Natural Science
Vol.4 No.5(2012), Article ID:19115,4 pages DOI:10.4236/ns.2012.45041
Theoretical DFT(B3LYP)/6-31+G(d) study on the prediction of the preferred interaction site of 3-methyl-4-pyrimidone with different proton donors
![]()
1Faculty of Sciences, University of Kinshasa, Kinshasa, Democratic Republic of Congo; *Corresponding Author: okumakasende@yahoo.com
2Department of Chemistry, University of Leuven, Leuven, Belgium
Received 11 December 2011; revised 13 January 2012; accepted 30 January 2012
Keywords: 3-Methyl-4-Pyrimidone; DFT/B3LYP Calculation; Proton-Donor Capability
ABSTRACT
Theoretical calculations were carried out using the DFT/B3LYP/6-31+G(d) methodology in an attempt to predict the preferred interaction site of a polyfunctional heterocyclic base 3-methyl-4- pyrimidone molecule with a series of proton donors of different acidic strength, i.e. water, methanol, phenol, 1-naphtol, 2,4,5 trichlorophenol, pentachlorophenol, picric acid and hydrogen chlordide. Computed H-bond interaction energies (ΔEc), internuclear and intermolecular distances r(O…H) and r(O…O), infrared frequency shifts (((C=O) and ((((OH) are proved to be reliable parameters for predicting the preferred interaction site of 3-methyl-4-pyrimidone. These computational data suggest that the O-H…O=C complex is preferred with water, methanol, phenol, 1-naphtol, 2,4,5 trichlorophenol and pentachlorophenol. However, for H-bonding with stronger acids such as picric acid or hydrochloric acid, the computational data suggest that the H-bonding occurs at the N1 ring atom of 3-methyl-4-pyrimidone. In the O-H…O=C complex, where the H-bond at the carbonyl O-atom can be oriented “anti” (Ha) and “syn” (Hb) with respect to the N3 atom, the same computational data suggest a higher stability of the “anti-O” compared to the “syn-O” orientation.
1. INTRODUCTION
Theoretical [DFT-B3LYP/6-31+G(d)] and experimental (matrix-isolation Infrared, denoted IR, spectroscopy) investigations are suitable to discriminate between O-H…N and O-H…O=C complexes of 3-methyl-4-pyrimidone
(3M4P) and proton donors [1-4]. Experimental frequency shifts of n(C=O), d(C=O), g(C=O) and n(N1=C) modes of 3-methyl-4-pyrimidone, which are sensitive to Hbonding, the values of the H-bond interaction energy (ΔEc) and the internuclear and intermolecular distances computed at the DFT(B3LYP)/6-31+G(d) level of theory have been shown that H-bonds between weak acids such as methanol are formed preferentially at the carbonyl group, whereas for stronger acids such as hydrochloric acid the complexation occurs on the N1 atom of the ring.
In spite of these exciting results, no other, detailed studies on reliable theoretical parameters on the preferred interaction site of the polyfunctional heterocyclic base 3M4P are available in the literature. In order to identify these parameters, it is necessary to widen the range of proton donors complexing 3M4P characterized by different acidic strengths. 1-Naphthol is used here because of its rotameric properties and its possibility to form a complex with a stacking structure supported by Hbonding. Ortho-substituted chlorophenols exhibit relatively strong intramolecular OH…Cl H-bonds, apart from their stacking ability. Therefore, in this case, there is a competition between intramolecular and intermolecular H-bonds in the 3M4P complex that can reduce the stability of the complex. In both cases the prediction of the preferred interaction site due to only the acidity of the proton donor can be modified.
Although H-bonding between weak acids and 3M4P occurs preferentially at the carbonyl group, the most stable configuration still remains unknown. Since the oxygen atom of the carbonyl group of 3M4P has two electronic pairs oriented in the “anti” (Ha) and “syn” (Hb) directions with respect to the N3 atom of the ring (Scheme 1), an H-bond formation with the carbonyl oxygen atom of the 3M4P can take place in both the “anti” Ha and “syn” Hb configurations, defined by the two electronic pairs of this oxygen atom.
The aim of the present study is to identify reliable

Scheme 1. 3-Methyl-4-pyrimidone.
theoretical parameters for the preferred interaction site in the 3M4P molecule with a series of proton donors with different proton-donors capabilities, and to predict the most stable configuration of the OH…O=C complexes, formed in most of the cases.
2. METHODOLOGY
Molecular properties such as geometries, energies and vibrational frequencies of the different monomers and H-bonded complexes were calculated by the Density Functional Theory (DFT) using the hybrid of Becke’s non-local three parameter exchange and correlation functional with the Lee-Yang-Parr functional (B3LYP) [5-7]. DFT methods provide an adequate compromise between the desired chemical accuracy and the computational cost. DFT/B3LYP methods have been demonstrated in former studies to produce quite accurate results for isolated molecules modeling heterocyclic bases [8-14]. Although it is well established that H-bonding involving nucleic acid bases is not fully correctly described by DFT methods, these methods remain useful. The reason is that interactions due to H-bonding are principally electrostatic and they are reasonably well accounted for by DFT or MP2 methods [15].
In H-bond studies, the standard 6-31+G(d), 6-31++G(d), 6-31++G(d,p) and 6-311++G(d,p) basis sets have frequently been used [16-19]. For the molecular orbital expansion we have initially used the 6-31++G(d,p) basis set. The choice of this basis set was based on the consideration that, in order to obtain reliable properties for hydrogen-bonded systems, it is essential to employ basis sets of orbitals which possess sufficient diffuseness and angular flexibility [20]. Since it appeared for methanol and its homo-associates that the calculated values of the electronic energies and vibrational frequencies with the basis set 6-31+G(d) were similar to those with the larger basis set 6-31++G(d,p) [21], we have only used the former set in the further study.
As has been demonstrated by one of us for larger molecules like as nucleic acid base derivatives, selective frequency scaling allows a reliable assignment of vibrational modes [22] the harmonic DFT frequencies were scaled with variable scaling factors, i.e. 0.95 for ν(X-H), 0.98 for out-of-plane modes and 0.975 for all other modes. The use of different scaling factors for DFT predicted frequencies has been proposed by several authors in the past [23-25]. The infrared frequencies, intensities, and the zero-point vibrational energy (ZPE) were subsequently computed using the analytical derivatives procedure incorporated in the GAUSSIAN 03 program.
Following these calculations, the total energies ET of the optimized structures were calculated as the sum of the electronic energy E(DFT) and 0.975 ZPE (ZPE determined with the DFT methodology). The H-bond interaction energy of each complex was computed as the difference between the energy of the complex (cluster) and the sum of the energies of the monomer 3M4P and the proton donor. These results were corrected for the basis set superposition error (BSSE) [26], by recalculating the monomer energies in the basis set of the hetero-dimers using the Boys-Bernardi counterpoise correction [27]. The corrected H-bond interaction energy of each complex (ΔEc) was finally computed as:
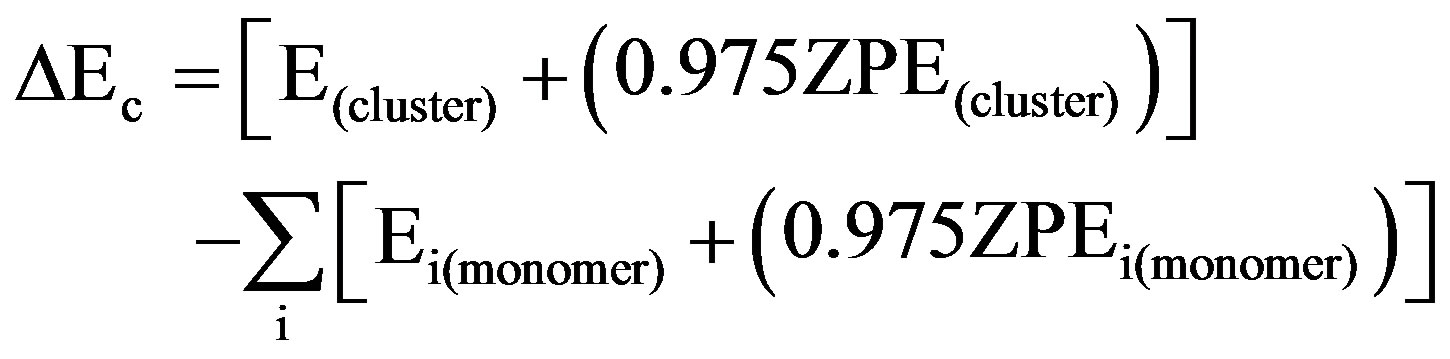
All calculations were performed using the Gaussian 03 ab initio software package [28].
3. RESULTS AND DISCUSSION
All structural and energetic parameters for H-bonded complexes between the studied proton donors and 3M4P computed at the DFT(B3LYP)/6-31+ G(d) level are summarized in Tables 1-4. The geometries of the H-bonded clusters are shown in Scheme 2.
3.1. Prediction of the Preferred Interaction Site of 3M4P with Different Proton Donors
The analysis of the H-bond interaction energies ΔEc in Tables 1-4 suggests that the complex between 3M4P and the weaker proton donors such as water, methanol, phenol, 1-naphtol, 2,4,5-trichlorophenol and pentachlorophenol, are more stable for about 2.5 to 3.0 kJ·mol–1 when the H-bond interaction takes place on the C=O group rather than the on the N1 atom of the base. For the complex between 3M4P and stronger acids such as picric acid or hydrochloric acid, the stability is higher when the interaction takes place on the N1 atom of the base rather than on the carbonyl group.
This stability difference of the complexes is confirmed by internuclear and intermolecular distances r(H…O)
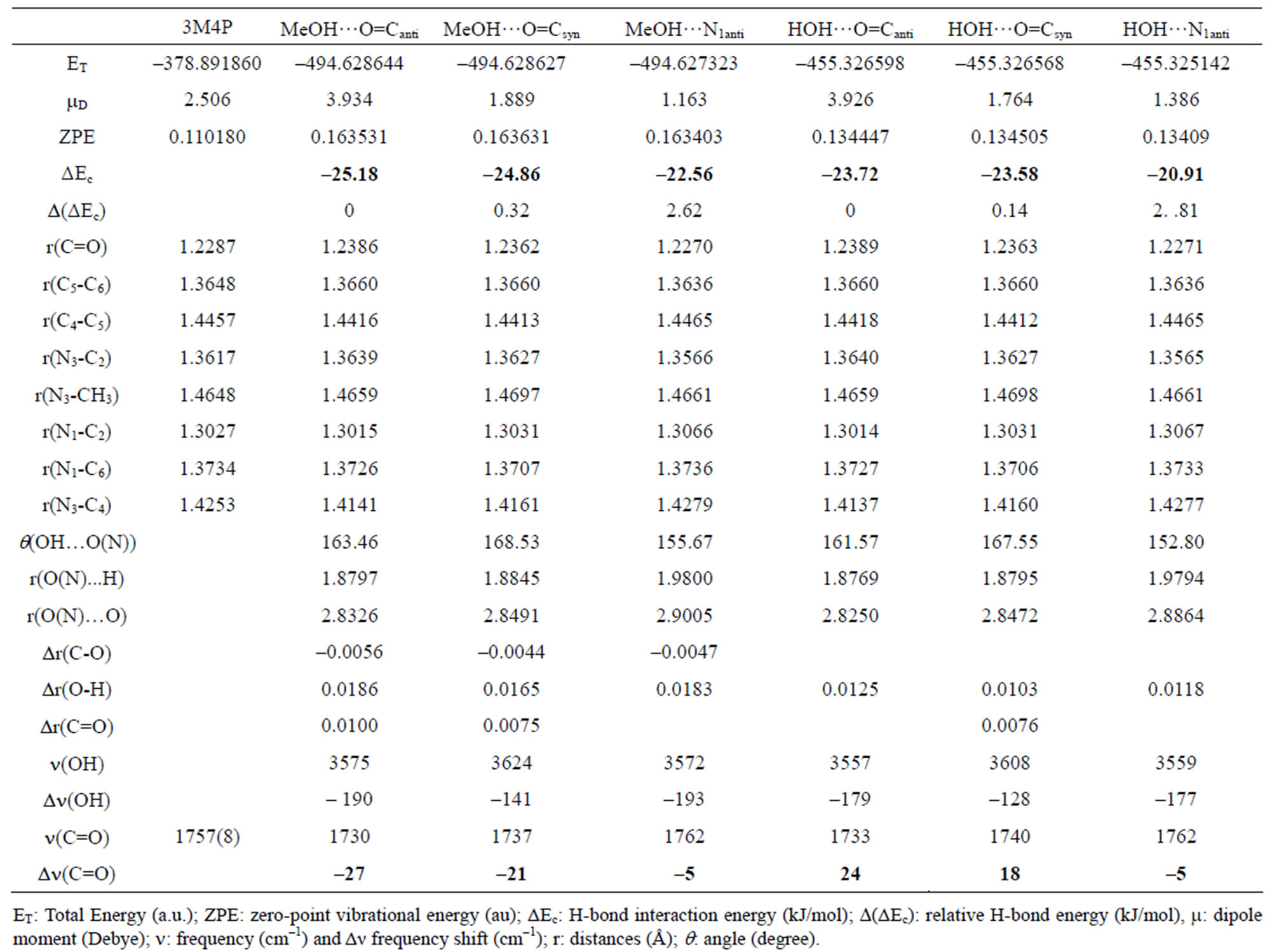
Table 1. Computed DFT (B3LYP)/6-31+G(d) energetic and structural parameters of the H-bonded complexes of 3MP4 with methanol or with water at the C=O or at the N1 interaction site of the base.
