Materials Sciences and Applications
Vol.3 No.2(2012), Article ID:17250,4 pages DOI:10.4236/msa.2012.32012
Chemical Analyses of Palm Kernel Oil-Based Polyurethane Prepolymer
![]()
1School of Chemical Sciences and Food Technology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Bangi, Malaysia; 2Polymer Research Center, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, Bangi, Malaysia.
Email: *kaybadri@ukm.my, *kaybadri@gmail.com
Received August 4th, 2011; revised November 17th, 2011; accepted December 29th, 2011
Keywords: Palm Kernel Oil; Prepolymerization; Polyurethane; Soft Segment; Hard Segment
ABSTRACT
Polyurethane (PU) was prepared from palm kernel oil-based monoester polyol (PKO-p) via prepolymerization method at NCO/OH ratio of 200/100, 150/100, 100/100, and 75/100 at ambient temperature under nitrogen gas atmosphere. The structure of the synthesized prepolymerized PKO-p PU was determined using FTIR and 13C NMR. The disapperance of NCO peak in the FTIR spectrum at 2270 cm–1 - 2250 cm–1 cm showed that MDI has completely reacted to form PU. The appearance of C=O peak at 1700 cm–1 indicated that hydrogen bonding was formed between the soft segmented chain of the PKO-p and the hard segmented MDI. Hence, urethane bond was the main polymeric chain in the PU.
1. Introduction
Polyurethane (PU) can be produced by different methods such as prepolymerization, single-step polymerization and quasiprepolymer. Single-step polymerization takes place when polyol, diisocyanate and catalyst are mixed together and the chain extension takes place in one step. As a result of the heat liberated, this method is best suited to make thin wall products [1]. Quasiprepolymer method is the reaction between polyol reacted with an excess of diisocyanate. Hence, the urethane prepolymer contains higher free isocyanate content (15% - 30%) and is called isocyanate quasiprepolymers or semiprepolymer because the diisocyanate molecules are partially reacted with polyol [2]. Preparation of polyurethane by prepolymerization on the other hand, involves formation of urethane polymer at the initial stage of reaction between polyol and diisocyanate. Then, the urethane prepolymer is chain-extended with diol or water to form polyurethane. To form urethane prepolymer, one of the isocyanate groups (NCO) will react with a hydroxyl group (OH) of the polyol and the other isocyanate group reacts with the second OH group. The resultant urethane prepolymer has two isocyanate groups on both ends of urethane prepolymer with urethane bonds. Prepolymers are normally produced with a mole ratio of 2 moles of diisocyanate to 1 mole of polyol. If the mole ratio of diisocyanate to polyol is higher (i.e. 3:1), the resultant product is called a quasiprepolymer. As the reaction proceeds, the chain length of urethane prepolymer will increase as the hydroxyl groups react with the terminal NCO end groups of the urethane prepolymers. Further chain extension of the chain can be carried out by reaction of the hydroxyl group of chain extender with either diisocyanate molecule or an isocyanate end groups from another urethane prepolymer chain.
In this research, monoester polyol was produced from palm kernel oil (PKO) as described in the studies carried out by Badri et al. [3]. Polyurethane produced in this research undergoes prepolymerization method which involves the use of specially designed prepolymers, namely structoset prepolymers. Structoset prepolymers are usually polymerized in the second step by the addition of a catalyst or other reactants. This is an advantage because it offers greater control of polymerization and crosslinking reaction. Generally, there are 2 categories in structoset prepolymers which are structoterminal prepolymers and structopendant prepolymers [4]. A structoterminal prepolymer has functional groups located at the ends of the prepolymer chains while a structopendant prepolymer has functional groups located along the prepolymer chain as shown in Figure 1.
Several reasons to make PU through prepolymerization method as opposed to the single step polymerization include lower isocyanate vapour levels and reduction of exotherm of the final reaction. When diisocyanate reacts with polyol, the molecular weight of urethane prepolymer increases with time, resulting in low vapor pressure.
