Engineering
Vol.07 No.09(2015), Article ID:59957,19 pages
10.4236/eng.2015.79053
Thermodynamic Equilibrium of the Saturated Fluid with a Free Surface Area and the Internal Energy as a Function of the Phase-Specific Volumes and Vapor Pressure
Albrecht Elsner
Am Mühlbach 14, D-85748, Garching, Germany
Email: alimeli.elsner@gmail.com
Copyright © 2015 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 July 2015; accepted 21 September 2015; published 25 September 2015
ABSTRACT
This study is concerned with describing the thermodynamic equilibrium of the saturated fluid with and without a free surface area A. Discussion of the role of A as system variable of the interface phase and an estimate of the ratio of the respective free energies of systems with and without A show that the system variables
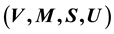 given by Gibbs suffice to describe the volumetric properties of the fluid. The well-known Gibbsian expressions for the internal energies of the two-phase fluid, namely
given by Gibbs suffice to describe the volumetric properties of the fluid. The well-known Gibbsian expressions for the internal energies of the two-phase fluid, namely
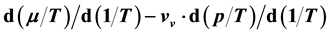 for the vapor and
for the vapor and
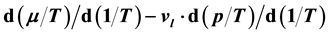 for the condensate (liquid or solid), only differ with respect to the phase-specific volumes
for the condensate (liquid or solid), only differ with respect to the phase-specific volumes
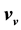 and
and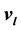 . The saturation temperature T, vapor presssure p, and chemical potential
. The saturation temperature T, vapor presssure p, and chemical potential
 are intensive parameters, each of which has the same value everywhere within the fluid, and hence are phase-independent quantities. If one succeeds in representing
are intensive parameters, each of which has the same value everywhere within the fluid, and hence are phase-independent quantities. If one succeeds in representing
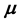 as a function of
as a function of
 and
and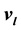 , then the internal energies can also be described by expressions that only differ from one another with respect to their dependence on
, then the internal energies can also be described by expressions that only differ from one another with respect to their dependence on
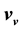 and
and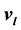 . Here it is shown that
. Here it is shown that
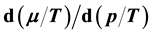 can be uniquely expressed by the volume function
can be uniquely expressed by the volume function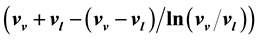 . Therefore, the internal energies can be represented explicitly as functions of the vapor pressure and volumes of the saturated vapor and condensate and are absolutely determined. The hitherto existing problem of applied thermodynamics, calculating the internal energy from the measurable quantities T, p,
. Therefore, the internal energies can be represented explicitly as functions of the vapor pressure and volumes of the saturated vapor and condensate and are absolutely determined. The hitherto existing problem of applied thermodynamics, calculating the internal energy from the measurable quantities T, p,
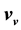 , and
, and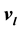 , is thus solved. The same method applies to the calculation of the entropy, chemical potential, and heat capacity.
, is thus solved. The same method applies to the calculation of the entropy, chemical potential, and heat capacity.
Keywords:
Fluid with Free Surface Area, Solution of Gibbs’s Internal Energy Equations, Chemical Potential Expression, Calculation of Entropy and Heat Capacity

1. Introduction
We are concerned here with electrically and magnetically neutral single-component matter under steady-state equilibrium conditions which are thermodynamically defined in the immediate vicinity of the critical point and below it. Between the critical gas temperature
 and the triple-point temperature
and the triple-point temperature
 (if it exists) the gas mass M is structureless and homogeneously distributed in the vessel volume V. It is assumed that M has the critical density
(if it exists) the gas mass M is structureless and homogeneously distributed in the vessel volume V. It is assumed that M has the critical density . Below the critical point, M is decomposed as condensed mass
. Below the critical point, M is decomposed as condensed mass
 with the density
with the density
 in the sub-volume
in the sub-volume
 and as vapor mass
and as vapor mass
 with the density
with the density
 in the sub-volume
in the sub-volume . This gas mass in thermodynamic equilibrium existing in two phases is called a saturated fluid. As thermodynamic theory teaches, the only independent variable of the saturated fluid that can be chosen is the saturation temperature T, since the other field variables possible, viz. vapor pressure p and chemical potential
. This gas mass in thermodynamic equilibrium existing in two phases is called a saturated fluid. As thermodynamic theory teaches, the only independent variable of the saturated fluid that can be chosen is the saturation temperature T, since the other field variables possible, viz. vapor pressure p and chemical potential , are unique functions of T.
, are unique functions of T.
The critical point, from the experimental perspective, is the first occurrence or vanishing of a free surface A observed in V, which separates the volumes
 and
and
 from one another. By variation of T and M or V and observing the occurrence of A, one can define and measure the critical values v,
from one another. By variation of T and M or V and observing the occurrence of A, one can define and measure the critical values v,
 , and
, and .
.
The work in Section 2 is concerned with describing the thermodynamic equilibrium of the real gas. The stationary equilibrium of M in V is known from Gibbs to be expressed by the fundamental equation
 which reproduces the functional relation among V, M, and the properties of the fluid, viz. entropy S, internal energy U, and free energy
which reproduces the functional relation among V, M, and the properties of the fluid, viz. entropy S, internal energy U, and free energy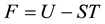 .
.
Section 3 firstly treats the equilibrium of the two-phase fluid without an internal free surface, i.e. . The distributions of the masses
. The distributions of the masses
 and
and
 in
in
 and
and
 are given as functions of T. Then the equilibrium in the case
are given as functions of T. Then the equilibrium in the case
 is discussed where there is a third fluid phase, called the interface phase. To it is assigned the free interface energy
is discussed where there is a third fluid phase, called the interface phase. To it is assigned the free interface energy , which is identified with
, which is identified with
 (
( surface tension), so that the ratio
surface tension), so that the ratio
 can be numerically estimated. Estimation and discussion of the role of A as system variable show that the system variables
can be numerically estimated. Estimation and discussion of the role of A as system variable show that the system variables
 given by Gibbs suffice to describe the volumetric properties of the fluid with and with- out a free surface area.
given by Gibbs suffice to describe the volumetric properties of the fluid with and with- out a free surface area.
In Section 4, it is reminded that the entropy and energy functions S,
 , and
, and
 and also U,
and also U,
 , and
, and
 assigned to the masses M,
assigned to the masses M,
 , and
, and
 are absolute temperature functions with thermodynamic zeros. This result, which can be deduced from internal energy functions being subject to Nernst’s theorem at absolute zero, is noteworthy, because the said quantities have been treated in Applied Thermodynamic Theory for more than a century as temperature functions with arbitrarily specified constants.
are absolute temperature functions with thermodynamic zeros. This result, which can be deduced from internal energy functions being subject to Nernst’s theorem at absolute zero, is noteworthy, because the said quantities have been treated in Applied Thermodynamic Theory for more than a century as temperature functions with arbitrarily specified constants.
Finally, in Section 5, the central task of this work, viz. finding an explicit thermodynamic expression for
 in
in , is tackled and then solved in Section 6. The energy functions u,
, is tackled and then solved in Section 6. The energy functions u,
 ,
,
 ,
,
 ,
,
 ,
,
 and
and , and the heat capacities c,
, and the heat capacities c,
 , and
, and
 can then be calculated from the measurable quantities
can then be calculated from the measurable quantities ,
,
 , and
, and .
.
By means of the expression given for the volume function
 many thermodynamic relations are verified in the Appendix, this in turn being evidence for the correctness of the volume function used.
many thermodynamic relations are verified in the Appendix, this in turn being evidence for the correctness of the volume function used.
2. Description of the Thermodynamic Equilibrium of the Real Gas
Thermodynamics uses intensive and extensive quantities to describe the equilibrium state of the gas mass M enclosed in the volume V. An intensive property of the gas is the same everywhere in the volume and is therefore independent of the mass. Intensive equilibrium state quantities are the temperature T, pressure p, and chemical potential . They are defined by the first partial derivatives of extensive quantities [1] , e.g., the internal energy U, which according to the fundamental equation is a function of entropy S, volume V, and mass M [2] :
. They are defined by the first partial derivatives of extensive quantities [1] , e.g., the internal energy U, which according to the fundamental equation is a function of entropy S, volume V, and mass M [2] :