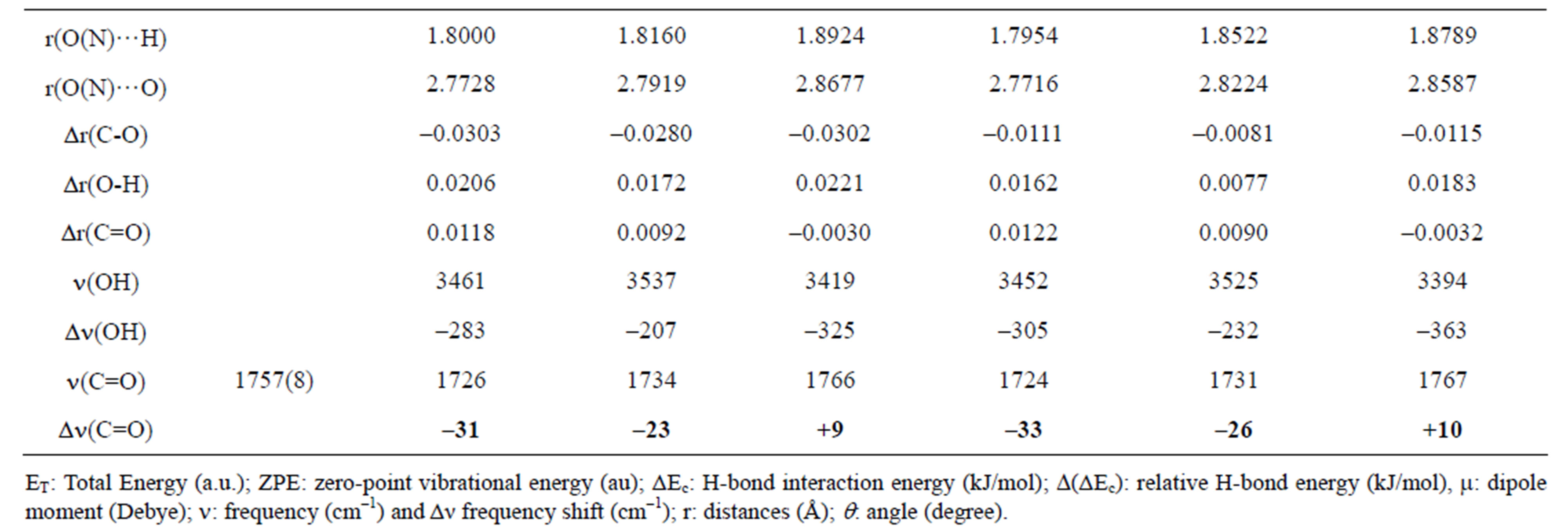
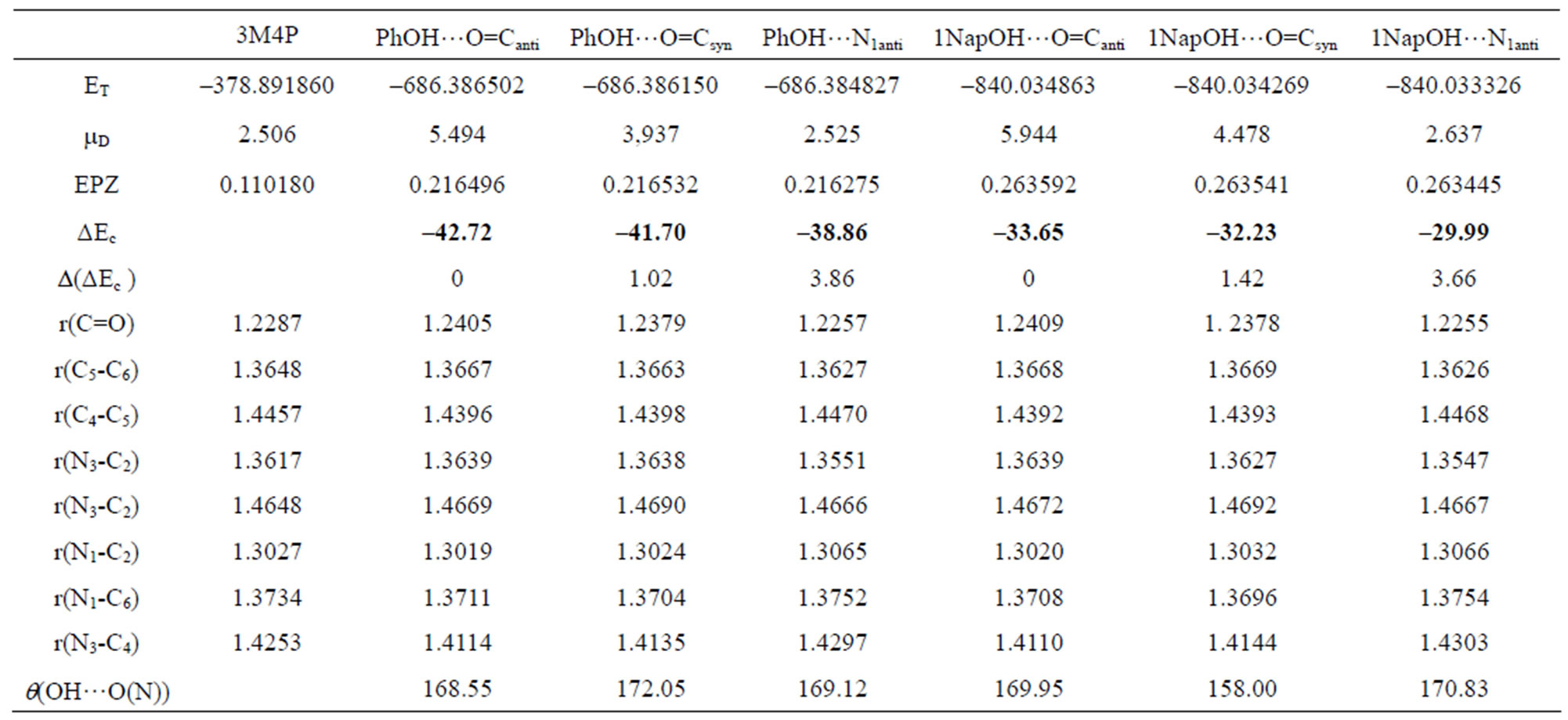
Table 2. Computed DFT(B3LYP)/6-31+G(d) energetic and structural parameters of the H-bonded complexes of 3MP4 with phenol or 1-naphthol at the C=O or at the N1 interaction site of the base.
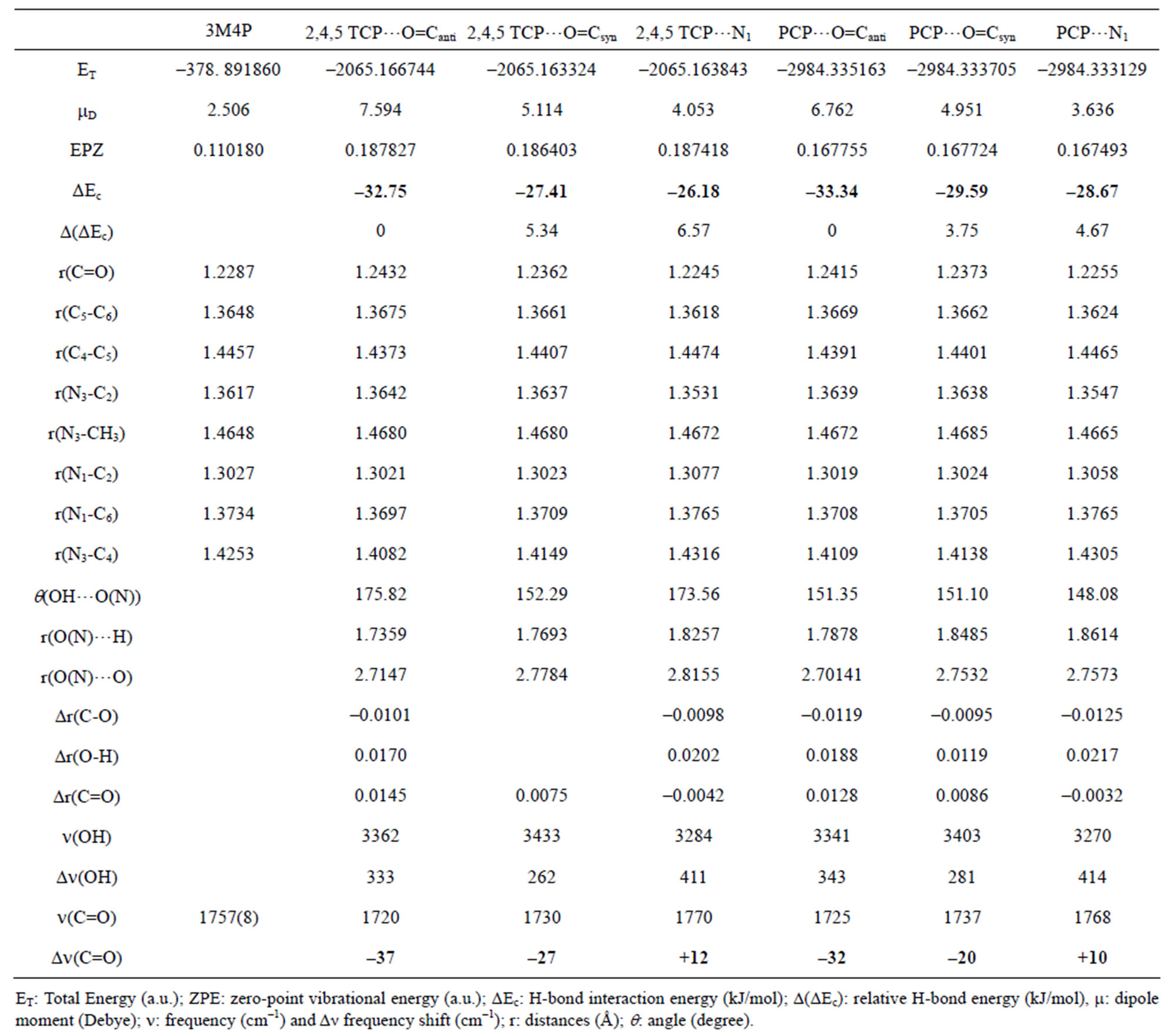
Table 3. Computed DFT (B3LYP)/6-31+G(d) energetic and structural parameters of the H-bonded complexes of 3MP4 with 2,4,5-trichlorophenol (2,4,5-TCP) or pentachlorophenol (PCP) at the C=O or at the N1 interaction site of the base.
