentrations suppresses doxorubicin-induced Akt phosphorylation [44] where Akt is a component of anti-apoptotic pathays [83] and influences glucose metabolism. Selenium is largely incapable of sensitizing cells to doxorubicin when Akt is constitutively activated (MCF-7 breast cancer cells) [44]. Doxorubicin-induced increases in Akt phosphorylation occurs simultaneously with elevations in the Akt phosphorylated substrates phospho-GSK3-β (glycogen synthase kinase-3-beta) and phosphor-FOXO3a (forkhead box O3) which each lose pro-apoptotic properties post phosphorylation (MCF-7 breast cancer cells) [44]. Selenium reduces phospho-GSK3beta abundance induced by doxorubicin, while chemical inhibition of GSK3beta biochemical activity mutes apoptotic responses created by selenium/doxorubicin combinations (MCF-7 breast cancer) [44]. Selenium is additionally believed to trigger apoptosis by increasing FOXO3a transcriptional factor activity [44] which occurs in concert with upregulation of Bim [44] and PUMA, or down-regulation of FLIP anti-apoptotic protein.
Interesting, in a slightly different context, selenium and α-tocopherol may both potentially improve internalization of chemotherapeutic-ligand conjugates designed to selectively “target” over-expressed receptor complexes known to undergo receptor-mediated endocytosis [89]. Such speculation is based on the observation that selenium and α-tocopherol deficiencies reduce receptor-mediated processes possibly due to greater levels of membrane oxidation and alterations in membrane fluidity. Selenium therefore holds promise as a form of adjunct therapy capable of increasing the cytotoxic anti-neoplastic potency of covalent anthracycline immunochemotherapeutics against aggressive and resistant forms of cancer.
5. Conclusion
Epirubicin-(C13-imino)-[anti-HER2/neu] exerted antineoplastic activity against chemotherapeutic-resistant mammary carcinoma populations that was greater than that detected for epiribicin at anthracycline-equivalent concentrations and these findings closely correlated with the efficacy of a 50/50 immunochemotherapeutic combination of epirubicin-(C3-amide)-[anti-HER2/neu] with epirubicin-(C3-amide)-[anti-EGFR] [10]. Maximum antineoplastic activity against chemotherapeutic-resistant mammary carcinoma SKBr-3 populations with covalent epirubicin-immunochemotherapeutics was ultimately achieved collectively through selective “targeted” epirubicin delivery at highly over-expressed membrane HER2/neu complexes that can facilitate internalization by ligand-induced receptor-mediated endocytosis. Selenium demonstrated an ability to further enhance the cytotoxic anti-neoplastic activity of epirubicin-(C13-imino)- [anti-HER2/neu] and epirubicin against chemotherapeutic-resistant mammary carcinoma.
The method described for the synthesis of epirubicin- (C13-imino)-[anti-HER2/neu] introduces a minimum number of cyclic ring structures into the final form of the covalent epirubicin-immunochemotherapeutic compared to other methodologies which has been proposed to minimize the induction of in-vivo humoral immune responses. Logistically, the scheme designed for the synthesis of epirubicin-(C13-imino)-[anti-HER2/neu] is relatively convenient to execute because the number of procedural steps required to adjust the pH of reaction buffers has been intentionally reduced [67]. Complementary attributes included minimal instrumentation requirements and the utilization of pre-sythesized (stock) sulfhydrylreactive maleimodocaproyl) hydrazone anthracycline (C13-imino) derivative in concert with thiolated antiHER2/neu which increased convenience and minized time requirements [19,28,47,89].
REFERENCES
- R. J. Pietras, M. D. Pegram, R. S. Finn, D. A. Maneval and D. J. Slamon, “Remission of Human Breast Cancer Xenografts on Therapy with Humanized Monoclonal Antibody to HER-2 Receptor and DNA-Reactive Drugs,” Oncogene, Vol. 17, No. 8, 1998, pp. 2235-2249. doi:10.1038/sj.onc.1202132
- R. Marches and J. W. Uhr, “Enhancement of the p27Kip1-Mediated Antiproliferative Effect of Trastuzumab (Herceptin) on HER2-Overexpressing Tumor Cells,” International Journal of Cancer, Vol. 112, No. 3, 2004, pp. 492-501. doi:10.1002/ijc.20378
- M. X. Sliwkowski, J. A. Lofgren, G. D. Lewis, T. E. Hotaling, B. M. Fendly and J. A. Fox, “Nonclinical Studies Addressing the Mechanism of Action of Trastuzumab (Herceptin),” Seminars Oncology, Vol. 26, No. 4, Suppl. 12, 1999, pp. 60-70.