 (1)
(1)
Variations in the extensive quantities S, V, and M lead to variations in the intensive quantities T, p, and
 [2] as follows:
[2] as follows:
 (2)
(2)
By means of Maxwell’s relations [2]
 ,
,
 ,
,
 ,
,  ,
, , and
, and
 the differentials (2) can be written in the form
the differentials (2) can be written in the form
 (3)
(3)
For the single-component gas the well-known Gibbs-Duhem relation between the intensive quantities in dif- ferential form reads [2] :
 (4)
(4)
Explicit writing gives
 (5)
(5)
Variations in S, V, and M can be performed independently of each other. For dV = 0 and dM = 0 one thus has
 or
or
 . Applying the identity
. Applying the identity
 , the entropy relation can be written as
, the entropy relation can be written as .
.
Using the Maxwell relations
 and
and , one arrives at the
, one arrives at the
Gibbs-Duhem form of the entropy:
 (6)
(6)
With
 and dM = 0 one obtains
and dM = 0 one obtains , whereas the conditions
, whereas the conditions

and
 give
give .
.
In the following, the properties of the real gas are investigated under the condition of constant mass M in given volume V. The entropy thus has the form (6). The internal-energy expression immediately follows from
the fundamental equation, i.e.
 or
or
 (7)
(7)
According to relations (6) and (7), the entropy and internal energy of the gas mass are defined by the temperature derivatives of the intensive quantities
 and p and the quantities M and V. The gas volume V has the significant property that it is proportional to the gas mass M since the gas of mean density
and p and the quantities M and V. The gas volume V has the significant property that it is proportional to the gas mass M since the gas of mean density
 assumes the equilibrium-state volume
assumes the equilibrium-state volume . The entropy S and internal energy U are thus quantities that are proportional to M and hence absolute quantities [3] . As mentioned above, extensive and intensive quantities, both of which are given with absolute figures, are used for describing the emquilibrium properties of the gas, no matter which gas phases exist.
. The entropy S and internal energy U are thus quantities that are proportional to M and hence absolute quantities [3] . As mentioned above, extensive and intensive quantities, both of which are given with absolute figures, are used for describing the emquilibrium properties of the gas, no matter which gas phases exist.
3. Two-Phase Equilibrium without and with a Free Surface Area
For every real gas there is a certain temperature , called critical temperature, at which the gas mass M decomposes into a low-density vapor-phase mass
, called critical temperature, at which the gas mass M decomposes into a low-density vapor-phase mass
 and a high-density condensed-phase mass
and a high-density condensed-phase mass
 (liquid or solid). The masses
(liquid or solid). The masses
 and
and
 are located in different subvolumes
are located in different subvolumes
 and
and , separated from one another by a free interface area A. If
, separated from one another by a free interface area A. If , the first occurrence of A in V defines the critical volume v and
, the first occurrence of A in V defines the critical volume v and , i.e. the critical point
, i.e. the critical point
 of the gas, which for temperatures
of the gas, which for temperatures
 is termed as a saturated fluid. Ignoring the existence of A, this fluid has the following properties:
is termed as a saturated fluid. Ignoring the existence of A, this fluid has the following properties:
 (8)
(8)
The relation
 states that the quantity X is proportional to the mass M, which means that each of the functions X (gas volume V, entropy S, internal energy U, free (Helmholtz) energy
states that the quantity X is proportional to the mass M, which means that each of the functions X (gas volume V, entropy S, internal energy U, free (Helmholtz) energy , enthalpy
, enthalpy , Gibbs energy
, Gibbs energy , and heat capacity
, and heat capacity ) ensures its uniqueness, has absolute value due to its thermodynamic zero, and is numerically interrelated to one another [3] .
) ensures its uniqueness, has absolute value due to its thermodynamic zero, and is numerically interrelated to one another [3] .
The decomposition of mass M into
 and
and
 below the critical point is intrinsicly described by [3]
below the critical point is intrinsicly described by [3]
 (9)
(9)
Taking ,
,
 and
and , Equation (9) gives
, Equation (9) gives
 (10)
(10)
It is seen, because of , that the mass ratios
, that the mass ratios
 are functions of v and T.
are functions of v and T.
The proof of
 is as follows: The positive functions
is as follows: The positive functions
 and
and
 are related by
are related by
 and
and . For
. For
 the functions
the functions
 and
and
 con- verge from above and below to the same limiting value
con- verge from above and below to the same limiting value . This yields
. This yields
 or
or .
.
Then equation
 represents the relation between the masses and specific volumes on incipient decomposition. With decreasing system temperature, more and more vapor particles con- dense into the liquid phase, i.e. the mass ratio
represents the relation between the masses and specific volumes on incipient decomposition. With decreasing system temperature, more and more vapor particles con- dense into the liquid phase, i.e. the mass ratio
 becomes less than 1 and decreases with decreasing
becomes less than 1 and decreases with decreasing
temperature:
 and
and
 for
for .
.
If one investigates the temperature dependence of the volumes
 and
and
 between the critical point and absolute zero, one can generally ascertain that the volume variations are smaller than those of the masses and take opposite directions. From
between the critical point and absolute zero, one can generally ascertain that the volume variations are smaller than those of the masses and take opposite directions. From
 one obtains
one obtains
 and
and
 , while from
, while from
 it follows that
it follows that
 and
and . Since for real gases
. Since for real gases
 the ratio
the ratio
 takes values between 1.5 and 3, with a jump occurring at the triple point. The following relations are valid:
takes values between 1.5 and 3, with a jump occurring at the triple point. The following relations are valid:
 (11)
(11)
Evaluation of relations (10) and (11) is given by means of published volume data for argon in Figure 1.
Let us now turn to the problem of thermodynamic treatment of the physics of the free interface surface A. As stated, the existence of A affords the possibility of distinguishing in V between two fluid phases of different mass densities
 and
and
 and defining the critical point. Thermodynamic theory teaches that the intensive quantities T, p and
and defining the critical point. Thermodynamic theory teaches that the intensive quantities T, p and
 have the same value everywhere in V, i.e. in the interface layer as well, in which the density decreases from
have the same value everywhere in V, i.e. in the interface layer as well, in which the density decreases from
 to