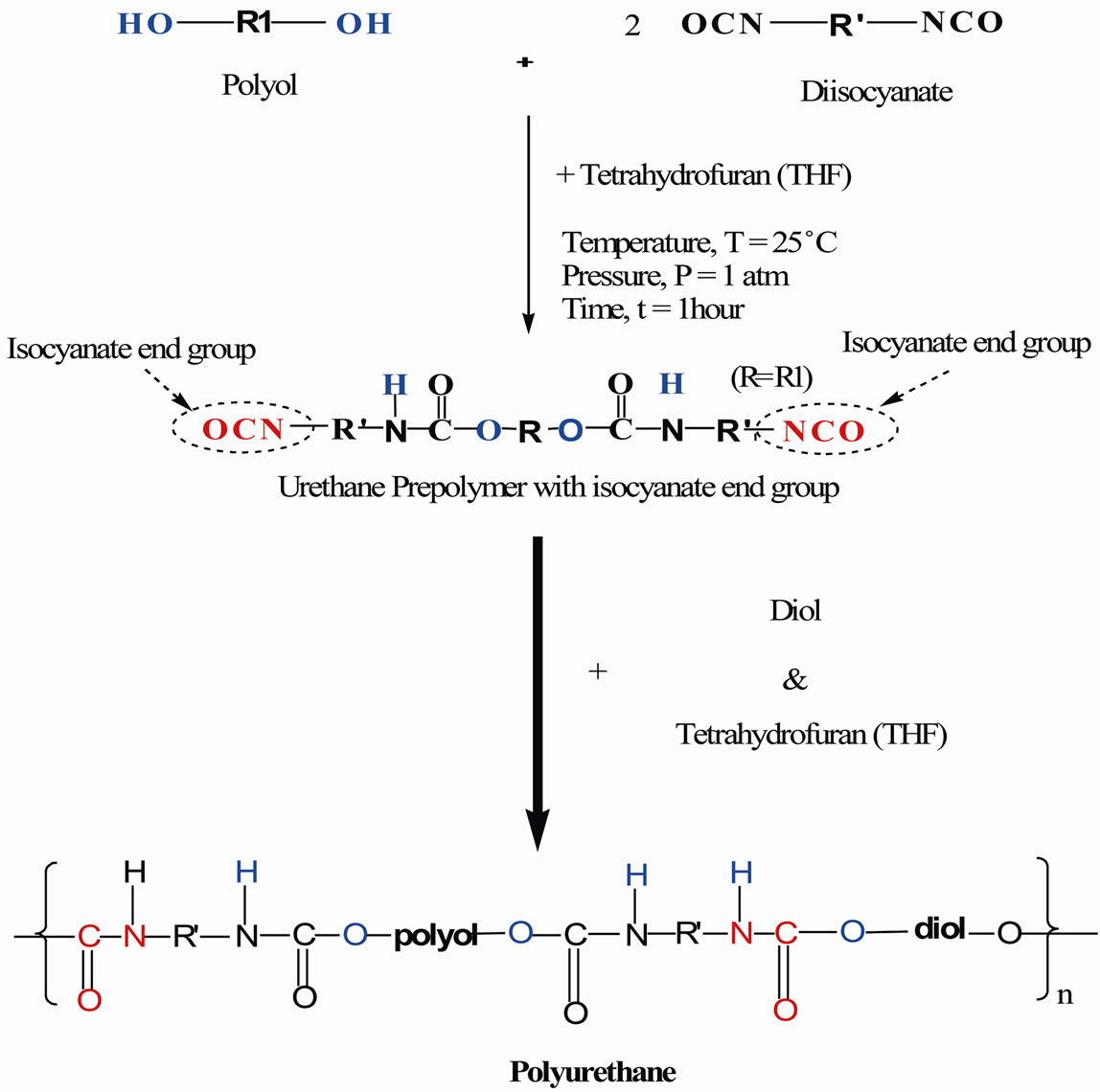
Figure 1. The chemical route of producing polyurethane via prepolymerization method.
A single step polymerization of polyurethane gives off heat when urethane bonds are formed and causes shrinkage problem. However, prepolymerization allows partial dissipation of the exotherm prior to the formation of PU. The significant of this research is to synthesize PU without heating as researched by others [5,6].