- N. U. Lin, L. A. Carey, M. C. Liu, J. Younger, S. E. Come and M. Ewend, “Phase II Trial of Lapatinib for Brain Metastases in Patients with Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer,” Journal of Clinical Oncology, Vol. 26 , No. 12, 2008, pp. 1993-1999. doi:10.1200/JCO.2007.12.3588
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl and L. Fehrenbacher, “Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer that Has Progressed After Chemotherapy for Metastatic Disease,” Journal of Clinical Oncology, Vol. 17, No. 9, 1999, pp. 2639-2648.
- C. L. Vogel, M. A. Cobleigh, D. Tripathy, J. C.Gutheil, L. N. Harris and L. Fehrenbacher, “Efficacy and Safety of Trastuzumab as a Single Agent in First-line Treatment of HER2-Overexpressing Metastatic Breast Cancer,” Journal of Clinical Oncology, Vol. 20, No. 3, 2002, pp. 719- 726. doi:10.1200/JCO.20.3.719
- G. C. Lewis Phillips, G. Li, D. L. Dugger, L. M. Crocker, K. L. Parsons and E. Mai, “Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an AntibodyCytotoxic Drug Conjugat,” Cancer Research, Vol. 68, No. 22, 2008, pp. 9280-9290. doi:10.1158/0008-5472.CAN-08-1776
- J. A. García-Sáenz, M. Martín, A. Calles, C. Bueno, L. Rodríguez and J. Bobokova, “Bevacizumab in Combination with Metronomic Chemotherapy in Patients with Anthracyclineand Taxane-Refractory Breast Cancer,” Journal of Chemotherapy, Vol. 20, No. 5, 2008, pp. 632- 639.
- D. J. Slamon, B. Leyland-Jone, S. Shak, H. Fuchs, V. Paton and A. Bajamonde, “Use of Chemotherapy plus Monoclonal Antibody against HER2 for Metastatic Breast Cancer that Overexpress HER2,” The New England Journal of Medicine, Vol. 344, No. 11, 2001, pp. 786-792. doi:10.1056/NEJM200103153441101
- C. P. Coyne, M. Ross, J. Bailey and T. Jones, “Dual Potency Anti-HER2/Neu and Anti-EGFR AnthracyclineImmune Conjugates in Chemotherapeutic-Resistant Mammary Carcinoma Combined with Cyclosporin-A and Verapamil P-Glycoprotein Inhibition,” Journal Drug Targeting, Vol. 17, 2009, pp. 474-489. doi:10.1080/10611860903012802
- A. Lau, G. Berube, C. H. J. Ford and M. Gallant, “Novel Doxorubicin-Monoclonal Anti-Carcinoembryonic Antigen Antibody Immunoconjugate Activity In-Vitro,” Bioorganic and Medicinal Chemistry, Vol. 3, No. 10, 1995, pp. 1305- 1312. doi:10.1016/0968-0896(95)00126-2
- G. L. Bidwell, A. N. Davis, I. Fokt, W. Priebe and D. Raucher, “A Thermally Targeted Elastin-like PolypeptideDoxorubicin Conjugate Overcomes Drug Resistance,” Investigational New Drugs, Vol. 25, 2007, pp. 313-326. doi:10.1007/s10637-007-9053-8
- C. Ryppa, H. Mann-Steinberg, I. Fichtner, H. Weber, R. Satchi-Fainaro and M. L. Biniossek, “In-Vitro and InVivo Evaluation of Doxorubicin Conjugates with the Divalent Peptide E-[c(RGDfK)2] that Targets Integrin aVb3,” Bioconjugate Chemistry, Vol. 19, 2008, pp. 1414- 1422. doi:10.1021/bc800117r
- G. Di Stefano, M. Lanza, F. Kratz, L. Merina and L. Fiume, “A Novel Method for Coupling Doxorubicin to Lactosaminated Human Albumin by an Acid Sensitive Hydrazone Bond: Synthesis, Characterization and Preliminary Biological Properties of the Conjugate,” European Journal of Pharmaceutical Sciences, Vol. 23, 2004, pp. 393- 397. doi:10.1016/j.ejps.2004.09.005
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab and P. Lazar, “Probing the Cysteine-34 Position of Endogenous Serum Albumin with Thiol-Binding Doxorubicin Derivatives. Improved Efficacy of an Acid-Sensitive Doxorubicin Derivative with Specific Albumin-Binding Properties Compared to that of the Parent Compound,” Journal of Medicinal Chemistry, Vol. 45, 2002, pp. 5523-5533. doi:10.1021/jm020276c