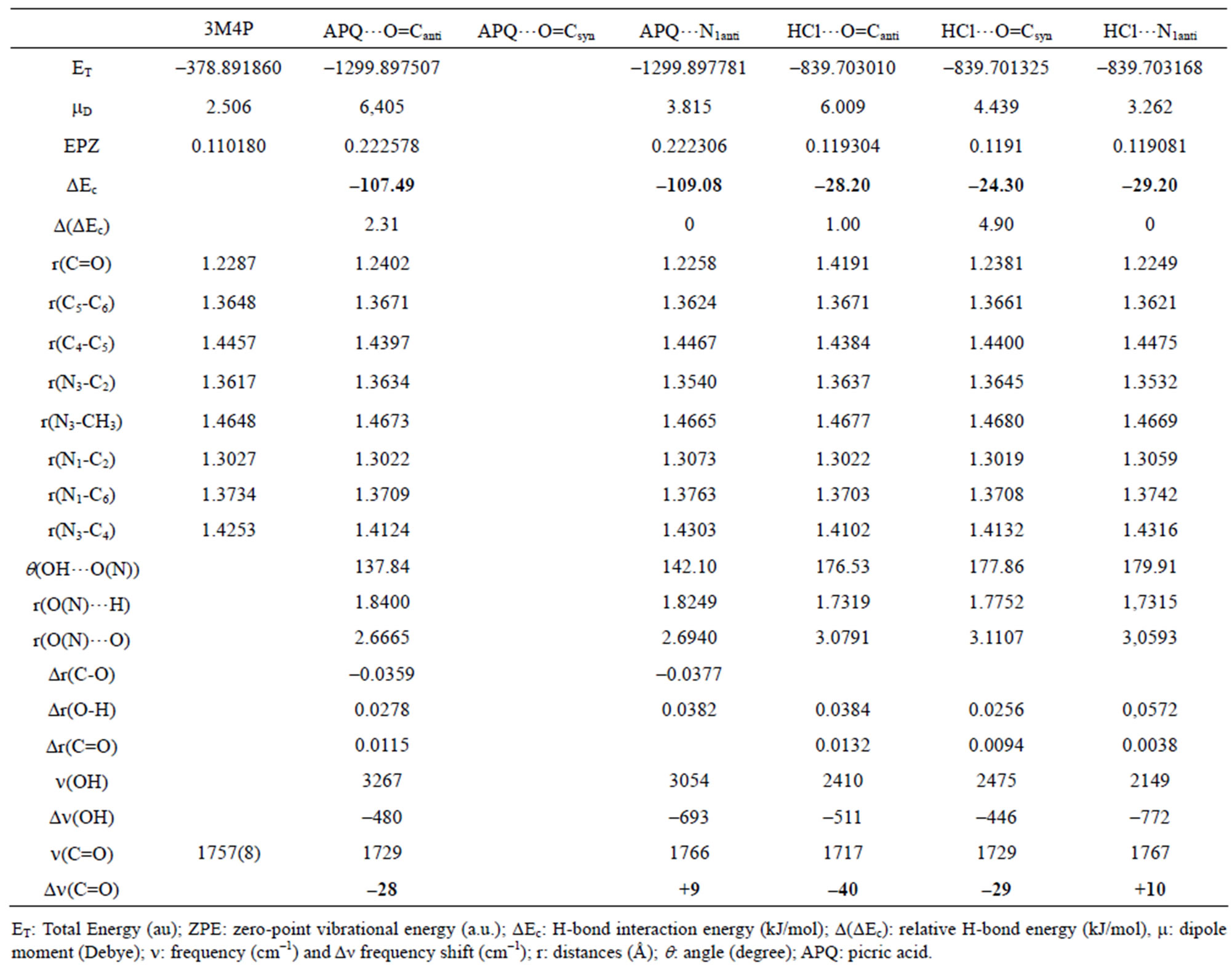
Table 4. Computed DFT(B3LYP)/6-31+G(d) energetic and structural parameters of the H-bonded complexes of 3MP4 with picric acid (APQ) or hydrochloric acid at the C=O or at the N1 interaction site of the base.
Water-3M4P (ΔEc, kJ/mol)
anti-O (–23.80) syn-O (–23.58)
syn-O (–23.58)
anti-N (–20.91)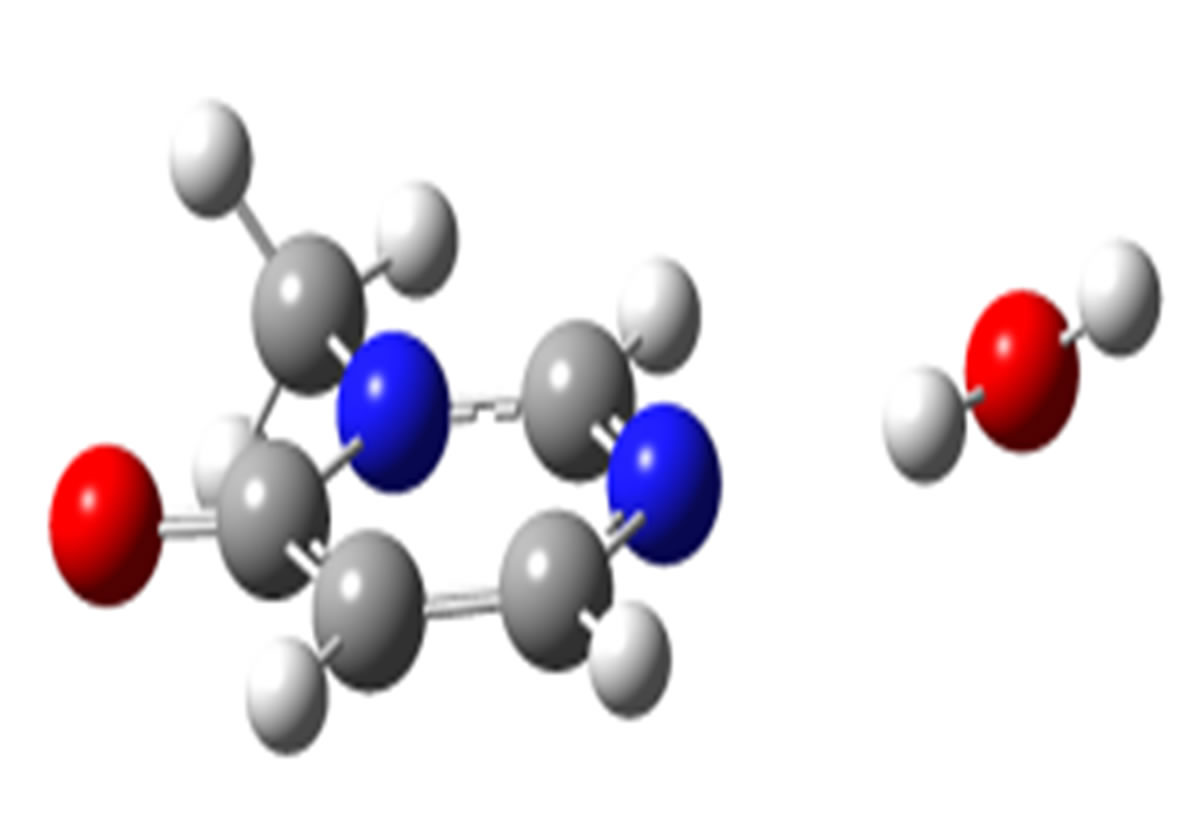
Methanol-3M4P (ΔEc, kJ/mol)
anti-O (–25.18)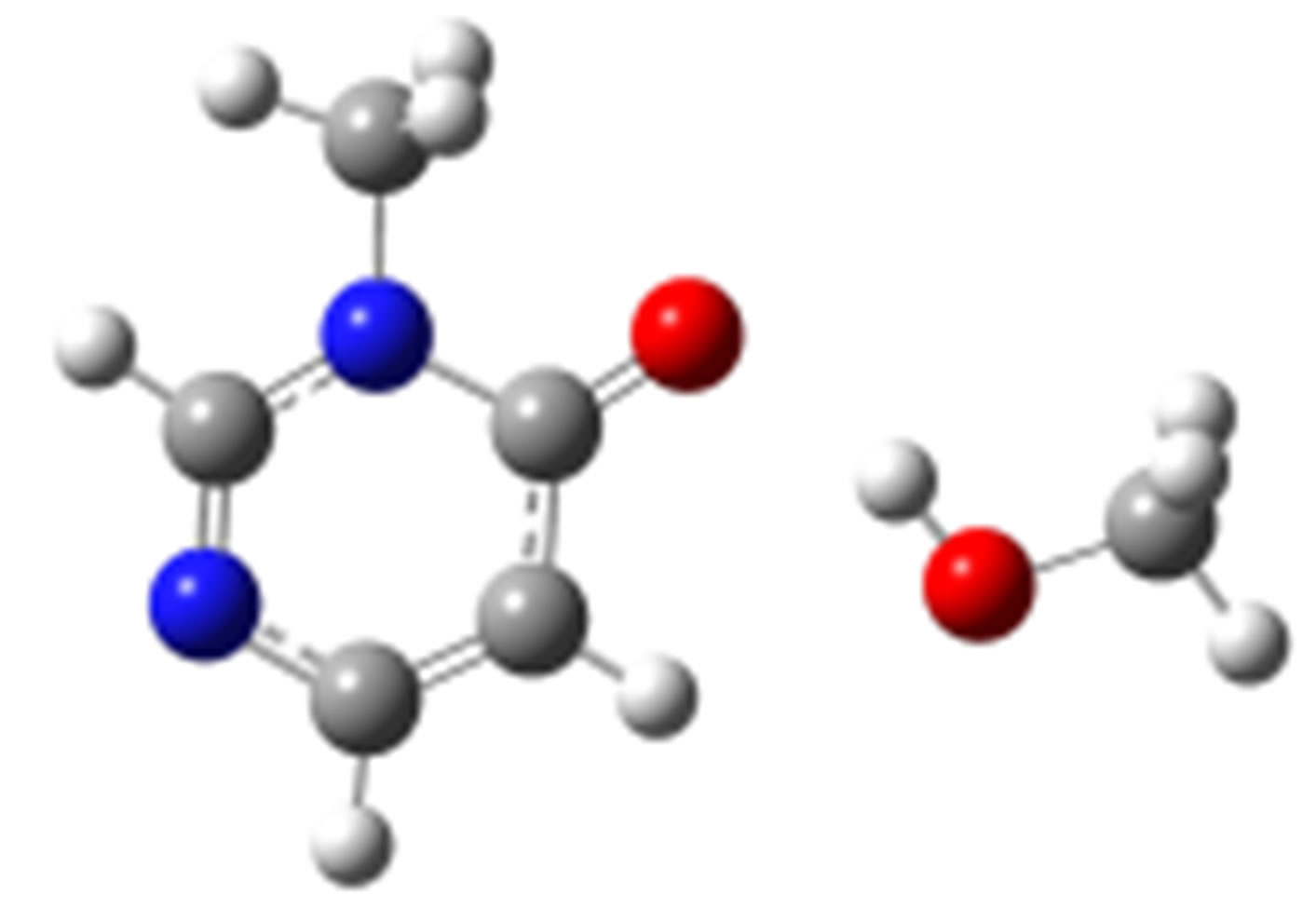 syn-O (–24.86)
syn-O (–24.86)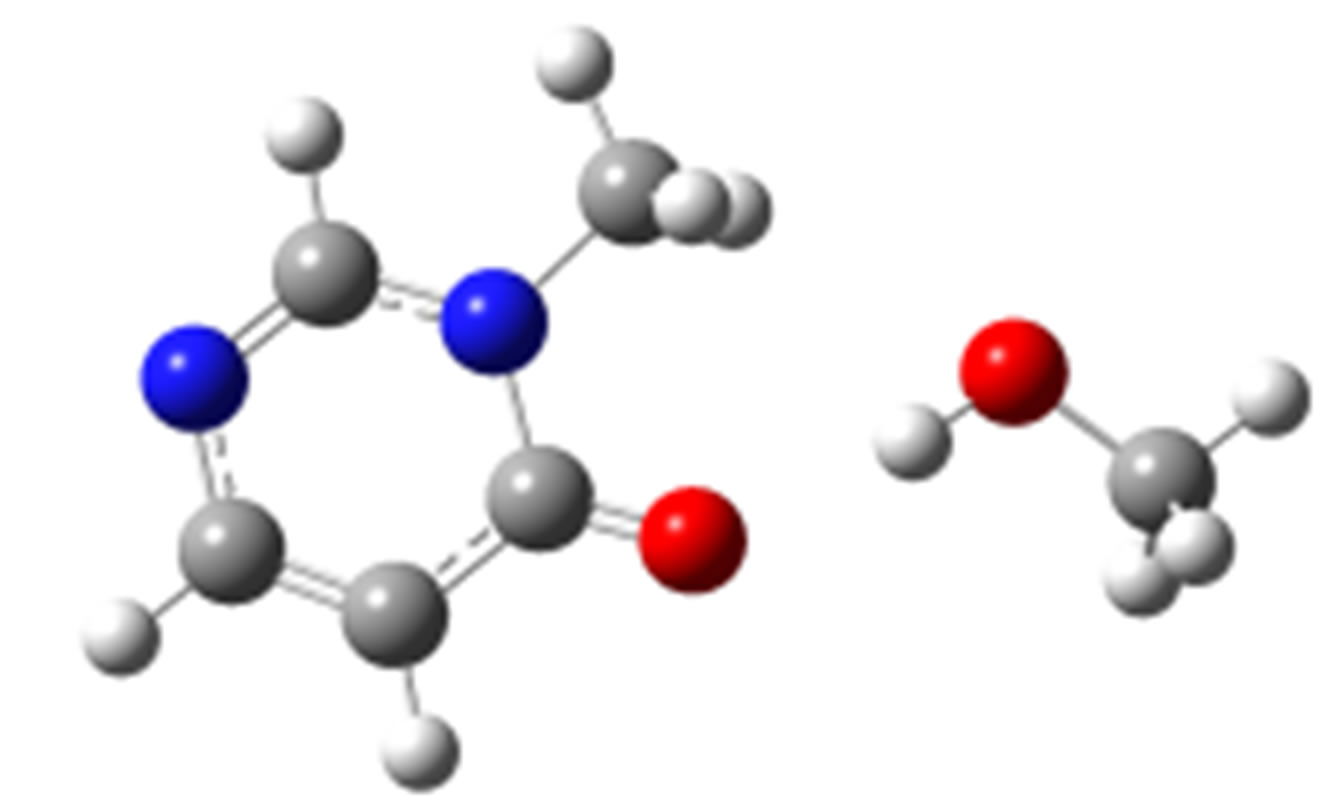
anti-N (–22.56) 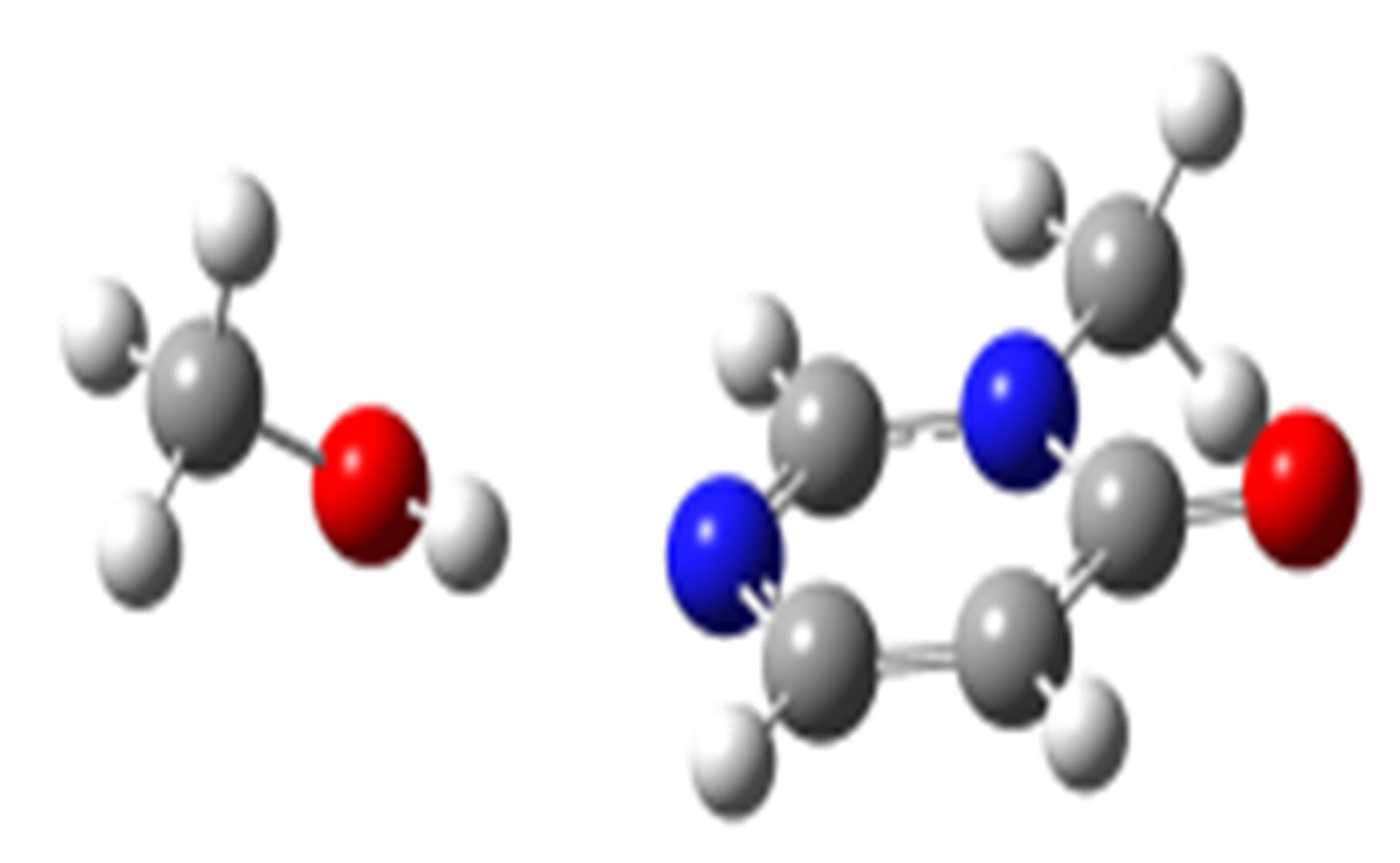
Phenol-3M4P (ΔEc, kJ/mol)
anti-O (–42.60)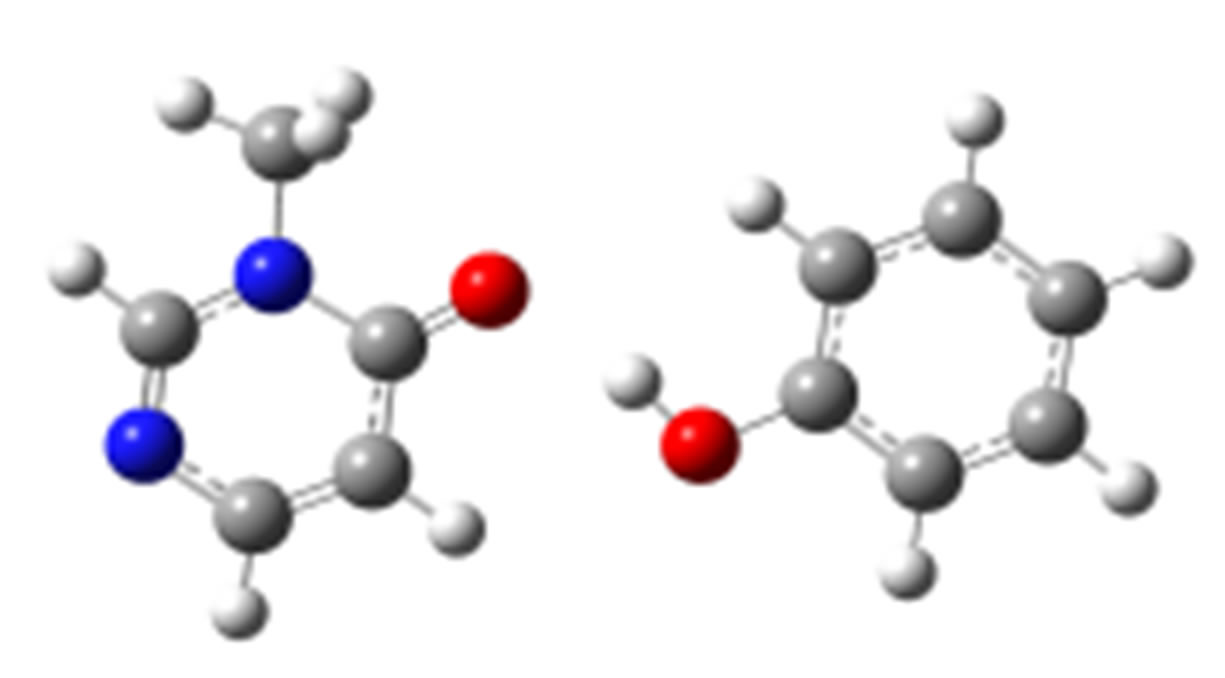 syn-O (–41.60)
syn-O (–41.60)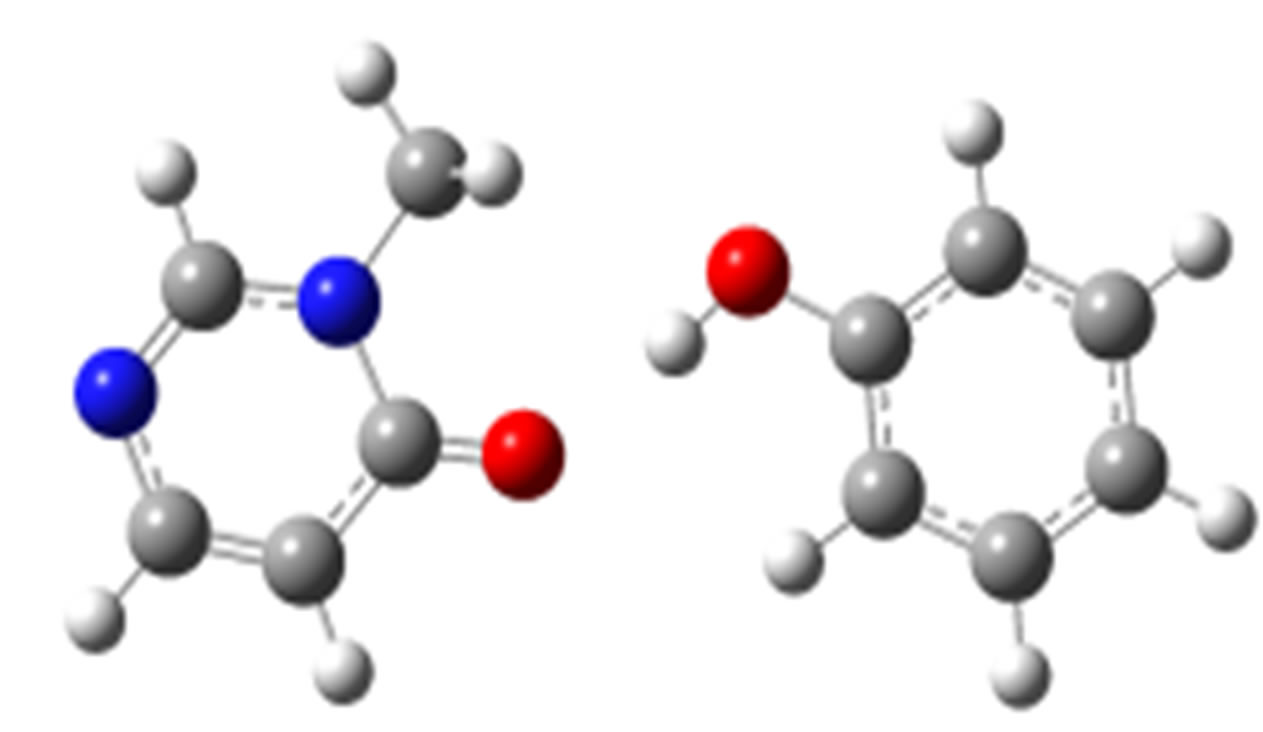
anti-N (–38.75)