2. Experimental Procedure
2.1. Materials
The palm kernel oil-based monoester polyol (PKO-p) was prepared as described elsewhere by Badri et al. [3]. 2,4- diphenylmethane diisocyanate (MDI) was obtained from Cosmopolyurethane (M) Sdn. Bhd., Klang, Malaysia. Tetrahydrofuran (THF) was supplied by Merck Sdn Bhd, Shah Alam, Malaysia. Diethylene glycol (DEG) was purchased from Fluka Chemie Sdn. Bhd.
2.2. Preparations of the Prepolymerised Polyurethane (PU)
The PKO-p was initially dissolved in THF before mixing with the MDI in a round-bottom flask under nitrogen gas atmosphere to form urethane prepolymer. DEG which acts as a chain extender was initially added at varying amount to the PKO-p as shown in Table 1. The mixture was agitated at 200 rpm for an hour at room temperature. It was then casted to a translucent film of about 50 microns thickness onto a Teflon plate. The film was dried in a vacuum oven at 55˚C for 24 hours to remove the solvent. All prepared samples were yellow, translucent and voidfree films.
Four types of samples labelled as PU1, PU2, PU3 and PU4 were prepared with NCO/OH ratios of 200/100, 150/100, 100/100 and 75/100 respectively. Observation was made onto the samples before and after the curing process. The FTIR spectroscopy and nuclear magnetic resonance spectroscopy analyses were carried out to determine the chemical changes occurred during the prepolymerization.
2.3. Measurements
The detection of important functional groups in the Fourier Transform Infrared (FTIR) spectrum of the PU was carried out using Perkin Elmer Spektrum BX spectrometer via Diamond Attenuation Total Reflectance (DATR) method. The presence of urethane carbonyl (C=O) group, carbamate (-CN) and amide (-NH) was identified.
The nuclear magnetic resonance spectroscopy 13C NMR spectra of PKO-p based polyurethane were recorded on a ECP 400 MHz spectrometer in dimethyl sulfoxide-d6 (DMSO-d6) with tetramethylsilane (Me4Si, TMS) as an internal standard at room temperature.
3. Results and Discussion
3.1. Physical Observations
PU1, PU2, PU3 and PU4 were physically checked for curing before and after oven-dried and the observations were tabulated in Table 2. All samples were sticky before the curing process. However, after the oven-drying process, PU1 and PU2 resulted in rigid and brittle films while PU3 and PU4 showed semi-rigid and flexible properties, respectively.
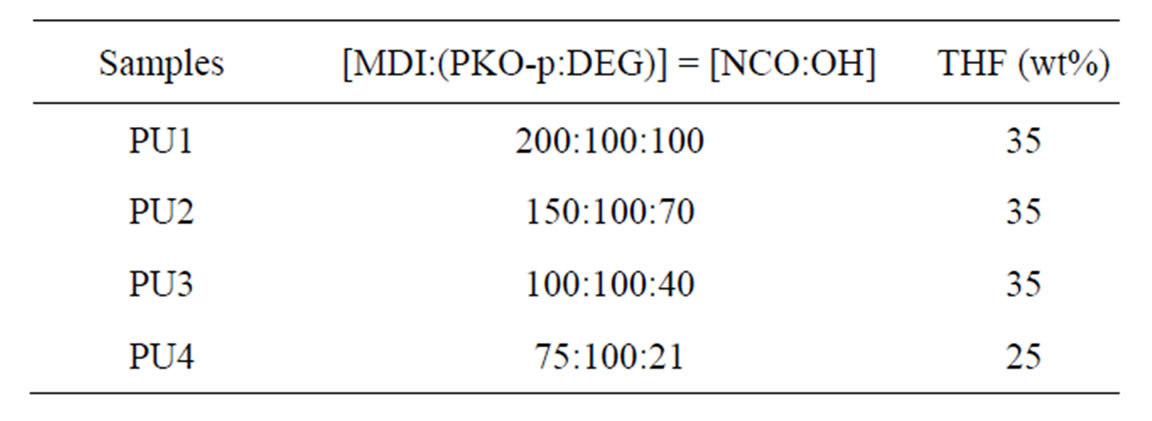
Table 1. The composition of the MDI to the PKO-p and DEG with THF as the solvent.