- C. Unger, B. Häring, M. Medinger, J. Drevs, S. Steinbild and F. Kratz, “Phase I and Pharmacokinetic Study of the (6-Maleimidocaproyl) Hydrazone Derivative of Doxorubicin,” Clinical Cancer Research, Vol. 13, No. 16, 2007, pp. 4858-4866. doi:10.1158/1078-0432.CCR-06-2776
- D. Y. Furgeson, M. R. Dreher and A. Chilkoti, “Structural Optimization of a ‘Smart’ Doxorubicin-Polypeptide Conjugate for Thermally Targeted Delivery to Solid Tumors,” Journal of Controled Release, Vol. 110, 2006, pp. 362-369. doi:10.1016/j.jconrel.2005.10.006
- T. Etrych, T. Mrkvan, B. Ríhová and K. Ulbrich, “StarShaped Immunoglobulin-Containing HPMA-Based ConJugates with Doxorubicin for Cancer Therapy,” Journal Control Release, Vol. 122, 2007, pp. 31-38. doi:10.1016/j.jconrel.2007.06.007
- T. Etrych, M. Jelínková, B. Ríhová and K. J. Ulbrich, “New HPMA Copolymers Containing Doxorubicin Bound Via pH-Sensitive Linkage: Synthesis and Preliminary In-Vitro and In-Vivo Biological Properties,” Control Release, Vol. 73, 2001, pp. 89-102. doi:10.1016/S0168-3659(01)00281-4
- P. C. Rodrigues, U. Beyer, P. Schumacher, T. Roth, H. H. Fiebig, C. Unger, L. Messori, P. Orioli, D. H. Paper, R. Mülhaupt and F. Kratz, “Acid-Sensitive Polyethylene Glycol Conjugates of Doxorubicin: Preparation, In-Vitro Efficacy and Intracellular Distribution.” Bioorganic Medicinal Chemistry, Vol. 7, 1999, pp. 2517-2524.
- Y. F. Huang, D. Shangguan, H. Liu, J. A. Phillips, X. Zhang and Y. Chen, “Molecular Assembly of an Aptamer-Drug Conjugate for Targeted Drug Delivery to Tumor Cells,” Chem Bio Chem, Vol. 10, 2009, pp. 862- 868. doi:10.1002/cbic.200800805
- U. Beyer, B. Rothen-Rutishauser, C. Unger, H. WunderliAllenspach and F. Kratz, “Difference in the Intracellular Distribution of Acid-Sensitive Doxorubicin-Protein Conjugates in Comparison to Free and Liposomal Formulated Doxorubicin as Shown by Confocal Microscopy,” Pharmaceutical Research, Vol. 18, 2001, pp. 29-38. doi:10.1023/A:1011018525121
- M. Kruger, U. Beyer, P. Schumacher, C. Unger, H. Zahn and F. Kratz. “Synthesis and Stability of Four Maleimide Derivatives of the Anti-Cancer Drug Doxorubicin for the Preparation of Chemoimmuneconjugates,” Chemical & Pharmaceutical Bulletin, Vol. 45, 1997, pp. 399-401.
- F. Kratz, G. Ehling, H. M. Kauffmann and C. Unger, “Acute and Repeat-Dose Toxicity Studies of the (6- Maleimi-docaproyl) Hydrazone Derivative of Doxorubicin (DoxoEmch), an Albumin-Binding Prodrug of the Anticancer Agent Doxorubicin,” Human & Experimental Toxicology, Vol. 26, 2007, pp. 19-35. doi:10.1177/0960327107073825
- D. Lebrecht, A. Geist, U. P. Ketelsen, J. Haberstroh, B. Setzer and F. Kratz, “The 6-Maleimidocaproyl Hydrazone Derivative of Doxorubicin (DOXO-EMCH) is Superior to Free Doxorubicin with Respect to Cardiotoxicity and Mitochondrial Damage,” International Journal of Cancer, Vol. 120, No. 4, 2007, pp. 927-934. doi:10.1002/ijc.22409
- P. A. Trail, D. Willner, J. Knipe, A. J. Henderson, S. J. Lasch and M. E. Zoeckler, “Effect of Linker Variation on the Stability, Potency and Efficacy of CarcinomaReactive BR64-Doxorubicin Immunoconjugates,” Cancer Research, Vol. 57, 1997, pp. 100-105. doi:10.1021/bc980100i
- H. D. King, D. Yurgaitis, D. Willner, R. A. Firestone, M. B. Yang and S. J. Lasch, “Monoclonal Antibody Conjugates of Doxorubicin Prepared with Branched Linkers: A Novel Method for Increasing the Potency of Doxorubicin Immunoconjugates,” Bioconjugate Chemistry, Vol. 10, No. 2, 1999, pp. 279-288.