1-Naphtol-3M4P (ΔEc, kJ/mol)
anti-O (–33.66)
anti-N (–30.00)
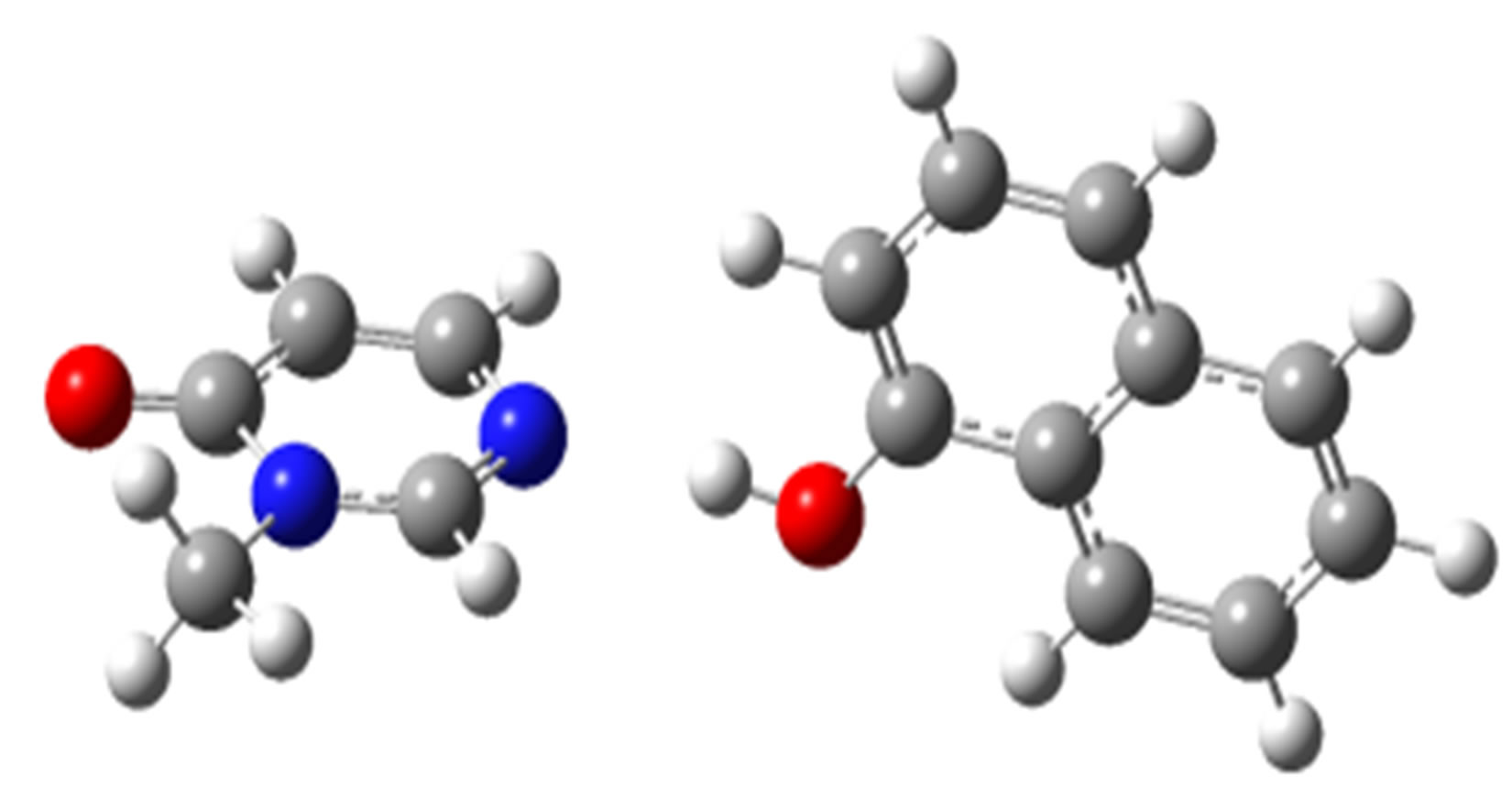
2,4,5-Trichlorophenol-3M4P (ΔEc, kJ/mol)
anti-O (–32.75)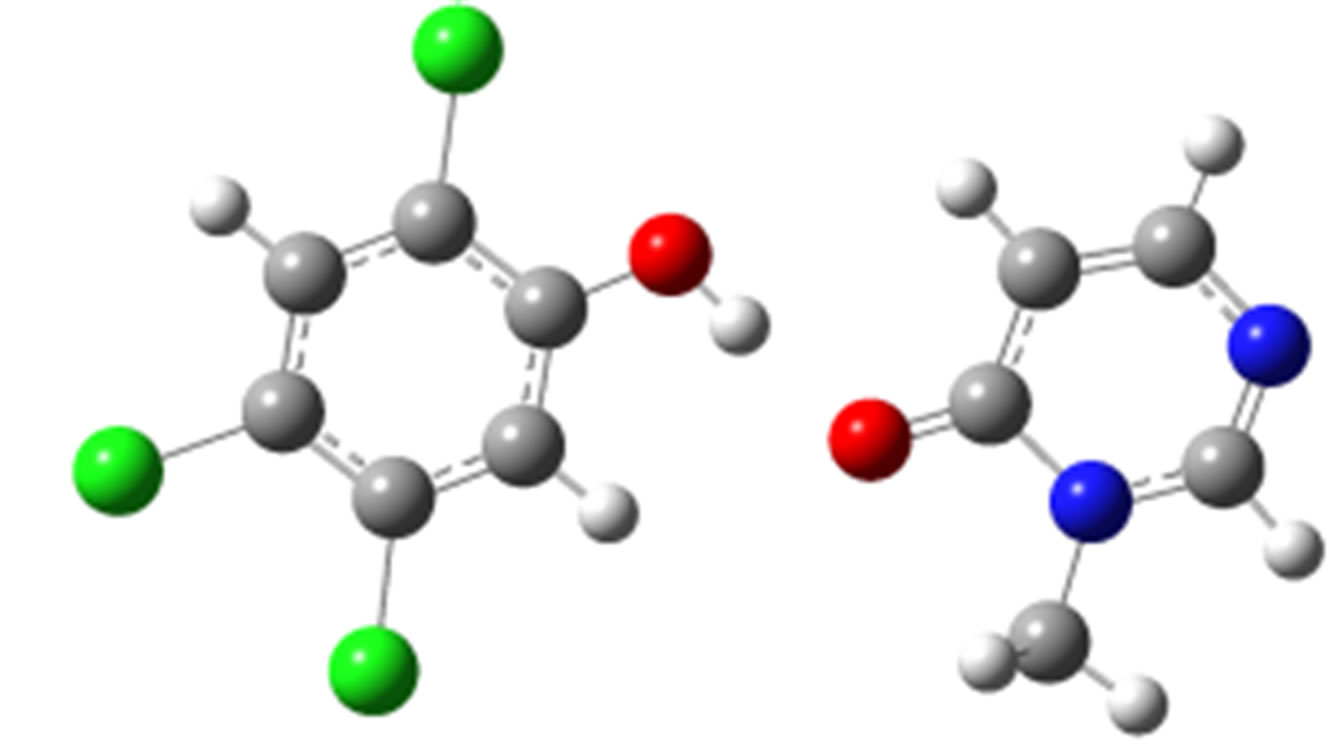 anti-N (–26.18)
anti-N (–26.18)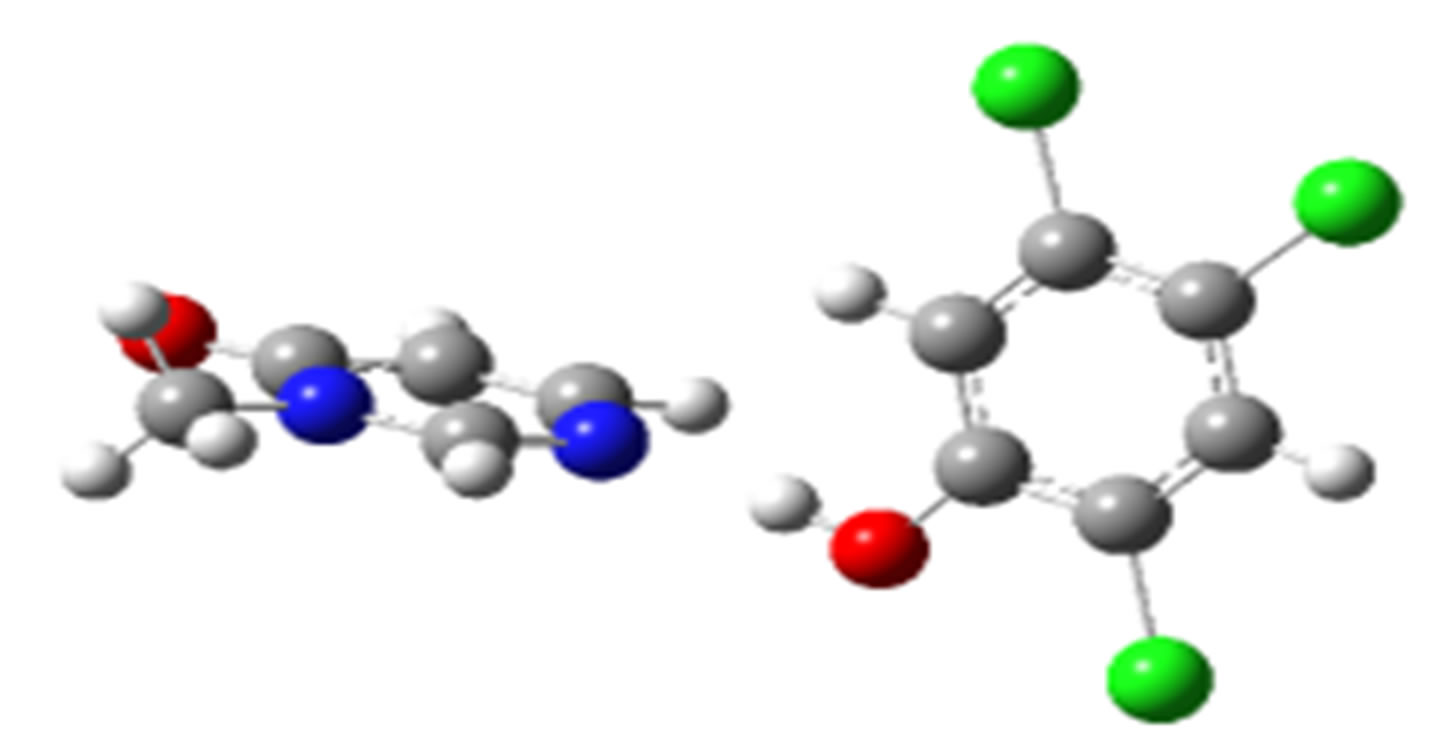
Pentachlorophenol-3M4P (ΔEc, kJ/mol)
anti-O (–33.34)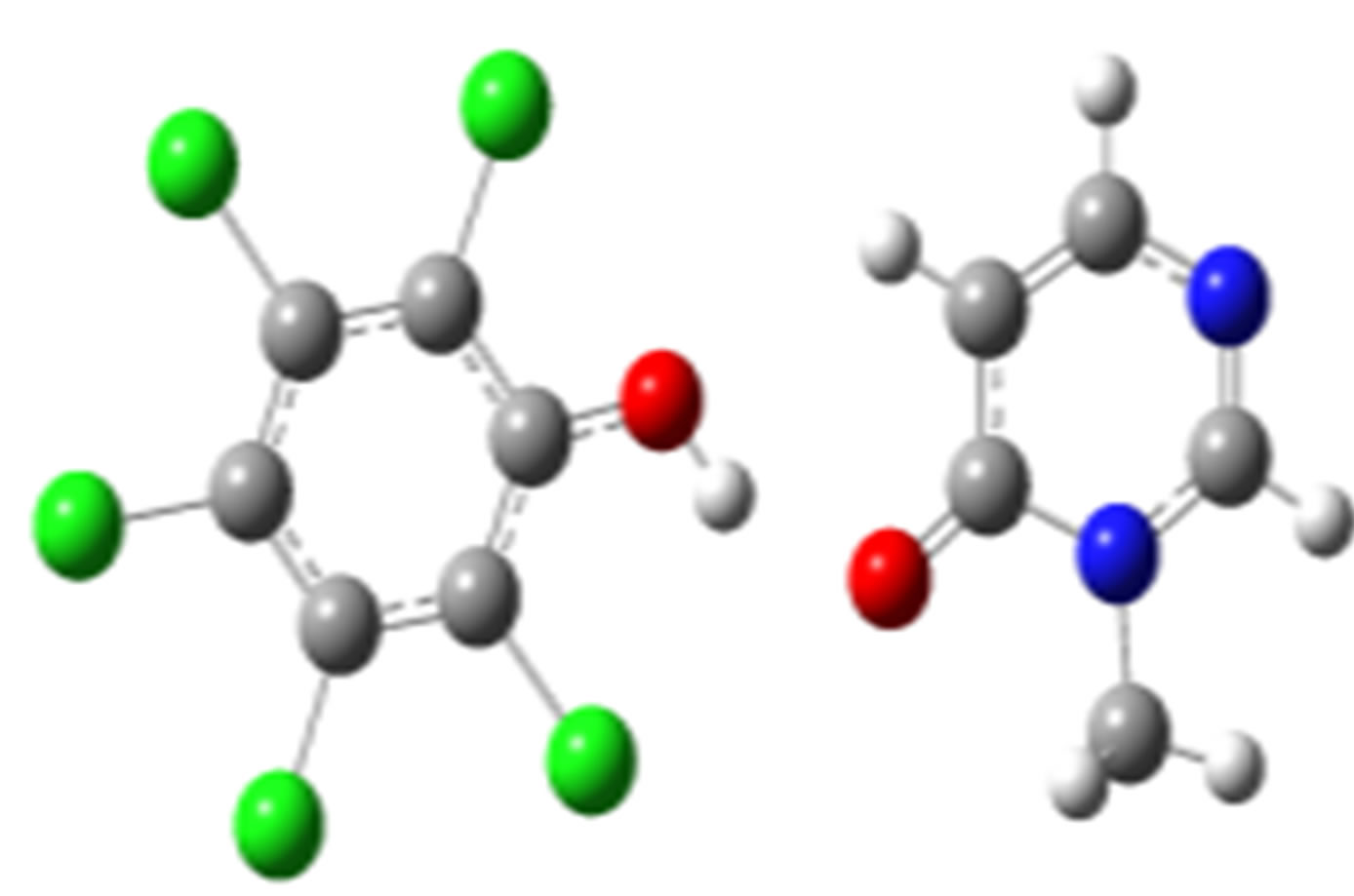 syn-O (–29.59)
syn-O (–29.59)
anti-N (–28.67)
Picric acid-3M4P (ΔEc, kJ/mol)
anti-O (–107.49) anti-N (–109.08)
anti-N (–109.08)
Hydrochloric acid-3M4P (ΔEc, kJ/mol)
anti-O (–28.20) syn-O (–24.30)
syn-O (–24.30)
anti-N (–29.20)
Scheme 2. Clusters of 3M4P with different proton donors (in red O, blue N, black C, green Cl, and grey H).
and r(O…O) which become shorter when the complex is more stable, i.e. when the H-bond interaction occurs at the C=O group with all proton donors, except for picric acid and hydrochloric acid. The frequency shift of the n(C=O) mode of a polyfunctional heterocyclic base such as 3M4P can also be used as an indication on the selectivity of the H-bond interaction in the base. A frequency decrease of n(C=O) suggests an interaction on the carbonyle site, whereas a frequency increase suggests a preferential interaction on the N1 atom of the base. The analysis of the calculated Dn(C=O) values in Tables 1-4 confirms the preference of 3M4P to interact with water, methanol, phenol, 1-naphtol, 2,4,5-trichlorophenol or pentachlorophenol by its C=O function, but with picric acid or hydrochloric acid by its N1 atom.