to . If the length of this decrease is denoted by the distance
. If the length of this decrease is denoted by the distance , the volume
, the volume
 can be assigned to the interface layer, in which the interface mass
can be assigned to the interface layer, in which the interface mass
 is located, where it holds that
is located, where it holds that
 and
and
 with
with . The fictitious quantities
. The fictitious quantities
 and
and , however, cannot be thermodynamically calculated, whereas both the minimum surface expansion A, resulting from intermolecular and acceleration forces, and the surface tension
, however, cannot be thermodynamically calculated, whereas both the minimum surface expansion A, resulting from intermolecular and acceleration forces, and the surface tension , which is a measure of the effectiveness of these forces at the surface, are measurable quantities,
, which is a measure of the effectiveness of these forces at the surface, are measurable quantities,
 being a positive quantity [5] . The force resulting from these two yields the direction of the surface normal of A. The expansion of A depends on the shape of the vessel V. If, for example, the shape of V is chosen such that arbitrary rotation about the center of gravity of M changes the location of the interface, i.e. the height and expansion, the values of p and
being a positive quantity [5] . The force resulting from these two yields the direction of the surface normal of A. The expansion of A depends on the shape of the vessel V. If, for example, the shape of V is chosen such that arbitrary rotation about the center of gravity of M changes the location of the interface, i.e. the height and expansion, the values of p and
 as measures of the energy density in
as measures of the energy density in
 and A remain unchanged, but the distribution of
and A remain unchanged, but the distribution of

Figure 1. Mass ratio
 and volume ratio
and volume ratio
 of argon as functions of the temperature. Data
of argon as functions of the temperature. Data
 and
and
 from Ref. [4] .
from Ref. [4] .
the interface mass
 in the newly formed volume
in the newly formed volume
 is changed and this can also be reversed isother- mally and isobarically, since the free energy measures the mechanical work done. On the other hand, the redistribution of the fluid mass is a result of the changed gravitation potential in V and this can, in principle, be determined as potental energy from the height differences of A before and after rotation and hence be thermodynmically expressed by the difference in the free energy F. Thus A can be regarded as an external thermodynamic variable, and the product
is changed and this can also be reversed isother- mally and isobarically, since the free energy measures the mechanical work done. On the other hand, the redistribution of the fluid mass is a result of the changed gravitation potential in V and this can, in principle, be determined as potental energy from the height differences of A before and after rotation and hence be thermodynmically expressed by the difference in the free energy F. Thus A can be regarded as an external thermodynamic variable, and the product
 is measurable and constitutes a thermodynamic quantity.
is measurable and constitutes a thermodynamic quantity.
Independently of prehistory, it holds for the saturated fluid that if the condition for forming a free surface between the liquid and vapor phases is given, then there is an interface particle layer, which represents a new equilibrium state described by a minimum internal energy U and simultaneously a maximum entropy S. Hence formation of the free interface surface A lowers the free energy of the fluid, F. This situation is formally taken into account by introducing the phase “interface” in keeping with the additive property of a variable X in addition to the phases “vapor” and “condensate” [6] :
 (12)
(12)
The energy term
 is interpreted as free interface energy and described by the function
is interpreted as free interface energy and described by the function ; the newly introduced function
; the newly introduced function
 is interpreted as surface energy, and the function
is interpreted as surface energy, and the function
 as surface entropy. These func- tions represent reversible interface quantities of the free surface A which vanish at
as surface entropy. These func- tions represent reversible interface quantities of the free surface A which vanish at
 and also at
and also at
 because
because
 and
and .
.
Then the enlarged Gibbs relations (6) and (7) read [6]
 (13)
(13)
Figure 2 shows the temperature dependence of ,
,
 , and
, and
 for argon. In turn, these functions multiplied by A represent the area-contributions to the negative internal and free energies and positive entropy of the saturated fluid.
for argon. In turn, these functions multiplied by A represent the area-contributions to the negative internal and free energies and positive entropy of the saturated fluid.
In order to put the interface quantity
 in thermodynamic relation to the above-mentioned quantities
in thermodynamic relation to the above-mentioned quantities
 and
and ,
,
 is set equal to
is set equal to . Assuming
. Assuming ,
,
 and a mean in-
and a mean in-
terface density , one obtains, because of
, one obtains, because of , a functional relation between surface
, a functional relation between surface
tension and interface quantities,
 (14)
(14)
which, however, remains numerically indeterminable owing to the hypothetical length . In turn, relation (14) allows the qualitative statement that
. In turn, relation (14) allows the qualitative statement that
 is continuously increasing from 0 at
is continuously increasing from 0 at
 to values of order
to values of order
 at
at
 (see Figure 2, Figure 4 and Figure 8).
(see Figure 2, Figure 4 and Figure 8).
The relative energy contribution of an interface quantity to the respective system quantity depends on the ratio of the numbers of interacting particles in the interface volume
 and system volume V, i.e.
and system volume V, i.e.
 , and is therefore extremely small. Despite the smallness of the order
, and is therefore extremely small. Despite the smallness of the order
 and less [7] [10] , surface effects play a great role in nature and technology. The smallness of an interface quantity shows, on the other hand, that ignoring it when studying volume properties of the fluid is completely justified. As the existence of a surface A does not change the mass M and volume V, the property of U, S, and F being extensive quantities is maintained.
and less [7] [10] , surface effects play a great role in nature and technology. The smallness of an interface quantity shows, on the other hand, that ignoring it when studying volume properties of the fluid is completely justified. As the existence of a surface A does not change the mass M and volume V, the property of U, S, and F being extensive quantities is maintained.
4. The Thermodynamic Zero of Thermodynamic Functions
According to the Gibbsian energy Equation (7), the mass- and phase-specific internal energies of the saturated