- D. Willner, P. A. Trail, S. J. Hofstead, H. D. King, S. J. Lasch and G. R. Braslawsky, “(6-Maleimidocaproyl) Hydrazone of Doxorubicin-A New Derivative for the Preparation of Immunoconjugates of Doxorubicin,” Bioconjugate Chemistry, Vol. 4, No. 6, 1993, pp. 521-527. doi:10.1021/bc00024a015
- J. Reményi, B. Balázs, S. Tóth, A. Falus, G. Tóth and F. Hudecz, “Isomer-Dependent Daunomycin Release and In-Vitro Antitumour Effect of Cis-Aconityl-Daunomycin,” Biochemical and Biophysical Research Communications, Vol. 303, No. 2, 2003, pp. 556-561. doi:10.1016/S0006-291X(03)00394-2
- J. R. Ogden, K. Leung, S. A. Kunda, M. W. Telander, B. P. Avner and S. K. Liao, “Immunoconjugates of Doxorubicin and Murine Antihuman Breast Carcinoma Monoclonal Antibodies Prepared via an N-Hydroxysuccinimide Active Ester Intermediate of Cis-Aconityl-Doxorubicin: Preparation and In Vitro Cytotoxicity,” Molecular Biotherapy, Vol. 1, No. 3, 1989, pp. 170-174.
- P. A. Trail, D. Willner, S. J. Lasch, A. J. Henderson, S. Hofstead and A. M. Casazza, “Cure of Xenografted Human Carcinomas by BR96-Doxorubicin Immunoconjugates,” Science, Vol. 261, 1993, pp. 212-215. doi:10.1126/science.8327892
- P. A. Trail, D. Willner, S. J. Lasch, A. J. Henderson, R. S. Greenfield and D. King, “Antigen-Specific Activity of Carcinoma-Reactive BR64-Doxorubicin Conjugates Evaluated In-Vitro and in Human Tumor Xenograft Models,” Cancer Research, Vol. 52, 1992, pp. 5693-5700.
- P. A. Trail, D. Willner, A. B. Bianchi, A. J. Henderson, M. D. Trail-Smith and E. Girit, “Enhanced Antitumor Activity of Paclitaxel in Combination with the Anticarcinoma Immunoconjugate BR96-Doxorubicin,” Clinical Cancer Research, No. 5, 1999, pp. 3632-3638.
- A. F. Wahl, K. L. Donaldson, B. J. Mixan, P. A. Trail and C. B. Siegall, “Selective Tumor Sensitization to Taxanes with the mAb-Drug Conjugate cBR96-Doxorubicin,” International Journal of Cancer, Vol. 93, No. 4, 2001, pp. 590-600. doi:10.1002/ijc.1364
- B. S. Hendriks, L. K. Opresko, H. S. Wiley and D. Lauffenburger, “Coregulaiton of Epidermal Growth Factor Receptor/Human Epidermal Growth Factor Receptor 2 (HER2) Levels and Locations: Quantitative Analysis of HER2 Overexpression Effects,” Cancer Research, Vol. 63, 2003, pp. 1130-1137.
- H. O. Sjögren, M. Isaksson, D. Willner, I. Hellström, K. E. Hellström and P. A. Trail, “Antitumor Activity of Carcinoma-Reactive BR96-Doxorubicin Conjugate against Human Carcinomas in Athymic Mice and Rats and Syngeneic Rat Carcinomas in Immunocompetent Rats,” Cancer Research, Vol. 57, No. 20, 1997, pp. 4530-4536.