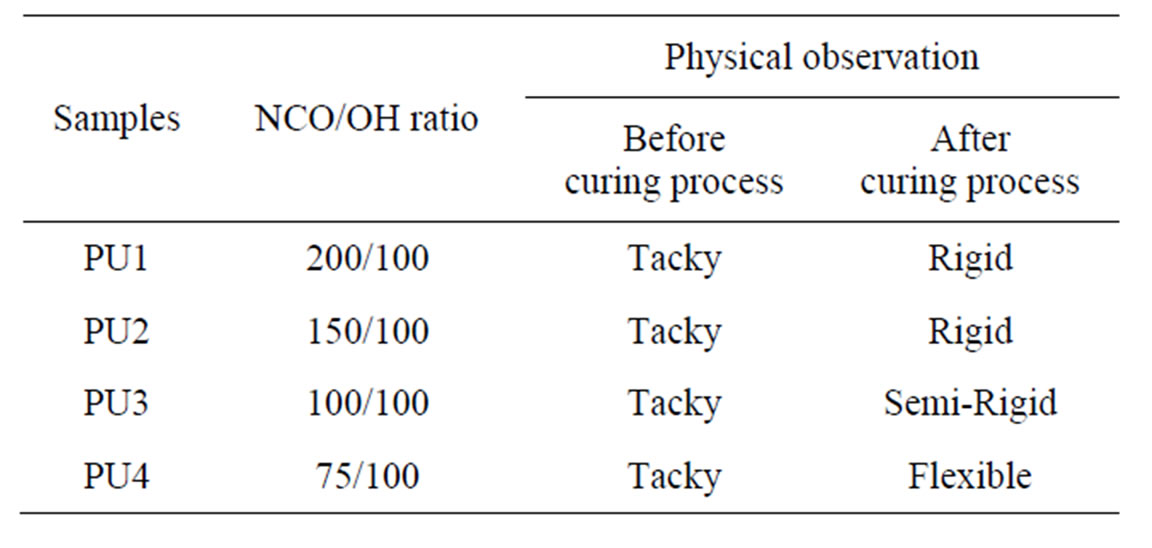
Table 2. Physical states of the PU at different NCO/OH ratios.
3.2. FTIR Spectroscopy Analysis
Figure 2 showed the FTIR spectra of standard MDI, isocyanate terminated urethane prepolymer for PU1 (as comparison) and standard PU. The NCO peak of MDI and urethane prepolymer is around 2270 cm–1 - 2250 cm–1. The intensity of the NCO peak of MDI is higher than the urethane prepolymer. The PKO-p reacted with diisocyanate to form urethane prepolymer in order to remove the free diisocyanate of MDI. However, there are still traces of PKO-p in the urethane polymer that are unreacted indicated by the C=O (ester) and the C-N (esteramide) at 1732 cm–1and 1611 cm–1 respectively. On the other hand, there is no traces of NCO peak detected in the FTIR spectrum of the PU formed via addition polymerization. This indicates that the diisocyanate has completely reacted with PKO-p to form polyurethane.
The FTIR spectrum of the synthesized PU prepolymers-PU1 to PU4 (Figure 3) is then analysed to determine degree of polymerization. The presence of the amide (-NH), carbonyl urethane group (-C=O), carbamate group (CN-H) and -C-O-C indicated the urethane linkages in the PU. The carbonyl peak (C=O) were detected around 1700 cm–1 which is a hydrogen bonded carbonyl urethane group. Clemitson (2008) identified peak at 1730 cm–1 as a non-hydrogen bonded carbonyl urethane group. There is less possibility of having urea as the side product since peak of C=O urea is absence at 1690 cm–1 - 1660 cm–1 [1]. The free NH band, hydrogenbonded NH with oxygen (ether) and hydrogen-bonded NH with oxygen (carbonyl) in the urethane are observed at 3450 cm–1, 3290 cm–1- 3310 cm–1 and 3300 cm–1 - 3350 cm–1 [7].