Figure 2. Analysis of free-surface quantities. From fitted published surface tension data
 of argon [8] [9] and setting
of argon [8] [9] and setting
 equal to the area-specific free energy, i.e.
equal to the area-specific free energy, i.e. , one gets the area-specific surface energy
, one gets the area-specific surface energy
 and the area-specific entropy
and the area-specific entropy . It holds that
. It holds that . Each of these functions vanishes at
. Each of these functions vanishes at .
.
fluid are interrelated as follows:
 (15)
(15)
Since
 and
and
 for
for , one has
, one has
 and
and
 for
for
 and
and
 at
at . By virtue of Nernst’s theorem
. By virtue of Nernst’s theorem
at absolute temperature zero it holds that ,
,

 and
and
 The vanishing energy value
The vanishing energy value
 gives the argument for binding the thermodynamic value 0 to the relations
gives the argument for binding the thermodynamic value 0 to the relations . The conditions
. The conditions
 ex- clude, however, the validity of the relations
ex- clude, however, the validity of the relations ,
,
 , and
, and
 for
for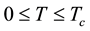 , and admit
, and admit
 only. Thus the unique solution for the internal energies is found:
only. Thus the unique solution for the internal energies is found:
 (16)
(16)
Relations (16) say that the internal vapor energy
 is not negative and the internal fluid energy u is equal or greater than the internal condensed matter energy
is not negative and the internal fluid energy u is equal or greater than the internal condensed matter energy
 and the two are not positive. At the crtical point, each of these energies vanishes.
and the two are not positive. At the crtical point, each of these energies vanishes.
In keeping with W. Gibbs [1] , it is hypothetically asserted in the literature (e.g. [11] -[13] ) that the temperature dependence of u,
 , and
, and
 is determined only up to an arbitrary constant a, i.e.
is determined only up to an arbitrary constant a, i.e.
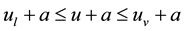 and
and . On the contrary, because of Nernst’s theorem the relations (16) state that
. On the contrary, because of Nernst’s theorem the relations (16) state that
 and thus confirm the universality of the Gibbsian entropy and energy expressions (6) and (7), which are given in thermodynamic terms without any shifts.
and thus confirm the universality of the Gibbsian entropy and energy expressions (6) and (7), which are given in thermodynamic terms without any shifts.
Moreover, some thermodynamic relations are mentioned in relation to the thermodynamic value 0:
 (17)
(17)
The chemical potential functions are given in explicit form as energy functions:
 (18)
(18)
Setting
 relations (9) yield
relations (9) yield
 (19)
(19)
The critical value is finite for
 and divergent for
and divergent for .
.
5. The Unsolved Problem in Applied Thermodynamics
Endeavors to publish data of the energy and entropy functions
 and
and
 are prominent in the current literature. Since the numerical solution
are prominent in the current literature. Since the numerical solution
 and the consequence, viz.
and the consequence, viz. , are known in the literature [3] , it is obvious from relations (15) that the task of finding an explicit thermodynamic expression for
, are known in the literature [3] , it is obvious from relations (15) that the task of finding an explicit thermodynamic expression for
 for
for
 should be tackled. Calcula- tion of
should be tackled. Calcula- tion of
 now occupies the center of interest in applied thermodynamics.
now occupies the center of interest in applied thermodynamics.
Solution of the problem is not trivial, as the following solution ansatzes for the volume function
 will show. The specified lower limit
will show. The specified lower limit
 does not constitute a solution for
does not constitute a solution for
Temperatures T < Tc, because this ansatz leads to a value of the condensate at absolute zero of , whereas
, whereas
 is the correct result there. The upper limit
is the correct result there. The upper limit
 is no solution of
is no solution of
 either, because in this case the function
either, because in this case the function
 would vanish identically.
would vanish identically.
In order to find a solution for , the obvious course is to consider the equilibrium relation that follows from relations (15):
, the obvious course is to consider the equilibrium relation that follows from relations (15):
 (20)
(20)
As the equations show, what is needed is a thermodynamic expression for . For temperatures
. For temperatures , the limits of the energies
, the limits of the energies
 in relation to the evaporation energy
in relation to the evaporation energy
 are known [3] :
are known [3] :
 (21)
(21)
Van der Waals showed that the volumes
 can be represented as functions of
can be represented as functions of . (Here
. (Here
 is expanded as a power function of the temperature distance from the critical point, where the temperature distance between
is expanded as a power function of the temperature distance from the critical point, where the temperature distance between
 and 0 is given by the expansion parameter
and 0 is given by the expansion parameter
 passing through the values from 0 to 1). Thus the functions
passing through the values from 0 to 1). Thus the functions
 can be analogously represented as functions of
can be analogously represented as functions of :
:
 (22)
(22)
At the critical point one gets
 and
and ,
,
 , and at absolute zero
, and at absolute zero
one gets
 and
and ,
, . The success of the van der Waals representation of
. The success of the van der Waals representation of
 as functions of
as functions of
 consists in giving a relation between
consists in giving a relation between
 and
and . As long as
. As long as
 is regarded as an independent variable, the ansatzes (22) merely state that the functions
is regarded as an independent variable, the ansatzes (22) merely state that the functions
 scale at the critical point as the function
scale at the critical point as the function . If, however, every value
. If, however, every value
 can be assigned a certain temperature value T, the values
can be assigned a certain temperature value T, the values
 are fixed. Therefore,
are fixed. Therefore,
 in Equation (22) is replaced as follows in order to characterize the phase of the energy functions
in Equation (22) is replaced as follows in order to characterize the phase of the energy functions
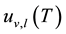 with temperature-dependent phase-specific functions
with temperature-dependent phase-specific functions
 and
and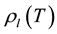 :
:
 (23)
(23)
Taking the difference
 yields the condition
yields the condition
 and calculating
and calculating
 and
and
 gives
gives
 (24)
(24)
Appropriate as variable of the phase functions
 is the volume ratio in the vapor and liquid phases,
is the volume ratio in the vapor and liquid phases,
 , because this numerical ratio at the same time inter-relates the effects of the interaction forces between the fluid particles in the respective phases. Just as the subscript v or l suffices to describe the phase of an energy function, a phase function
, because this numerical ratio at the same time inter-relates the effects of the interaction forces between the fluid particles in the respective phases. Just as the subscript v or l suffices to describe the phase of an energy function, a phase function
 is adequately described either by the subscript
is adequately described either by the subscript
 or
or
 again or else just by specifying the variables
again or else just by specifying the variables
 and z. The definition of z and phase functions
and z. The definition of z and phase functions
 and
and
 is thus
is thus
 (25)
(25)
A phase function
 represents a state function in that it contains information on the density and internal energy distribution in the respective phase. This becomes particularly clear when the ratio
represents a state function in that it contains information on the density and internal energy distribution in the respective phase. This becomes particularly clear when the ratio
 is formed,
is formed,
which can also be expressed by the ratio . It holds that
. It holds that
 at absolute zero and
at absolute zero and
that
 at the critical point, whence
at the critical point, whence . Furthermore, it holds that
. Furthermore, it holds that