- U. Beyer, T. Roth, P. Schumacher, G. Maier, A. Unold, A. W. Frahm, H. H. Fiebig, C. Unger and F. Kratz, “Synthesis and In-Vitro Efficacy of Transferring Conjugates of the Anticancer Drug Chlorambucil,” Journal of Medicinal Chemistry, Vol. 41, 1998, pp. 2701-2708. doi:10.1021/jm9704661
- L. C. Xu, M. Nakayama, K. Harada, A. Kuniyasu, H. Nakayama, S. Tomiguchi, A. Kojima, M. Takahashi, M. Ono, Y. Arano, H. Saji, Z. Yao, H. Sakahara, J. Konishi and Y. Imagawa, “Bis(hydroxamamide)-Based Bifunctional Chelating Agent for 99mTc Labeling of Polypeptides,” Bioconjugate Chemistry, Vol. 10, 1999, pp. 9-17. doi:10.1021/bc980024j
- Y. Arano, T. Uezono, H. Akizawa, M. Ono, K. Wakisaka, M. Nakayama, H. Sakahara, J. Konishi and A. Yokoyama, “Reassessment of Diethyl-enetriamine-pentaacetic Acid (Dtpa) as a Chelating Agent for Indium-111 Labeling of Polypeptides Using a Newly Synthesized Monoreactive Dtpa Derivative,” Journal of Medicinal Chemistry, Vol. 39, 1996, pp. 3451-3460. doi:10.1021/jm950949+
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, “HPMA Copolymers with Ph-Controlled Release of Doxorubicin: In-Vitro Cytotoxicity and In-Vivo Antitumor Activity,” Journal of Controlled Release, Vol. 87, 2003, pp. 33-47. doi:10.1016/S0168-3659(02)00348-6
- G. Hempel, P. Schulze-Westhoff, S. Flege and N. Laubrock, “Therapeutic Drug Monitoring of Doxorubicin in Paediatric Oncology Using Capillary Electrophoresis,” Journal of Electrophoresis, Vol. 19, No. 16-17, 1998, pp. 2939-2943. doi:10.1002/elps.1150191624
- S. Li, Y. Zhou, Y. Dong and C. Ip, “Doxorubicin and Selenium Cooperative Induce Fas Signaling in the Absence of Fas/Fas Ligand Interaction,” Anticancer Research, Vol. 27, 2007, pp. 3075-3082.
- S. Li, Y. Zhou, R. Wang, H. Zhang, Y. Dong and C. Ip, “Selenium Sensitizes Mcf-7 Breast Cancer Cells to Doxorubicin-Induced Apoptosis through Modulation of Phospho-akt and Its Downstream Substrates,” Molecular Cancer Therapeutics, Vol. 6, No. 3, 2007, pp. 1031-1038. doi:10.1158/1535-7163.MCT-06-0643
- S. Juliger, H. Goenaga-Infante, T. A. Lister, J. Fitzgibbon and S. P. Joel, “Chemosensitization of B-Cell Lymphomas by Methylseleninic Acid Involves Nuclear Factor-Kb Inhibition and the Rapid Generation of Other Selenium Species, Cancer Research, Vol. 67, No. 22, 2007, pp. 10984-10992. doi:10.1158/0008-5472.CAN-07-0519
- K. Jonsson-Videsater, L. Jborkhem-Bergman, A. Hossain, A. Soderberg, L. C. Eriksson and C. Paul, “Selenite-Induced Apoptosis in Doxorubicin-Resistant Cells and Effects on the Thioredoxin System,” Biochemical Pharmacology, Vol. 67, 2004, pp. 513-522. doi:10.1016/j.bcp.2003.09.021
- R. S. Greenfield, T. Kaneko T, A. Daues, M. A. Edson, K. A. Fitzgerald and L. J. Olech, “Evaluation In-Vitro of Adriamycin Immunoconjugates Synthesized Using an Acid-Sensitive Hydrazone Linker,” Cancer Research, Vol. 50, No. 20, 1990, pp. 6600-6607.
- J. A. Sinkule, S. T. Rosen and J. A. Radosevich, “Monoclonal Antibody 44-3a6 Doxorubicin Immunoconjugates: Comparative In-Vitro Anti-Tumor Efficacy of Different Conjugation Methods,” Tumour Biology, Vol. 12, No. 4, 1991, pp. 198-206. doi:10.1159/000217705
- H. Hu, C. Jiang, G. Li and J. Lü, “PKB/AKT and ERK Regulation of Caspase-Mediated Apoptosis by Methylseleninic Acid in Lncap Prostate Cancer Cells,” Carcinogenesis, Vol. 26, No. 8, 2005, pp. 1374-1381. doi:10.1093/carcin/bgi094
- R. O. Dillman, D. E. Johnson, J. Ogden and D. Beidler, “Significance of Antigen, Drug, and Tumor Cell Targets in the Preclinical Evaluation of Doxorubicin, Daunorubicin, Methotrexate, and Mitomycin-C Monoclonal Antibody Immunoconjugates,” Molecular Biotherapy, Vol. 1, 1989, pp. 250- 255.
- G. P. Sivam, P. J. Martin, R. A. Reisfeld and B. M. Mueller, “Therapeutic Efficacy of a Doxorubicin Immunoconjugate in a Preclinical Model of Spontaneous Metastatic Human Melanoma,” Cancer Research, Vol. 55, No. 11, 1995, pp. 2352-2356.