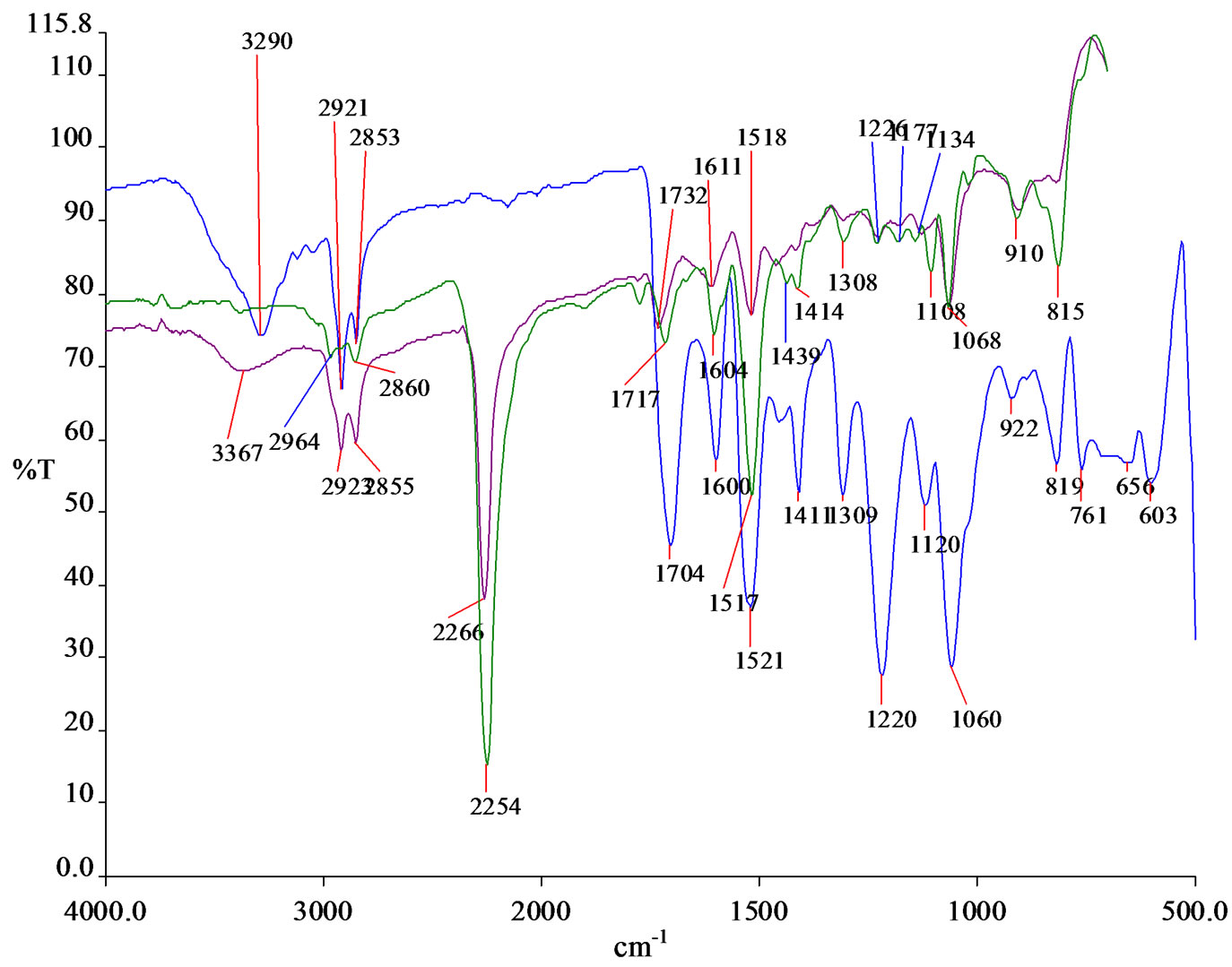
Figure 2. The FTIR spectrum of MDI, urethane prepolymer (PU1 as reference) and PU.