and , and hence
, and hence ; it thus follows that
; it thus follows that
 or
or , i.e. the function
, i.e. the function
 increases strictly
increases strictly
monotonically as z and, at the same time, the function
 decreases strictly monotonically. The domain of
decreases strictly monotonically. The domain of
the function
 is
is , because the value
, because the value
 tends to zero when the value of z grows beyond all limits.
tends to zero when the value of z grows beyond all limits.
What is now needed is a solution of the functional equation for
 with subsidiary conditions:
with subsidiary conditions:
 (26)
(26)
The general solution is of the form
 (27)
(27)
Proof: Suppose
 being a real (composed) function,
being a real (composed) function,
 for
for . One has
. One has
 with
with
and .
.
Of the mathematical solutions possible the following (with
 at the critical point
at the critical point ) is selected:
) is selected:
 (28)
(28)
This equation yields the physically relevant solution. It is noted that the solution ρ according to Equation (28) can be represented as a convergent Taylor series. Figure 3 shows the functions
 and
and .
.
Before tackling the important investigation of the uniqueness of this solution, one should consider the method of solution that uses the variable . Equations (23) and (24) yield
. Equations (23) and (24) yield
 (29)
(29)
Admittedly, the latter equation does not yield a direct solution , but it does give the interesting dependence
, but it does give the interesting dependence
 in the form of
in the form of
 with the solutions
with the solutions
 and
and . The equality of
. The equality of
 and
and
 only exists at the critical point and marks the start of the single-fluid phase.
only exists at the critical point and marks the start of the single-fluid phase.
With
 and
and
 the variable
the variable
 has the following dependence on z:
has the following dependence on z:

Figure 3. ,
,
 , and
, and
 as functions of z for
as functions of z for .
.
 (30)
(30)
and one calculates
 for
for
 and
and
 for
for .
.
According to Equations (20) and (22) one obtains
 (31)
(31)
Inserting the solution for
 in Equation (31) gives the symmetric form:
in Equation (31) gives the symmetric form:
 (32)
(32)
Let us now turn to the uniqueness of the solution (28). It is immediately seen that the functions
 and
and
 with
with
 may also be regarded as solutions of Equation (27). Any discussion of values
may also be regarded as solutions of Equation (27). Any discussion of values
 leads, however, to contradictions in the physical behavior of
leads, however, to contradictions in the physical behavior of .
.
It is claimed that every solution
 is expandable into a convergent Taylor series for all
is expandable into a convergent Taylor series for all ; this condition is probably contained in the theory of Yang and Lee [14] stating that the equation of state of a one-phase system or a system with possible phase transition is represented by an analytic function of a complex argument Z for all Z in the corresponding region, which contains a segment of the real positive axis. As stated above, solution (28) can be expanded into a convergent Taylor series for all
; this condition is probably contained in the theory of Yang and Lee [14] stating that the equation of state of a one-phase system or a system with possible phase transition is represented by an analytic function of a complex argument Z for all Z in the corresponding region, which contains a segment of the real positive axis. As stated above, solution (28) can be expanded into a convergent Taylor series for all
 and meets, for the case of a complex argument Z instead of z, the condition according to Yang and Lee. One now looks for further solutions. Let a solution be assumed in the form
and meets, for the case of a complex argument Z instead of z, the condition according to Yang and Lee. One now looks for further solutions. Let a solution be assumed in the form

It follows that


Expanding
 into a convergent Taylor series in
into a convergent Taylor series in , one now obtains
, one now obtains


If , all the
, all the
 for
for
 must be zero in order to satisfy the condition above,
must be zero in order to satisfy the condition above, .
.
Then
 also holds, because the condition must be satisfied for every value z. Thus
also holds, because the condition must be satisfied for every value z. Thus
 vanishes identically and the solution
vanishes identically and the solution
 is unique.
is unique.
6. Explicit Expression for

The expression proposed [15] [16] for the volume function
 is
is
 (33)
(33)
It is symmetric in the variables and linear in both
 and
and , and at the critical point it yields v. Figure 4
, and at the critical point it yields v. Figure 4
shows the temperature dependence of
 for argon and the boundaries
for argon and the boundaries
 and
and ,
,
which are set by relations (31).