According to Allen [29], for the same H-bond enthalpy values, the Δn(OH) value is larger for OH…N than for OH…O=C H-bonds, which is due to a greater charge transfer contribution in OH…N than in OH…O=C bonds. This is illustrated in Figure 1, which relates Dn(OH) with pKa values of the different proton donors. Thus, a value of the shift Dn(OH) as large as –650 to –700 cm–1 suggests a H-bond interaction on the N atom. For the complexes between 3M4P and picric or hydrochloric acid, the calculated Dn(OH) values of –693 and –772 cm–1, respectively, undoubtedly demonstrate HC…N and OH…N interactions. The frequency shift difference D[Dn(OH)] = [Dn(OH)N – Dn(OH)O] can be used as a more precise indication for the selectivity of the H-bond interaction in heterocyclic polyfunctional bases. The shift differences D[Dn(OH)] listed in Table 5 appear to be smaller than –100 cm–1 for all complexes of 3M4P at the C=O group, whereas the values are larger than –200 cm–1 for the complexes at the N1 atom.
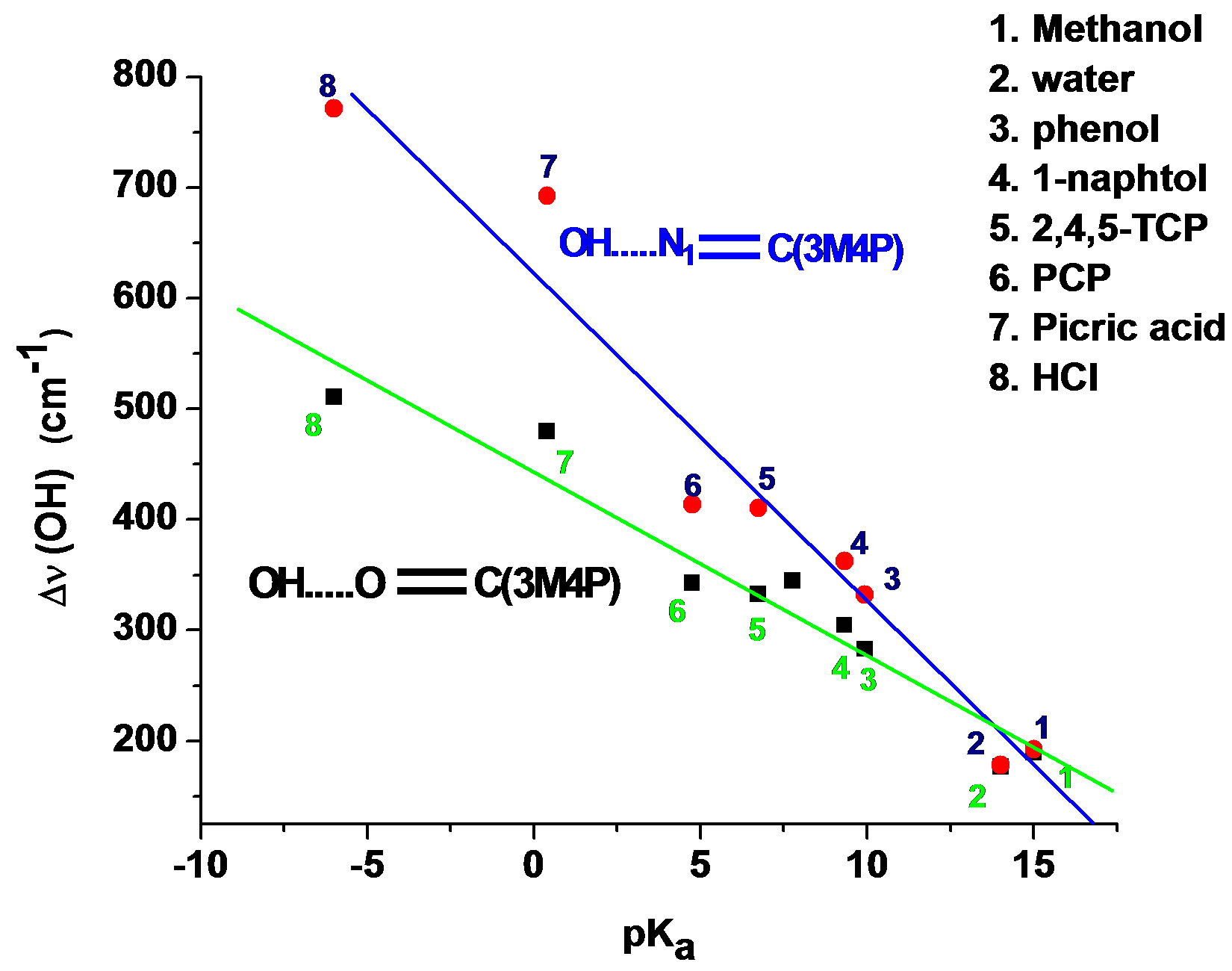
Figure 1. Δn(OH) values of 3M4P complexes against pKa values of the proton donors.
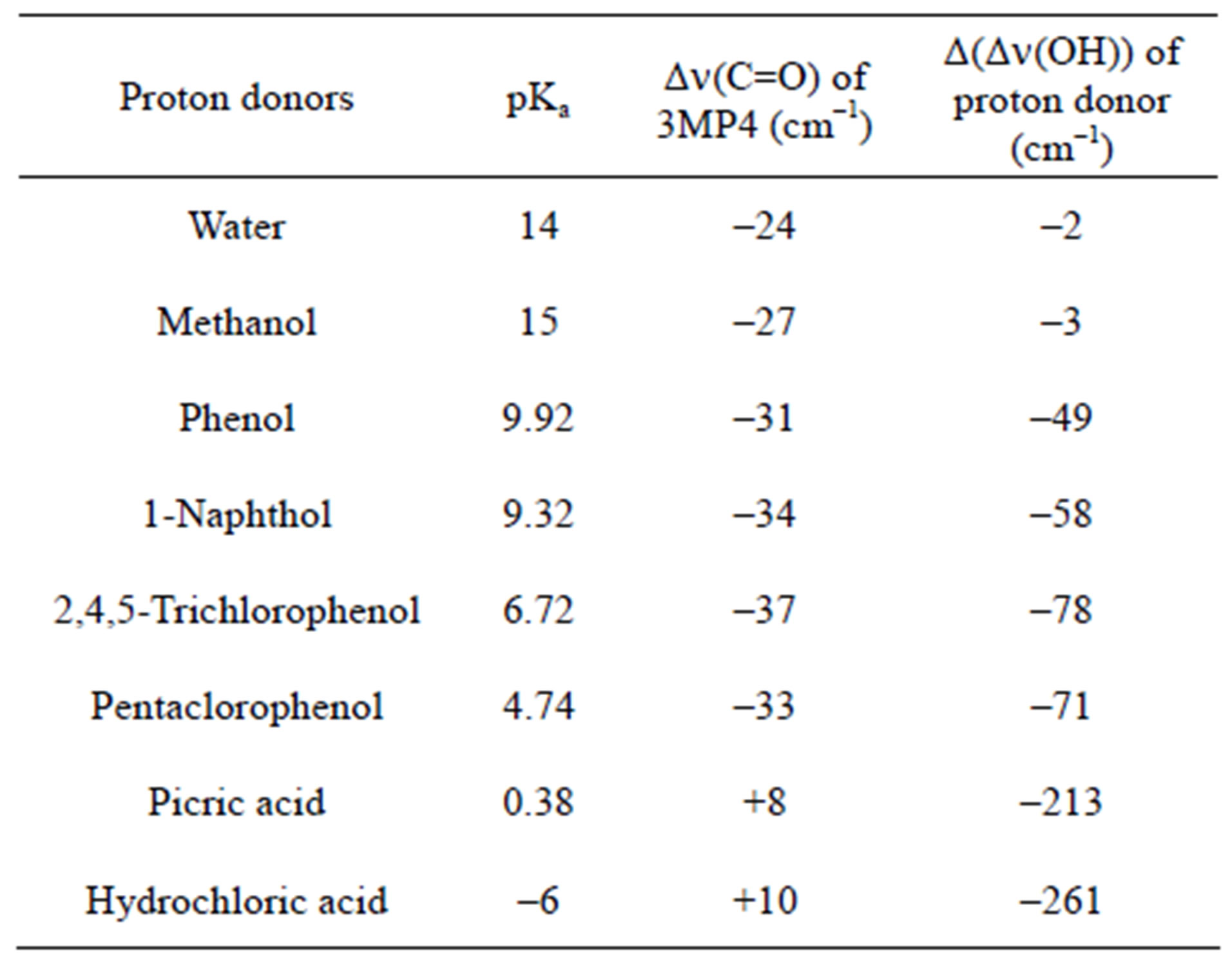
Table 5. Computed DFT(B3LYP)/6-31+G(d) frequency shifts Δn(C=O) and shift differences D[Dn(OH)] of the most stable complexes of 3MP4 with different proton donors.
For the complexes which are more stable when the H-bond interaction takes place on the carbonyl site rather than on the N1 atom of the base, a plot of Dn(C=O) against pKa of the proton donors is illustrated in Figure 2. It is observed that the parameter Dn(C=O) increases with increasing acidity of proton donor, until a pKa value of around 6.7 and then decreases with increasing acidity. The sequence of the Δn(C=O) values are: water < methanol < phenol < 1-naphtol < 2,4,5-trichlorophenol > pentachlorophenol. Other parameters, e.g. the shift Δn(OH) and the relative energy between both sites [D(ΔEc)] vary in a fairly similar way.
As shown in Figure 2, the pentachlorophenol which is classified as a strong proton donor by its small pKa value (4.6), has a weak value of Δn(C=O), suggesting a unexpected weaker interaction with 3M4P.
OH…Cl intramolecular and OH…O=C intermolecular H-bonds in the pentachlorophenol-3M4P complex, which reduces the stability of the latter complex. Since the pentachlorophenol is twicely ortho-subsituted with Cl, it is not surprising to observe strong intramolecular OH…Cl H-bonding.
3.2. Stability of “anti-O” and “syn-O” Conformations
For the complexes between 3M4P and weak proton donors, which are formed preferentially at the carbonyl group, the comparison of H-bond interaction energies ΔEc in Table 5 illustrates that the “anti-O” complexes are more stable than the “syn-O” complexes. The H-bond interaction energies ΔEc decrease in the order anti-O > syn-O > anti-N. On the other hand, for the complexes between 3M4P and stronger acids such as picric acid or hydrochloric acid the H-bond interaction energies ΔEc decrease in the order anti-O > syn-O > anti-N. For the stronger donors, the polarization of the O-H bond becomes more accentuated and induces a strong electronic repulsion between the proton donor O atom and the N3 atom of the base in the “syn-O” orientation (Scheme 3). This polarization of the O-H bond becomes too small for the weak proton donors, resulting in a negligible electronic repulsion.