- P. Sapra, R. Stein, J. Pickett, Z. Qu, S. V. Govindan and T. M. Cardillo, “Anti-CD74 Antibody-Doxorubicin Conjugate, IMMU-110, in a Human Multiple Myeloma Xenograph and in Monkeys,” Clinical Cancer Research, Vol. 11, No. 14, 2005, pp 5257-5264. doi:10.1158/1078-0432.CCR-05-0204
- H. M. Yang and R. A. Reisfeld, “Doxorubicin Conjugated with Monoclonal Antibody Directed to a Human Melanoma-Associated Proteoglycan Suppresses the Growth of Established Tumor Xenografts in Nude Mice,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 85, 1988, pp. 1189-1193. doi:10.1073/pnas.85.4.1189
- K. Ulbrich, T. Etrych, P. Chytil, M. Jelinkova and B. Rihova, “Anti-body Targeted Polymer-Doxorubicin Conjugates with pH-Controlled Activation. HPMA Copolymers with pH-Controlled Release of Doxorubicin. In-Vitro Cytotoxicity and In-Vivo Antitumor Activity,” Journal of Drug Targeting, Vol. 12, 2004, pp. 477-489. doi:10.1080/10611860400011869
- G. L. Griffiths, M. J. Mattes, R. Stein, S. V. Govindan, I. D. Horak and H. J. Hansen, “Cure of SCID Mice Bearing Human B-lymphoma Xenografts by an Anti-CD74 Antibody-An-Thracycline Drug Conjugate,” Clinical Cancer Research, Vol. 9, No. 17, 2003, pp. 6567-6571.
- J. Liu, H. Zhao, K. J. Volk, S. E. Klohr, E. H. Kerns and M. S. Lee, “Analysis of Monoclonal Antibody and Immunoconjugate Digests by Capillary Electrophoresis and Capillary Liquid Chromatography,” Journal of Chromatography A, Vol. 735, 1996, pp. 357-366. doi:10.1016/0021-9673(95)01054-8
- K. Inoh, H. Muramatsu, S. Torii, S. Ikematsu, M. Oda and H. Kumai, “Doxorubicin-Conjugated Anti-Midkine Monoclonal Antibody as a Potential Anti-tumor Drug,” Japanese Journal of Clinical Oncology, Vol. 36, No. 4, 2006, pp. 207-211. doi:10.1093/jjco/hyl004
- G. M. Dubowchik, S. Radia, H. Mastalerz, M. A. Walker, R. A. Firestone and H. Dalton King, “Doxorubicin Immunoconjugates Containing Bivalent, Lysosomally-Cleavable Dipeptide Linkages,” Bioorganic & Medicinal Chemistry Letters, Vol. 12, No. 1, 2002, pp. 1529-1532. doi:10.1016/S0960-894X(02)00194-4
- L. B. Shih, D. M. Goldenberg, H. Xuan, H. W. Lu, M. J. Mattes and T. C. Hall, “Internalization of an Intact Doxorubicin Immunoconjugate,” Cancer Immunol Immunother, Vol. 38, No. 2, 1994, pp. 92-98. doi:10.1007/BF01526203
- Y. T. Zhang, N. Q. Wang, N. Li, T. Liu and Z. W. Dong, “The Antitumor Effect of Adriamycin Conjugated with Monoclonal Antibody against Gastric Cancer In-Vitro and In-Vivo,” Acta Pharmaceutica Sinica, Vol. 27, 1992, pp. 325-330.
- H. J. Hansen, G. L. Ong and H. Diril, “Internalization and Catabolism of Radiolabeled Antibodies to the MHC Class-II Invariant Chain by B-cell Lymphomas,” Biochemical Journal, Vol. 320, 1996, pp. 293-300.
- D. A. Johnson, S. L. Briggs, M. C. Gutowski and R. Barton, “Anti-Tumor Activity of CC49-Doxorubicin Immunoconjugates,” Anticancer Research, Vol. 15, No. 4, 1995, pp. 1387-1393.
- C. Herbert, K. Norris and J. J. Sauk, “Targeting of Human Squamous Carcinomas by SPA470-Doxorubicin Immunoconjugates,” Journal of Drug Targeting, Vol. 11, No. 2, 2003, pp. 101-107. doi:10.1080/1061186031000121478
- M. V. Pimm, M. A. Paul, T. Ogumuyiwa and R. W. Baldwin, “Biodistribution and Tumour Localization of a Daunomycin-Monoclonal Antibody Conjugate In Nude Mice and Human Tumour Xenografts,” Cancer Immunology Immunotherapy, Vol. 27, No. 3, 1988, pp. 267-271. doi:10.1007/BF00205450
- A. C. Stan, D. L. Radu, S. Casares, C. A. Bona and T. D. Brumeanu, “Antineoplastic Efficacy of Doxorubicin Enzymatically Assembled on Galactose Residues of a Monoclonal Antibody Specific for the Carcinoembryonic Antigen,” Cancer Research, Vol. 59, 1999, pp. 115-121.