Figure 4. Volume function
 of argon versus the temperature between
of argon versus the temperature between
 and
and . Curve 1 represents Equation (33), curve 2 gives the lower boundary
. Curve 1 represents Equation (33), curve 2 gives the lower boundary , and curve 3 the upper boundary
, and curve 3 the upper boundary .
.
Expression (33) is the only one thermodynamically possible and it alone satisfies all known thermodynamic conditions (see Appendix).
The description of the two-phase state of the saturated fluid by the expression (33) admits further formu- lations of the two-phase equilibrium.
Relations (15) and (33) yield the result of the ambitious task of representing the phase-specific internal en- ergies in terms of phase-specific volumes and vapor pressure, i.e. measurable quantities:
 (34)
(34)
The positive term
 can be written in agreement with Equations (19), (20), and (33) as
can be written in agreement with Equations (19), (20), and (33) as
 (35)
(35)
Rearranging this to
 yields the following very interesting
yields the following very interesting
thermodynamic equations valid for :
:
 (36)
(36)
Equation (36) are valid for temperatures
 to
to
 and can serve as a criterion for calculated data
and can serve as a criterion for calculated data
 of a gas, if the experimental data
of a gas, if the experimental data
 and
and
 are considered to be trustworthy. In turn, from Equa- tion (36) one obtains
are considered to be trustworthy. In turn, from Equa- tion (36) one obtains
 (37)
(37)
which is in agreement with Equation (34). Figure 5 shows some energy functions of saturated argon.
As mentioned by relation (35), the term
 gives the weighted volume value
gives the weighted volume value

Figure 5. Energies of saturated argon: 1 , 2
, 2 , 3 u, 4
, 3 u, 4 , 5
, 5 .
.
 . Let us recall here the mean-value theorem of the differential calculus, which, when applied to the function
. Let us recall here the mean-value theorem of the differential calculus, which, when applied to the function , yields the result
, yields the result
 (38)
(38)
The term
 can thus be replaced by the function
can thus be replaced by the function , where the function
, where the function
 depends only on the volume ratio
depends only on the volume ratio
 or
or . With the definitions
. With the definitions
 (39)
(39)
it holds that
 (40)
(40)
The function
 varies strictly monotonically from the value
varies strictly monotonically from the value
 at
at
 to the asymptotic
to the asymptotic
value 1 for , the increase being greatest with
, the increase being greatest with
 at
at , and
, and
 decreases mono-
decreases mono-
tonically to 0. The physical meaning of
 is discussed in Ref. [16] . There the phase-specific energies
is discussed in Ref. [16] . There the phase-specific energies
 and
and
 are represented as a product function,
are represented as a product function,
 , composed of the one term
, composed of the one term , which denotes the phase, and the term
, which denotes the phase, and the term , which specifies the temperature dependence. The phase-specific term is related to the local interaction potential of fluid particles in the vapor space and in the liquid. With a phase change of fluid particles, the phase-specific energy value, say
, which specifies the temperature dependence. The phase-specific term is related to the local interaction potential of fluid particles in the vapor space and in the liquid. With a phase change of fluid particles, the phase-specific energy value, say , becomes
, becomes
 in a manner that can be des- cribed simply by interchanging the phase indices. This yields the following equations:
in a manner that can be des- cribed simply by interchanging the phase indices. This yields the following equations:
 (41)
(41)
The procedure in Ref. [16] is the exact opposite of that described here: There Equation (41) serves as starting point to derive relation (40) and find their solution (38). The energy ratio
 depends only on the volume ratio
depends only on the volume ratio
 and with the same value z is equal for all gases, i.e. is universal:
and with the same value z is equal for all gases, i.e. is universal:
 (42)
(42)
It is found that
 is equal to
is equal to
 and varies with values between 0 and
and varies with values between 0 and
 as z varies from high values to 1.
as z varies from high values to 1.
The ratios of the phase-specific internal energies to the evaporation energy are likewise universal and for temperatures
 it holds that
it holds that
 (43)
(43)
Finally, the integral
 with the heat-capacity function [17] (see Figure 6),
with the heat-capacity function [17] (see Figure 6),
 (44)
(44)
yields, of course, the temperature value of the fluid internal energy,
 (45)
(45)
The entropy value
 is defined by the integral
is defined by the integral
 because of
because of . Since
. Since
 one gets
one gets . From this and from Equations (1) and (4) it
. From this and from Equations (1) and (4) it
follows that
 (46)
(46)
According to Equations (10), (34), and (45), one has . In gen- eral, it holds that [3]
. In gen- eral, it holds that [3]

Figure 6. Heat capacity
 of saturated argon ac- cording to Equation (44) for
of saturated argon ac- cording to Equation (44) for .
.
 (47)
(47)

Thermodynamic equations for
 of the saturated fluid are listed in Table 1.
of the saturated fluid are listed in Table 1.
7. Results and Discussion
In treating the thermodynamic equilibrium of a fluid mass M in a volume V, a distinction is made between an equilibrium state with a free surface area
 among the phase volumes, vapor and condensate, and a state without a free surface. In the case
among the phase volumes, vapor and condensate, and a state without a free surface. In the case , according to Gibbs the internal energy U and entropy S of the fluid are functions with minimum equilibrium value
, according to Gibbs the internal energy U and entropy S of the fluid are functions with minimum equilibrium value
 and maximum equilibrium value
and maximum equilibrium value . As the quantities V, U, and S are proportional to the mass M, they are absolute quantities with the known thermodynamic zeros
. As the quantities V, U, and S are proportional to the mass M, they are absolute quantities with the known thermodynamic zeros
 at the critical point and
at the critical point and
 at the absolute temperature zero. Equations (16) to (19) and (59) afford examples of absolute order of relations between different thermodynamic functions. Extensive quantities have the additive property
at the absolute temperature zero. Equations (16) to (19) and (59) afford examples of absolute order of relations between different thermodynamic functions. Extensive quantities have the additive property , where
, where . The intensive
. The intensive