In addition to the H-bond interaction energy ΔEc, the shifts of the n(C=O) and n(OH) modes as well as the internuclear and intermolecular distances r(O…H) and r(O…O) listed in Table 6 confirm the higher energetical stability of the “anti-O” complexes with 3M4P as compared to the “syn-O” complexes. For the complexes between 3M4P and weak proton donors the shifts Δn(C=O) and Dn(OH) decrease as anti-O > syn-O while the internuclear and intermolecular distances r(O…H) and r(O…O) increase as anti-O < syn-O < anti-N. A reverse effect is found for the complexes between 3M4P and the stronger acids, the internuclear and intermolecular
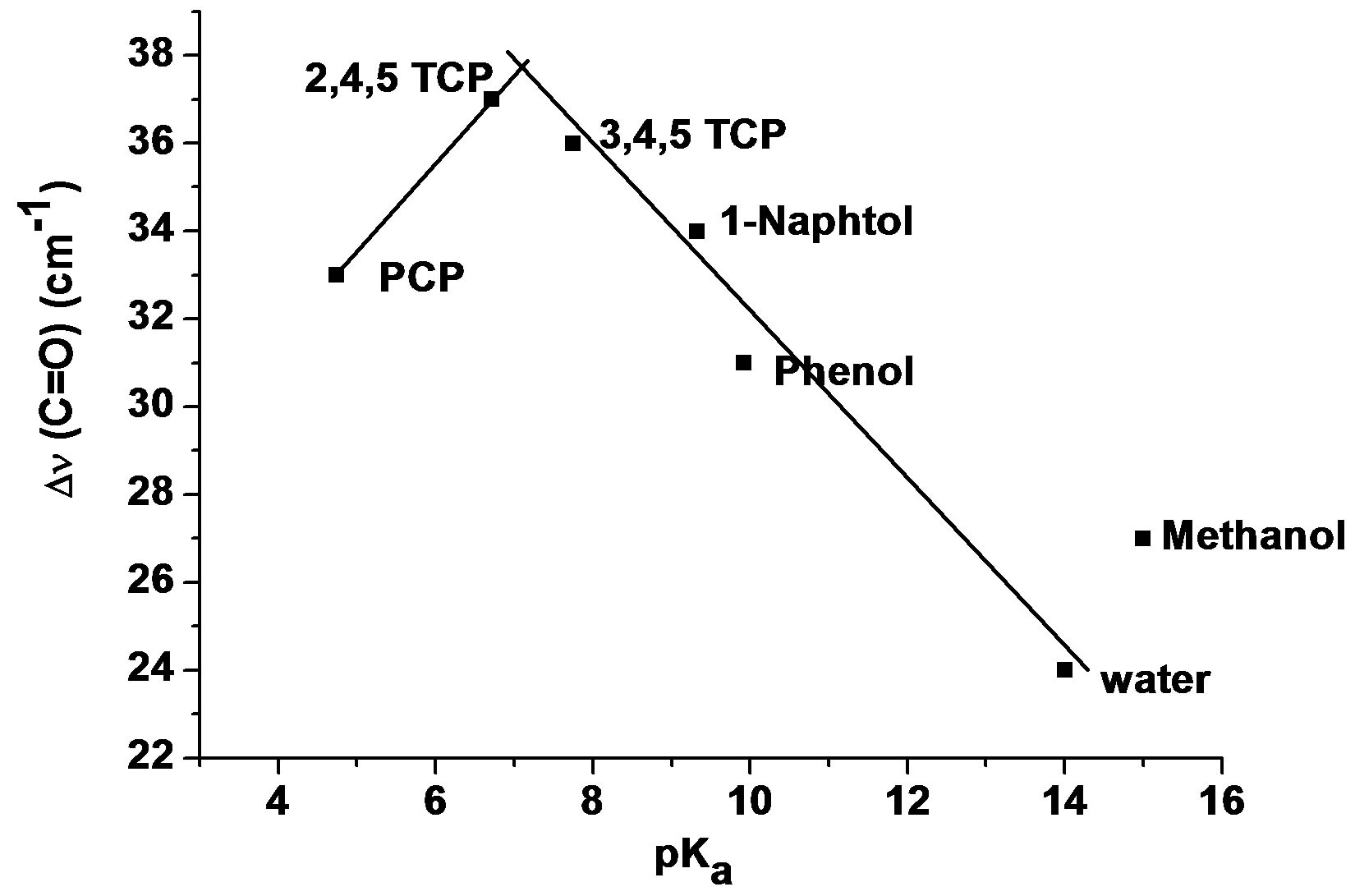
Figure 2. Δn(C=O) of 3M4P against the pKa value of the proton donors for C=O…H-O complexes of 3M4P 2,4,5-TCP: 2,4,5-trichlorophenol; 3,4,5-TCP: 3,4,5- trichlorophenol; PCP: pentachlorophenol.
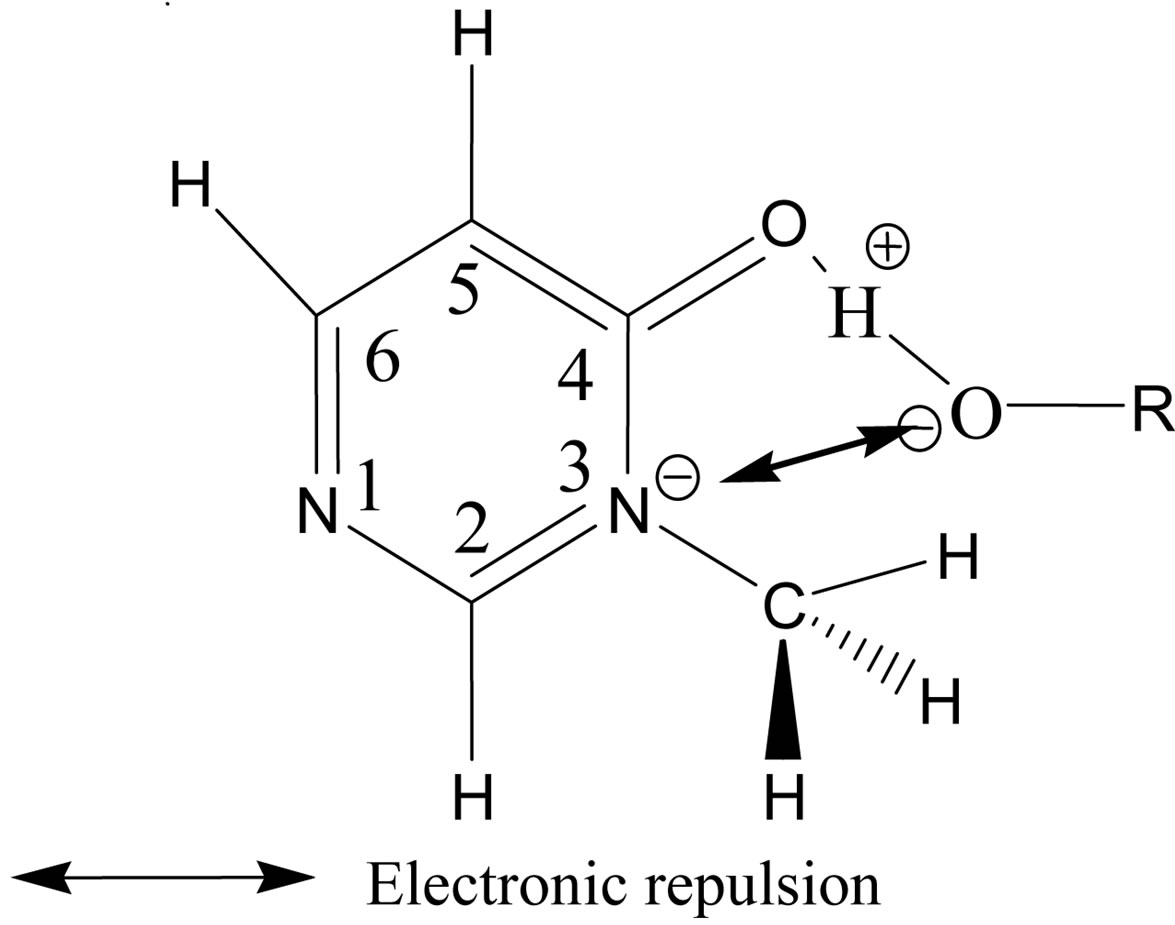
Scheme 3. Electronic repulsion in “syn” (Hb) C=O…H-O complexes.
distances r(O…H) and r(O…O) increasing as anti-N1 < anti-O < syn-O.
4. CONCLUSION
Theoretical (DFT-B3LYP/6-31+G(d)) calculations were performed to predict the preferred interaction site of the polyfunctional heterocyclic base 3-methyl-4-pyrimidone with proton donors of different acidic strength (water, methanol, phenol, 1-naphtol, 2,4,5-trichlorophenol, pentachlorophenol, picric acid and hydrochloric acid). Special attention has been paid to the H-bond interaction energy (ΔEc), the internuclear and intermolecular distances, the shifts Dn(C=O) and Dn(OH) and the shift difference parameter D[Dn(OH)]. The computational data were proved to be reliable for predicting the preferred interaction site of the 3-methyl-4-pyrimidone molecule with proton donors. The complexes of 3-methyl-4-pyrimidone and water, methanol, phenol, 1-naphtol,2,4,5- trichlorophenol and pentachlorophenol are more stable when the H-bond interaction takes place on the C=O function rather than on the N1 atom of the base. At the contrary, for the complex between 3-methyl-4-pyrimidone and the stronger acids picric acid and hydrochloric acid, the stability is higher when the interaction takes place on the N(1) atom of the base. For the complexes
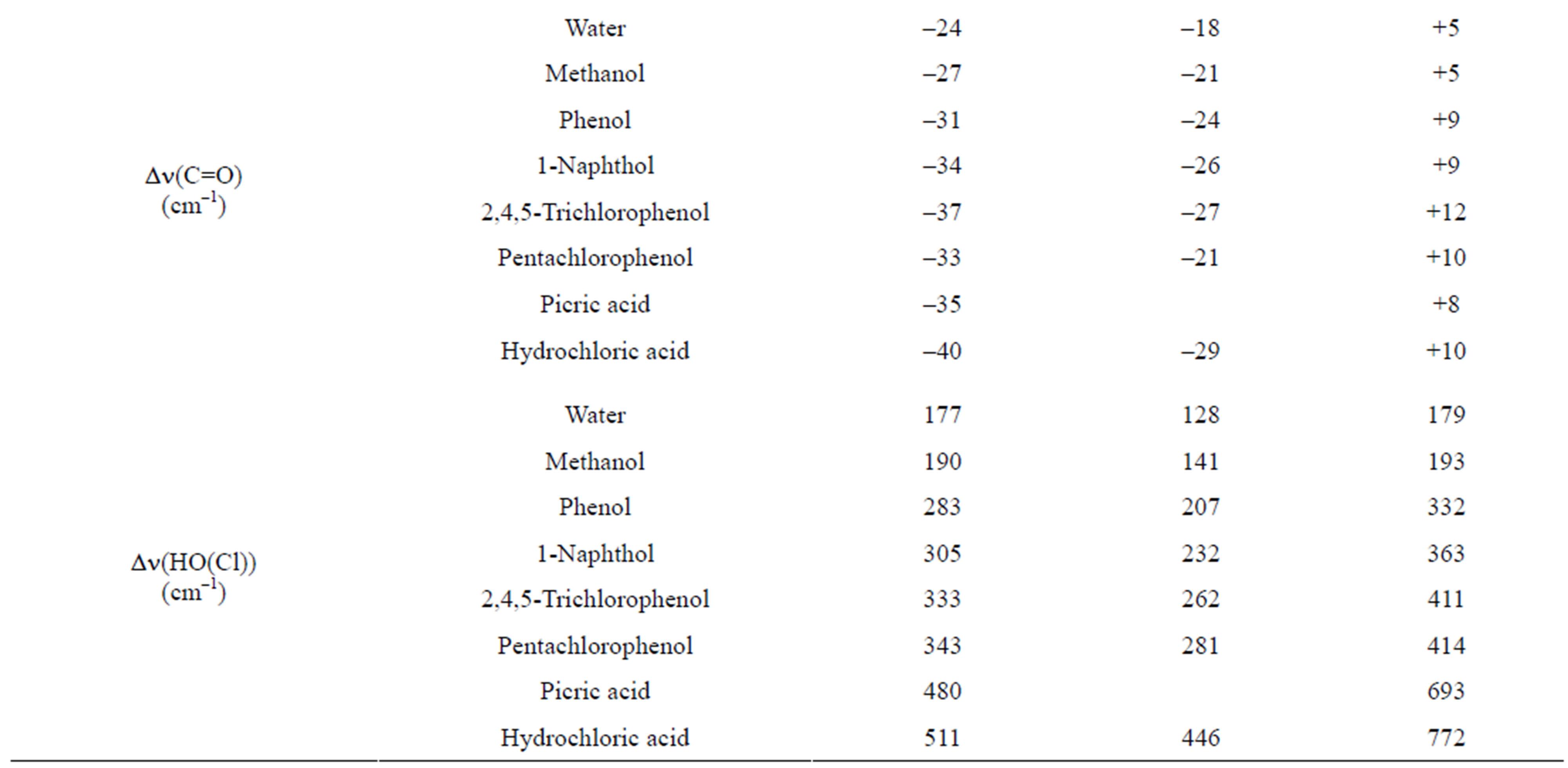
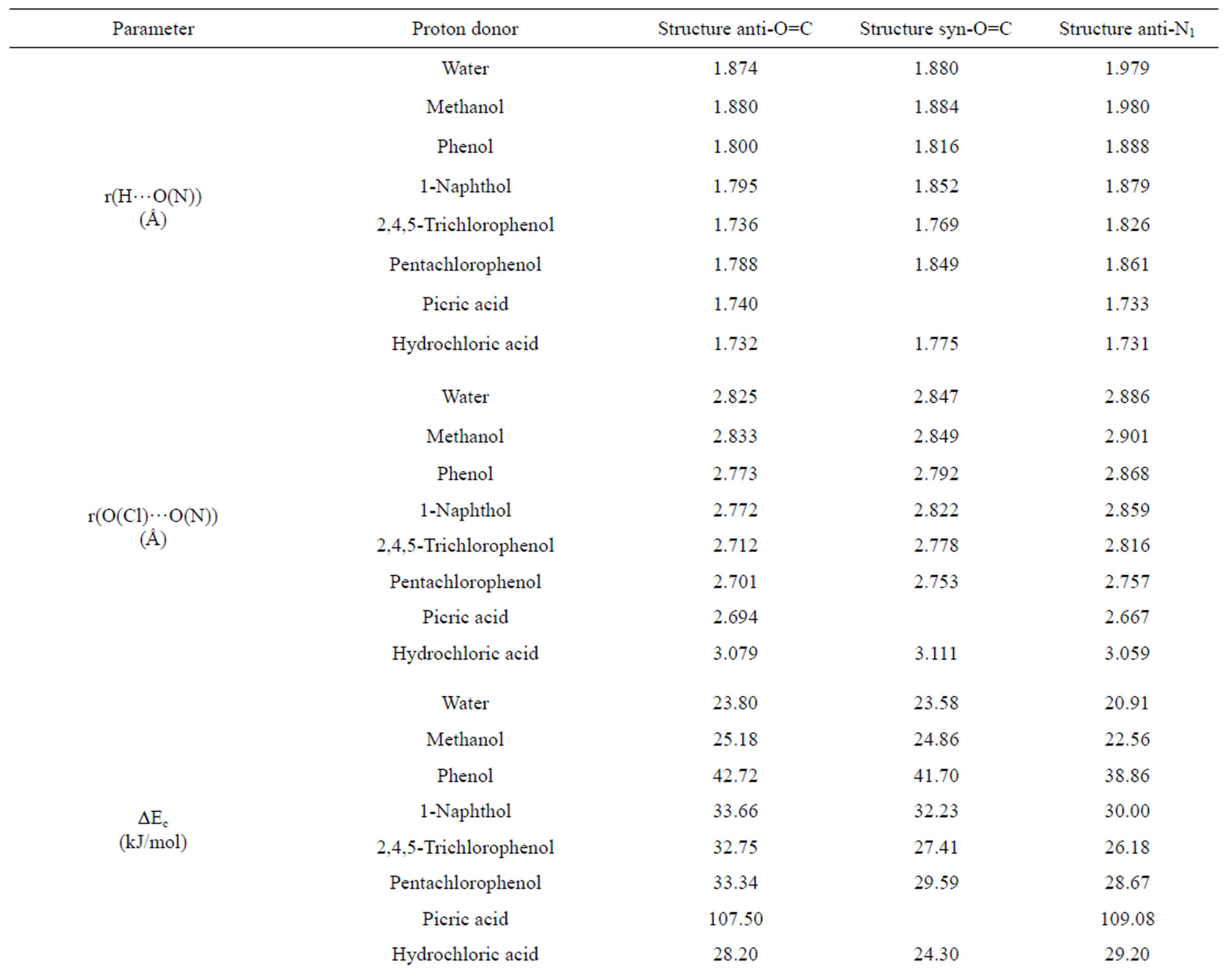
Table 6. Summary of computed DFT(B3LYP)/6-31+G(d) energetic and structural parameters of the H-bonded complexes of 3MP4 with proton donors at the C=O or at the N1 interaction site of the base.
between weak acids and 3-methyl-4-pyrimidone, which are formed preferentially at the carbonyl group, “anti” and “syn” complexes are possible, since the O atom of the group has two electronic pairs oriented in the directions (Ha and Hb) of the N3 atom of the ring. The H-bond interaction energies ΔEc, the shifts Δn(C=O) and Dn(OH) and the internuclear and intermolecular distances r(O…H) and r(O…O) all suggest a larger stability of the “anti-O” than of the “syn-O” complexes.
5. ACKNOWLEDGEMENTS
The authors acknowledge the Department of Chemistry of KULeuven for financial support. Two of us (Kasende, O. E. and Muzomwe, M.) would like to thank Professor Maes, G. for his hospitality. The support of the stay of one of us (O.E. Kasende) as Visiting Fellow at KULeuven by the Interfaculty Council for the Development Cooperation Specialisation Programme is appreciated. This research was partly conducted utilizing the high performance computational resources provided by the University of Leuven, http://ludit.kuleuven.be/hpc.
REFERENCES
- Kasende, O. and Zeegers-Huyskens, Th. (1981) Hydrogen-bonding and the protonation site in 3-methyl-4-pyrimidone. Journal of Molecular Structure, 75, 201-207. doi:10.1016/0022-2860(81)85234-9
- Kasende, O. and Zeegers-Huyskens, Th. (1984) Infrared study of hydrogen-bonded complexes involving phenol derivatives and polyfunctional bases. 2.3-methyl-4-pyrimidone, 1-methyl-2-pyrimidone, 1,4, 4-trimethylcytosine, and 1,3-dimethyluracil. Journal of Physical Chemistry, 88, 2636-2642. doi:10.1021/j150656a042
- Kasende, O. and Zeegers-Huyskens, Th. (1984) Infrared spectra of protonated pyrimidine-derivatives in the solidstate. Spectroscopy Letters, 17, 783-801. doi:10.1080/00387018408075706
- Muzomwe, Boeckx, B., Maes, G. and Kasende, O. (2011) Discrimination between OH..N and O-H..O=C complexes of 3-methyl-4 pyrimidone and methanol: A matrixisolation FT-IR and theoretical DFT/B3LYP investigations. South African Journal of Chemistry, 64, 23-33.
- Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti conelation energy formula into a functional of the electron density. Physical Review B, 37, 785- 792. doi:10.1103/PhysRevB.37.785
- Becke, A.D. (1993) Density functional thermochemistry. III The role of exact exchange. Journal of Chemical Physics, 93, 5648-5648. doi:10.1063/1.464913