- J. H. Liu, L. Cao, P. G. Luo, S. T. Yang, F. Lu and H. Wang, “Fullerene-Conjugated Doxorubicin in Cells.” ACS Applied Mater Interfaces, Vol. 2, No. 5, 2010, pp. 1384-1389.
- M. Haas, F. Moolenaar, A. Elsinga, E. A. van der Wouden, P. E. De Jong and D. K. F. Meijer, “Targeting of Doxorubicin to the Urinary Bladder of the Rat Shows Increased Cytotoxicity in the Bladder Urine Combined with An Absence of Renal Toxicity,” Journal Drug Targeting, Vol. 10, 2002, pp. 81-89.
- H. M. Yang and R. A. Reisfeld, “Pharmacokinetics and Mechanism of Action of a Doxorubicin-Monoclonal Antibody 9.2.27 Conjugate Directed to a Human Melanoma Proteoglycan,” Preview Journal of the National Cancer Institute, Vol. 80, No. 14, 1988, pp. 1154-1159.
- S. V. Lutsenko, N. B. Feldman and S. E. Severin, “Cytotoxic and Antitumor Activities of Doxorubicin Conjugates with the Epidermal Growth Factor and Its Receptor-Binding Fragment,” Journal Drug Targeting, Vol. 10, No. 7, 2002, pp. 567-571.
- E. Aboud-Pirak, E. Hurwitz, F. Bellot, J. Schlessinger and M. Sela, “Inhibition of Human Tumor Growth in Nude Mice by a Conjugate of Doxorubicin with Monoclonal AntiBodies to Epidermal Growth Factor Receptor,” PNAS, Vol. 86, No. 10, 1989, pp. 3778-3781.
- H. Mueller, M. U. Kassack and M. Wiese, “Comparison of the Usefulness of the MTT, ATP and Calcein Assays to Predict the Potency of Cytotoxic Agents in Various Human Cancer Cell Lines.” Biomolecular Screening, Vol. 9, No. 6, 2004, pp. 506-515.
- E. Ulukaya, F. Ozdikicioglu, A. Y. Oral and M. Dermirci, “The MTT Assay Yields a Relatively Lower Result of Growth Inhibition than the ATP Assay Depending on the Chemotherapeutic Drug Tested,” Toxicology In-Vitro, Vol. 22, No. 1, 2008, pp. 232-239.
- M. Varache-Lembège, S. Larrouture, D. Montaudon, J. Robert and A. Nuhrich A, “Synthesis and Antiproliferative Activity of Aryland Heteroaryl-Hydrazones Derived from Xanthone Carbaldehydes,” European Journal Medicinal Chemistry, Vol. 43, No. 6, 2008, pp. 1336-1343.
- M. D. Kars, O. D. Iseri, U. Gunduz and J. Molnar, “Reversal of Multidrug Resistance by Synthetic and Natural Compounds in Drug-Resistant MCF-7 Cell Lines,” Chemotherapy, Vol. 54, No. 3, 2008, pp. 194-200.
- H. Huang, E. Pierstorff, E. Osawa and D. Ho, “Active Nanodiamond Hydrogels for Chemotherapeutic Delivery,” Nano Letter, Vol. 7, No. 11, 2007, pp. 3305-3314.
- M. C. Dery, C. van Themsche, D. Provencher, A. M. Mes-Masson and E. Asselin, “Characterization of EN- 1078D, a Poorly Differentiated Human Endometrial Carcinoma Cell Line: A Novel Tool to Study Endometrial Invasion In-Vitro,” Reproductive Biology Endocrinology, Vol. 5, 2007, pp. 38-39.
- L. Tan, X. Jia, X. Jiang, Y. Zhang, H. Tang and S. Yao. “In-Vitro Study on the Individual and Synergistic Cytotoxicity of Adriamycin and Selenium Nanoparticles against Bel7402 Cells with a Quartz Crystal Microbalance,” Biosens Bioelectron, Vol. 24, No. 7, 2009, pp. 2268-2272.
- J. van Wijngaarden, E. van Beek, G. van Rossum, C. van der Bent, K. Hoekman and G. van der Pluijm, “Celecoxib Enhances Doxorubicin-Induced Cytotoxicity in MdaMb231 Cells by Nf-Kappab-Mediated Increase of Intracellular Doxorubicin Accumulation,” European Journal of Cancer, Vol. 43, No. 2, 2007, pp. 433-442.