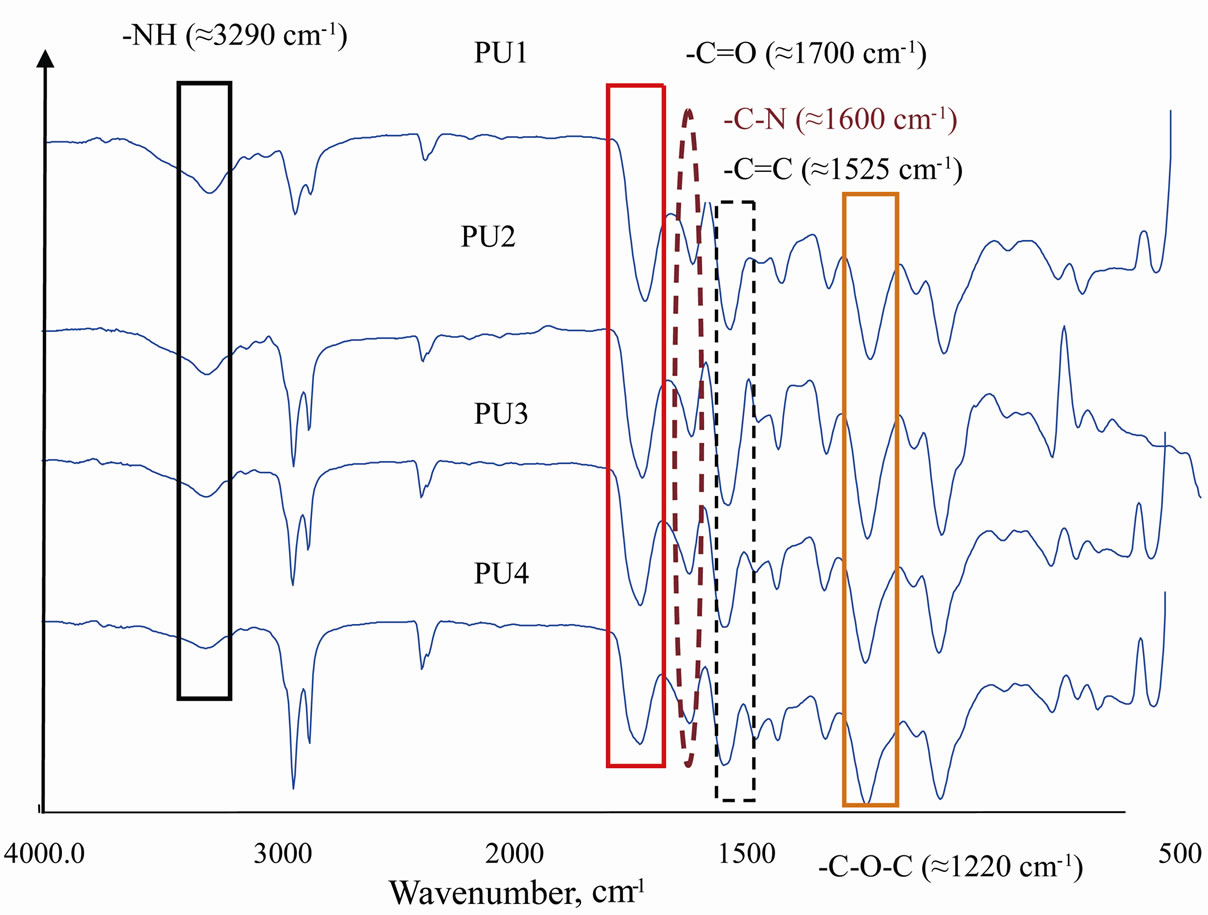
Figure 3. FTIR spectrum of PU at various NCO/OH ratios.
This can be further illustrated by the mechanism shown in Figure 4. In the hard segment, the urethane groups have electrostatic charges at hydrogen, oxygen and nitrogen atoms and these charged atoms form dipoles which attract another atom of opposite charge [8]. In this research, the hydrogen bond formed by NH acted as proton donor while C=O acts as proton acceptor. C=O accepts proton from NH from urethane group. So hydrogen bond is formed.
When the NCO/OH ratio decreases from PU1 to PU4, the carbonyl urethane group -C=O peak shifts to the left. Excess of PKO-P reduced the tendency of forming the urethane linkage. With lower isocyanate content in the PU system, lesser urethane bond is formed resulting in weak interaction between the PU chain. The formation of the imide group is connected with the disappearance of the characteristic absorption band of the isocyanate group. Moreover an FTIR absorption maximum showed up at 1778 cm–1 and 1718 cm–1 due to the imide-carbonyl, at 1373 cm–1 due to C-N stretching and at 720 cm–1 due to imide ring deformation. Furthermore, FTIR spectra of modified polyimides do not show any peak near 2250 cm–1 which indicates the absence of isocyanate groups. Moreover an FTIR absorption maximum showed up at 1778 cm–1 and 1718 cm–1 due to the imide-carbonyl, at 1373 cm–1 due to C-N stretching and at 720 cm–1 due to imide ring deformation.