Table 1. Chemical potential relations [3] .
quantities of the fluid are the temperature , pressure
, pressure , and the chemical
, and the chemical
potential , which are related to one another because of
, which are related to one another because of
 and
and . The thermodynamic equilibrium is described by the so-called fundamental equation
. The thermodynamic equilibrium is described by the so-called fundamental equation .
.
When a free surface
 first transpires among the phase volumes, vapor and condensate, one has the possibility by varying T and M or V, of experimentally determing the critical point of a fluid and measuring the critical values
first transpires among the phase volumes, vapor and condensate, one has the possibility by varying T and M or V, of experimentally determing the critical point of a fluid and measuring the critical values ,
,
 , and
, and . The presence of a free surface area shows at the same time that between the volumes
. The presence of a free surface area shows at the same time that between the volumes
 and
and , in which fluid particles are homogeneously distributed, there exists an interface phase with inhomogeneous particle distribution. Unlike in volume phases, fluid particles in the interface phase are not exposed to isotropically effective interaction potentials and therefore interface particles are endowed with a surface energy
, in which fluid particles are homogeneously distributed, there exists an interface phase with inhomogeneous particle distribution. Unlike in volume phases, fluid particles in the interface phase are not exposed to isotropically effective interaction potentials and therefore interface particles are endowed with a surface energy
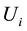 and entropy
and entropy . When a free surface is increasing, the redis- tribution of the fluid particles in V proceeds in the direction of decreasing U and increasing S; in other words, in the direction of decreasing F by virtue of decreasing interface free energy
. When a free surface is increasing, the redis- tribution of the fluid particles in V proceeds in the direction of decreasing U and increasing S; in other words, in the direction of decreasing F by virtue of decreasing interface free energy . Surprisingly, its value
. Surprisingly, its value
 can be macroscopically determined. With
can be macroscopically determined. With
 as surface tension, the effect of the interaction potential of all interface particles is measurably available. The Gibbs’ Equations (6) and (7) are enlarged by the interface terms and are given by Equation (13). The interface terms are evaluated in Figure 2 for fluid argon. The ratio of free interface energy
as surface tension, the effect of the interaction potential of all interface particles is measurably available. The Gibbs’ Equations (6) and (7) are enlarged by the interface terms and are given by Equation (13). The interface terms are evaluated in Figure 2 for fluid argon. The ratio of free interface energy
 and free volume energy
and free volume energy
 yields with the assumptions
yields with the assumptions ,
,
 and averaged values of
and averaged values of
 (see Figure 2 and Figure 8) the approximate number
(see Figure 2 and Figure 8) the approximate number
 . This estimate explains why the free energy at the free surface A in
. This estimate explains why the free energy at the free surface A in
relation to its value in V can be completely ignored if volumetric considerations are to the fore, as in this work.
Here it is investigated whether there is a definitely specifiable functional relation between phase-specific energies
 and the phase-invariant energy term
and the phase-invariant energy term , which is given by Gibbs in the energy Equation (15) for the saturated fluid. The solution
, which is given by Gibbs in the energy Equation (15) for the saturated fluid. The solution
 , Equation (33), has been known in the literature [15] [16] for many years but has remained neglected, presumably because proof of its uniqueness was lacking. This is made up for here in Section 5 by showing that the dimensionless phase-specific temperature functions
, Equation (33), has been known in the literature [15] [16] for many years but has remained neglected, presumably because proof of its uniqueness was lacking. This is made up for here in Section 5 by showing that the dimensionless phase-specific temperature functions
 and
and
 in the ansatzes for the internal energies
in the ansatzes for the internal energies
 and
and
 have the physically relevant and unique solutions
have the physically relevant and unique solutions
 and
and
 (see Equations (24)-(28) and Figure 3). Numerical evaluation of
(see Equations (24)-(28) and Figure 3). Numerical evaluation of
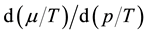 is presented in Figure 4. The presentation of
is presented in Figure 4. The presentation of
 in terms of
in terms of
 and p (see Equation (34)) allows evaluating internal energy functions as shown in Figure 5 for fluid argon. As is known, with the solutions described and proved here one can represent and calculate all thermodynamic volumetric functions as functions of
and p (see Equation (34)) allows evaluating internal energy functions as shown in Figure 5 for fluid argon. As is known, with the solutions described and proved here one can represent and calculate all thermodynamic volumetric functions as functions of
 and
and . In Table 1, for example, expressions for
. In Table 1, for example, expressions for
 are listed, and in the Appendix thermodynamically derived relations are investigated for their validity by means of
are listed, and in the Appendix thermodynamically derived relations are investigated for their validity by means of
 and verified.
and verified.
Acknowledgements
The author would like to thank Max Planck Institute of Plasma Physics (IPP), Garching, for providing computing facilities. He is also grateful to A. Kechriniotis and H. Tasso for helpful discussions concerning the unique solution of the phase function
 introduced here to describe the fluid state, and to A. M. Nicol for the English translation.
introduced here to describe the fluid state, and to A. M. Nicol for the English translation.
Cite this paper
AlbrechtElsner, (2015) Thermodynamic Equilibrium of the Saturated Fluid with a Free Surface Area and the Internal Energy as a Function of the Phase-Specific Volumes and Vapor Pressure. Engineering,07,577-596. doi: 10.4236/eng.2015.79053
References
- 1. Gibbs, J.W. (1973) A Method of Geometrical Representation of the Thermodynamic Properties of Substances by Means of Surfaces. Transactions of the Connecticut Academy, 2, 382-404.
- 2. Callen, H.B. (1960) Thermodynamics. John Wiley & Sons, New York.
- 3. Elsner, A. (2012) Applied Thermodynamics of the Real Gas with Respect to the Thermodynamic Zeros of the Entropy and Internal Energy. Physica B: Condensed Matter, 407, 1055-1067.
http://dx.doi.org/10.1016/j.physb.2011.12.118 - 4. Tegeler, C., Span, R. and Wagner, W. (1999) Thermodynamic Properties of Saturated Argon. Journal of Chemical and Physical Reference Data, 28, 779-850.
- 5. Landau, L.D., Lifshitz, E.M. and Pitaevskii, L.P. (1980) Statistical Physics: Course of Theoretical Physics. Volume 5, Pergamon Press, Oxford.
- 6. Elsner, A. (2014) Thermodynamic Fit Functions of the Two-Phase Fluid and Critical Exponents. Engineering, 6, 789- 826.
http://dx.doi.org/10.4236/eng.2014.612076 - 7. Stierstadt, K. (1989) Physik der Materie. VCH Verlagsgesellschaft, Weinheim, 235.
- 8. Stansfield, D. (1958) The Surface Tensions of Liquid Argon and Nitrogen. Proceedings of the Physical Society, 72, 854-866.
http://dx.doi.org/10.1088/0370-1328/72/5/321 - 9. Sprow, F.B. and Prausnitz, J.M. (1966) Surface Tensions of Simple Liquids. Transactions of the Faraday Society, 62, 1097-1104.
http://dx.doi.org/10.1039/tf9666201097 - 10. Dörfler, H.D. (1994) Chapter 2: Grenzflächen und Kolloidchemie. VCH Verlagsgesellschaft, Weinheim.
- 11. Hahne, E. (1993) Technische Thermodynamik. Addison-Wesley Publishing Company, Boston, 66.
- 12. Hudson, J.B. (1996) Thermodynamics of Materials. John Wiley & Sons, New York, 22, 145, 185.
- 13. Lauth, J.G. and Kowalczyk, J. (2015) Thermodynamik. Springer Verlag, Berlin, 67, 68, 115, 135, 138-142, 199-211, 365.
http://dx.doi.org/10.1007/978-3-662-46229-4 - 14. Huang, K. (1987) Statistical Mechanics. John Wiley & Sons, New York.
- 15. Elsner, A. (1987) Temperature Dependence of Absolute Fluid-State Parameters. Cryogenics, 27, 189-201.
http://dx.doi.org/10.1016/0011-2275(87)90018-X - 16. Elsner, A. (1988) Thermodynamic Representation of the State of a Saturated Fluid. Physics Letters A, 130, 225-233.
http://dx.doi.org/10.1016/0375-9601(88)90598-1 - 17. Elsner, A. (1993) Specific Heats of Saturated Water Vapor and Liquid. Physical Review E, 48, 1839-1843.
http://dx.doi.org/10.1103/PhysRevE.48.1839
Appendix: Test Functions for the Volume Function