- S. Hashitani, M. Urade, N. Nishimura, T. Maeda, K. Takaoka and K. Noguchi, “Apoptosis Induction and Enhancement of Cytotoxicity of Anticancer Drugs by Celecoxib, a Selective Cyclooxygenase-2 Inhibitor, in Human Head and Neck Carcinoma Cell Lines,” International Journal of Oncology, Vol. 23, No. 3, 2003, pp. 665-672.
- K. R. Roy, G. V. Reddy, L. Maitreyi, S. Agarwal, C. Achari and S. Vali, “Apoptosis Induction and Enhancement of Cytotoxicity of Anticancer Drugs by Celecoxib, a Selective Cyclooxygenase-2 Inhibitor, in Human Head and Neck Carcinoma Cell Lines. Celecoxib Inhibits mdr1 Expression through COX-2-Dependent Mechanism in Human Hepatocellular Carcinoma (hepg2) Cell Line,” Cancer Chemotherapy and Pharmacology, Vol. 65, No. 5, 2010, pp. 903-911.
- W. M. Awara, A. E. El-Sisi, M. E. El-Sayad and A. E. Goda, “Apoptosis Induction and Enhancement of Cytotoxicity of Anticancer Drugs by Celecoxib, a Selective Cyclooxygenase-2 Inhibitor, in Human Head and Neck Carcinoma Cell Linesthe Potential Role of Cyclooxygenase-2 Inhibitors in the Treatment of Experimentally-Induced Mammary Tumour: Does Celecoxib Enhance the Anti-Tumour Activity of Doxorubicin?” Pharmacology Research, Vol. 50, No. 5, 2004, pp. 487-498.
- K. W. Last, V. Cornelius, T. Delves, C. Sieniawska, J. Fitzgibbon and A. Norton, “Presentation Serum Selenium Predicts for Overall Survival, Dose Delivery, and First Treatment Response in Aggressive Non-Hodgkin’S Lymphoma,” International Journal of Oncology, Vol. 21, No. 12, 2003, pp. 2335-2341.
- L. Guan, B. Han, J. Li, Z. Li, F. Huang and Y. Yang, “Exposure of Human Leukemia Nb4 Cells to Increasing Concentrations of Selenite Switches the Signaling From Pro-Survival to Pro-Apoptosis,” Annals of Hematology Vol. 88, No. 8, 2009, pp. 733-742.
- P. Suchocki, I. Misiewicz, K. Skupinska, K. Waclawek, Z. Fijalek and T. Kasprzycka-Guttman, “The Activity of Selol in Multidrug-Resistant and Sensitive Human Leukemia Cells,” Oncology Report, Vol. 18, No. 4, 2007, pp. 893- 899.
- M. Popovic, J. Kolarovic, M. Mikov, S. Trivic and B. Kaurinovic, “Anthracycline-Based Combined Chemotherapy in the Mouse Model,” European Journal of Drug Metab Pharmacokinet, Vol. 32, No. 2, 2007, pp. 101-108.
- J. V. Vadgama, Y. Wu, D. Shen, S. Hsia and J. Block, “Effect of Selenium in Combination with Adriamycin or Taxol on Several Different Cancer Cells,” Anticancer Research, Vol. 20, No. 3A, 2000, pp. 1391-1414.
- S. Cao, F. A. Durrani and Y. M. Rustum, “Selective Modulation of the Therapeutic Efficacy of Anticancer Drugs by Selenium Containing Compounds against Human Tumor Xenografts,” Clinical Cancer Research, Vol. 10, 2004, pp. 2561-2569.
- M. Lopez-Lazaro, E. Willmore, S. L. Elliott and C. A. Austin, “Selenite Induces Topoisomerase I and II Dna Complexes in K562 Leukemia Cells,” International Journal Cancer, Vol. 123, 2008, pp. 2217-2221. doi:10.1002/ijc.23783
- G. M. Pighetti, M. L. Eskew, C. C. Reddy and L. M. Sordillo, “Selenium and Vitamin E Deficiency Impair Transferrin Receptor Internalization but Not Il-2, Il-2 Receptor, or Transferrin Receptor Expression,” Journal of Leukocyte Biology, Vol. 63, 1998, pp. 131-137.
- T. Kaneko, D. Willner, J. O. Knipe, G. R. Braslawsky, R. S. Greenfield and D. M. Vyas, “New Hydrazone Derivatives of Adriamycin and Their Immunoconjugates – A Correlation between Acid Stability and Cytotoxicity,” Bioconjugate Chemistry, Vol. 2, 1991, pp. 133-141. doi:10.1021/bc00009a001