- Parr, R.G. and Yang, W. (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York.
- Latajka, Z. and Bouteiller, Y. (1994) Application of Density functional methods for the study of hydrogen-bonded systems: The hydrogen fluoride dimmer. Journal of Chemical Physics, 101, 9793-9799. doi:10.1063/1.467944
- Novoa, J.J. and Sosa, C. (1995) Evaluation of the density approximation on the compoutation of hydrogen bond interactions. Journal of Physical Chemistry, 99, 15837- 15845. doi:10.1021/j100043a023
- Planas, M., Lee, C. and Novoa, J.J. (1996) Kinetics of the proton transfer in X…(H2O)4 clusters (X=H2O, NH3, H2S and HCl): Evidence of a concerted mechanism. Journal of Physical Chemistry, 100, 16495-16501. doi:10.1021/jp960789t
- Lundell, J. and Latajka, Z. (1997) Density functional study of hydrogen-bonded systems: The H2O-CO complex. Journal of Physical Chemistry, 101, 5004-5009. doi:10.1021/jp963727b
- Mo, O., Yanez, M. and Elguero, J. (1997) Study of the methanol trimer potential energy surface. Journal of Chemical Physics, 107, 3592-3601. doi:10.1063/1.474486
- Dkhissi, A., Adamowicz, L. and Maes, G. (2000) Density functional theory study of the hydrogen-bonded pyridine-H2O complex: A comparison with RHF and MP2 methods with experimental data. Journal of Physical Chemistry, 104, 2112-2119. doi:10.1021/jp9938056
- Dkhissi, A., Adamowicz, L. and Maes, G. (2000) Hybrid density functional and ab initio studies of 2-Pyridone- (H2O) and 2-Pyridone-(H2O)2. Chemical Physics Letters, 324, 127-136. doi:10.1016/S0009-2614(00)00474-7
- Boyd, S.I. and Boyd, R.J. (2007) A density functional study of methanol clusters. Journal of Chemical Theory and Computation, 3, 54-61. doi:10.1021/ct6002912
- Parra, R.D. and Zeng, X.Z. (1999) hydrogen bonding and cooperative effects in mixed dimmers and trimers of methanol and trifluoromethanol: An ab itio study. Journal of Chemical Physics, 110, 6329-6338. doi:10.1063/1.478537
- Bing, D., Kuo, J.-L., Suhara, K.-I., Fujii, A. and Mikami, N. (2009) Proton switch correlated with the morphological development of the hydrogen bond network in H+(CH3OH)m(H2O)1 (m = 1-9): An theoretical and infrared spectroscopic study. Journal of Physical Chemistry A, 113, 2323-2332. doi:10.1021/jp900066u
- Mejia, S.M., Espinal, J.F. and Mondragon, F.-S. (2009) Cooperative effect on the structure and stability of (ethanol)3-water, (methanol)3-water heterotetramers and (ethanol)4, (methanol)4 tetramers. Journal of Molecular Structure (Theochem), 901, 186-193. doi:10.1016/j.theochem.2009.01.027
- Palafox, M.A., Iza, N., De la Fuente, M. and Navarro, R. (2009) Simulation of the first hydration shell of nucleosides D4T and thymidine: Structures obtained using MP2 and DFT methods. Journal of Physical Chemistry B, 113, 2458-2476. doi:10.1021/jp806684v
- Muzomwe, M. (2011) Influence of cooperativity, regioselectivity and stereoselectivity on the stability of complexes formed between methylpyrimidones bases and proton donors acids: A DFT/B3LYP/6-31 + G(d) and FTIR in inert argon matrix study. Ph.D. Thesis, University of Kinshasa, Congo.
- Chalasinski, G. and Szczesniak, M. (1994) Origins of structure and energetics of van der waals clusters from ab initio calculations. Chemical Reviews, 94, 1723-1765. doi:10.1021/cr00031a001
- Ramaekers, R., Maes, G., Adamowicz, L. and Dkhissi, A. (2001) Matrix-isolation FT-IR study and theoretical calculations of the vibrational, taufomeric and H-bonding properties of hypoxanthine. Journal of Molecular Structure, 560, 205-221. doi:10.1016/S0022-2860(00)00733-X
- Halls, M.D., Velkowski, J. and Schleger, H.B. (2001) Harmonic frequency scaling factors for hartree-fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theoretical Chemistry Accounts, 105, 413-421. doi:10.1007/s002140000204
- Rauhut, G. and Pulay, P. (1995) Transferable scaling factors for density functional derived vibrational force fields. Journal of Physical Chemistry, 99, 3093-3100. doi:10.1021/j100010a019
- Florián, J., Baumruk, V. and Leszczyński, J. (1996) IR and Raman spectra, tautomeric stabilities, and scaled quantum mechanical force fields of protonated cytosine. Journal of Physical Chemistry, 100, 5578-5589. doi:10.1021/jp953284w
- Gutowski, M., Van Duijneveldt, F.B., Van Duijneveldtvan de Rijdt, G.C.M. and Van Lenthe, J.H. (1993) Accuracy of the boys and Bernardi function counterpoise method. Journal of Chemical Physics,, 98, 4728-4738. doi:10.1063/1.465106
- Boys, S.F. and Bernardi, F. (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Molecular Physics, 19, 553-566. doi:10.1080/00268977000101561
- Frisch, M.J.T.G.W., Schlegel, H.B, et al. (2004) Gaussian 03, Revision D.02, Version. Gaussian, Inc., Wallingford.
- Allen, L.C. (1975) Simple model of hydrogen bonding. Journal of the American Chemical Society, 97, 6921- 6940. doi:10.1021/ja00857a001