The correctness of Equation (33) is now demonstrated in a few test cases. With , the volume function (33) can be written as
, the volume function (33) can be written as . Since
. Since , one has
, one has
 (48)
(48)
From
 one obtains the relations
one obtains the relations
 (49)
(49)
which shows the symmetry of the volume function in respect of its variables
 and
and .
.
At this point it is appropriate to comment on the argument of the logarithm in expression (33). As can be seen, the relations of the last line can be transformed to the relations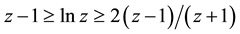 , which are known to describe the asymptotic behavior of
, which are known to describe the asymptotic behavior of
 in the vicinity of
in the vicinity of . This shows that the argument of the logarithm must be exactly the volume ratio
. This shows that the argument of the logarithm must be exactly the volume ratio
 and cannot be, for example, the entropy ratio
and cannot be, for example, the entropy ratio . An entropy ratio
. An entropy ratio
 as argument of the logarithm would, admittedly, yield the required symmetry property
as argument of the logarithm would, admittedly, yield the required symmetry property , but relations
, but relations
 could not be numerically satisfied, since one has
could not be numerically satisfied, since one has
 for
for .
.
A particularly critical test for the correctness of Equation (33) is afforded by the internal relations (16) and (34). As it holds that
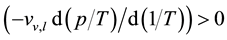 for
for , it is only the terms
, it is only the terms
 (50)
(50)
that have to be investigated. It is in fact found that
 for
for
 and
and
 for
for
 since
since


According to relation (20) it is postulated that the sum
 is not be positive. This can be confirmed since
is not be positive. This can be confirmed since
 (51)
(51)
It should be emphasized that the energy sum , which according to Equation (34) can be written as
, which according to Equation (34) can be written as
 and hence evaluated as a function of the measurable quantities
and hence evaluated as a function of the measurable quantities
 and p for every gas, is a negative function of the temperature, which increases strictly monotonically as the temperature and is convex in the very immediate vicinity of the critical point (see Figure 5).
and p for every gas, is a negative function of the temperature, which increases strictly monotonically as the temperature and is convex in the very immediate vicinity of the critical point (see Figure 5).
The fluid energy
 is not positive and given by
is not positive and given by
 (52)
(52)
It is negative and greater than
 for
for
 since
since
 and
and

The difference between the energy sum
 and
and
 must not be negative according to Equations (16) and (52). The energy Equation (15) give
must not be negative according to Equations (16) and (52). The energy Equation (15) give
 (53)
(53)
This difference is not negative if
 (54)
(54)
see also Figure 7. Thermodynamically correct density data confirm the validity of condition (54). For example, the formulae for
 and
and
 in Ref. [4] allows one to prove the correctness of condition (54) up to the critical point. On the other hand, the relations
in Ref. [4] allows one to prove the correctness of condition (54) up to the critical point. On the other hand, the relations
 and
and
 with
with
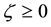 are valid in the critical
are valid in the critical
region. They lead to
 and
and
 . The last expression is greater than
. The last expression is greater than ,
,
which means that condition (54) is satisfied and that the expression
 instead of
instead of
 confirms the thermodynamically derived result
confirms the thermodynamically derived result .
.
The next example investigated is the ratio
 given by Equation (42). It is found that
given by Equation (42). It is found that
 is equal to
is equal to
 (see Figure 3) with values between 0 and -1:
(see Figure 3) with values between 0 and -1:

Finally, the ratio of the phase-specific internal energies to the evaporation energy is considered, for which the theory yields according to relation (21) . This ratio is likewise universal and the result can be confirmed with the solution (34) since
. This ratio is likewise universal and the result can be confirmed with the solution (34) since
 (55)
(55)
Chemical potential relations: with the identity

as starting point and by means of the expression for the volume function
 according to
according to

Figure 7. Functions
 and
and
 of argon for temperatures between the triple point and critical point (logarithmic representation).
of argon for temperatures between the triple point and critical point (logarithmic representation).
Equation (33),
 and the two temperature derivatives
and the two temperature derivatives
 and
and
 are calculated as functions of p and
are calculated as functions of p and :
:
 (56)
(56)
 (57)
(57)
 (58)
(58)
and (see Figure 8)
 (59)
(59)

Figure 8. Functions ,
,
 ,
,
 , and
, and
 of argon for temperatures between the triple point and critical point.
of argon for temperatures between the triple point and critical point.