The reaction mechanism in the formation of the urethane linkage through prepolymerization method is by nucleophilic substitution reactions [9]. Figures 5(a) and (b) are the nucleophilic substitution reaction with amine as the nucleophile. Amine attacks the carbonyl of the isocyanate (MDI) to form two resonance structures of the intermediate complexes A and B. The intermediate complex B has a greater tendency to react with PKO-p due toits carbonyl bond (C=O) which is stronger than the C=N bond in the intermediate complex A. Hence, formation of intermediate complex B is more stable than intermediate complex A. Furthermore, nitrogen (N) is more electropositive than oxygen, that -CN bond is attracted more to the cation (H+) compared to -CO [10]. PKO-p then reacts with intermediate complex B to form the urethane linkage. Figure 6 is another nucleophilic substitution reaction but takes place with DEG. The oxygen of DEG nucleophile attacks diisocyanate of MDI to form two intermediate complexes A and B. Again, the intermediate complex B then reacts with PKO-p to form the urethane linkage.
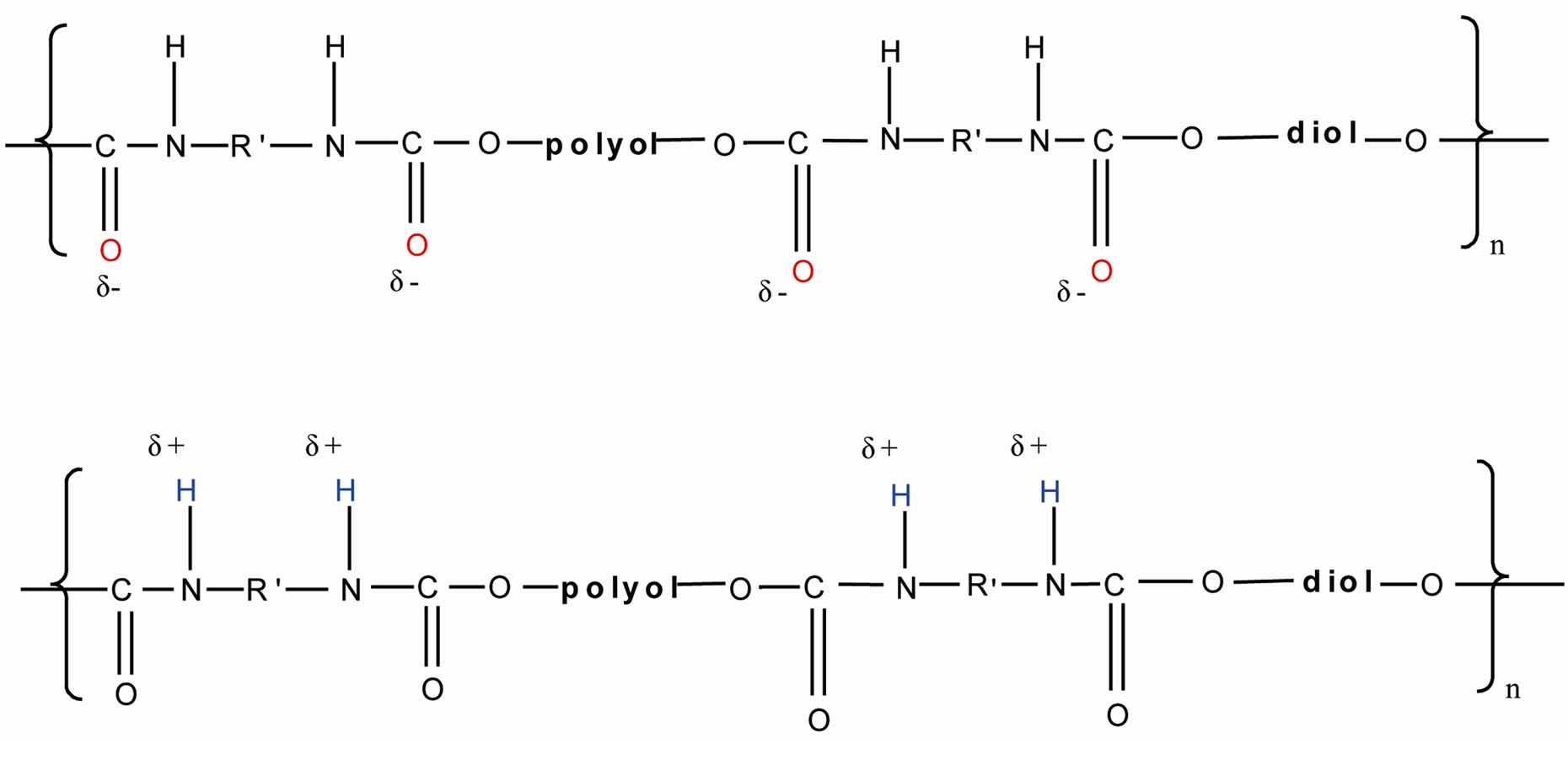
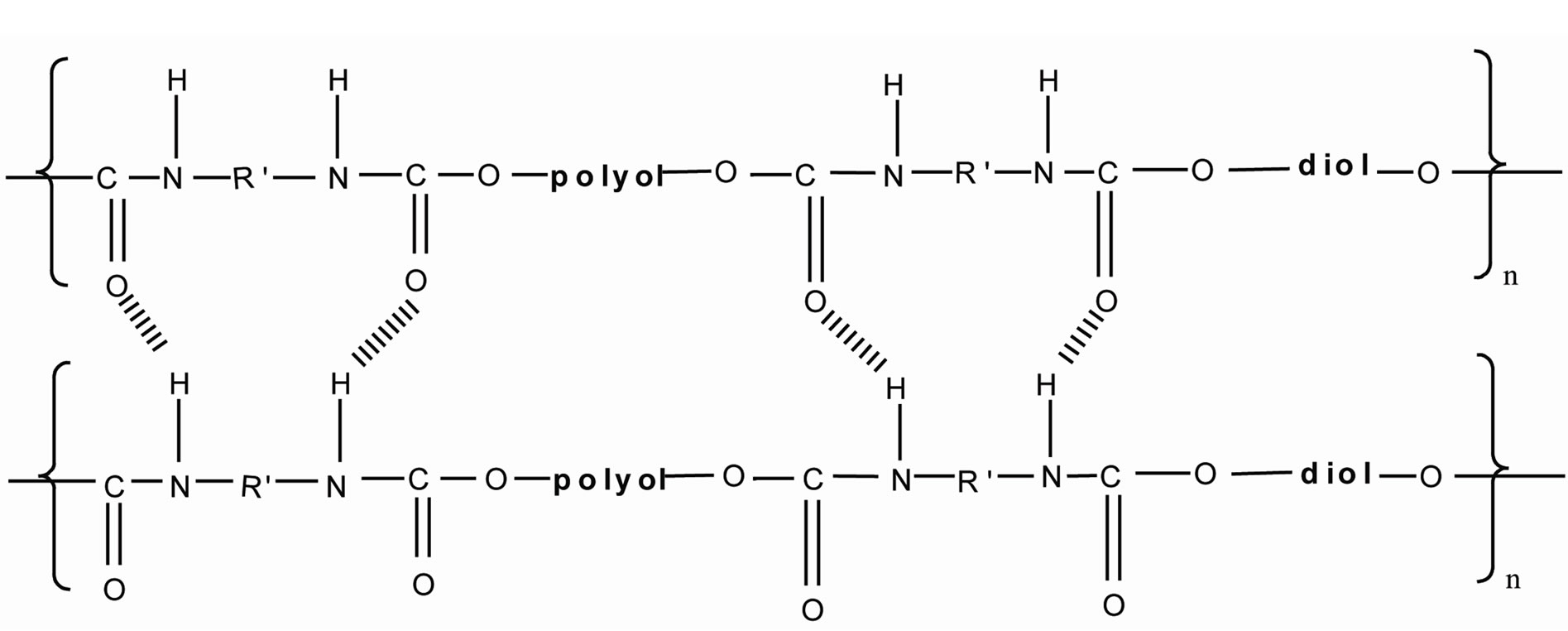
Figure 4. The formation of hydrogen bonding by the NH and C=O of the urethane group.
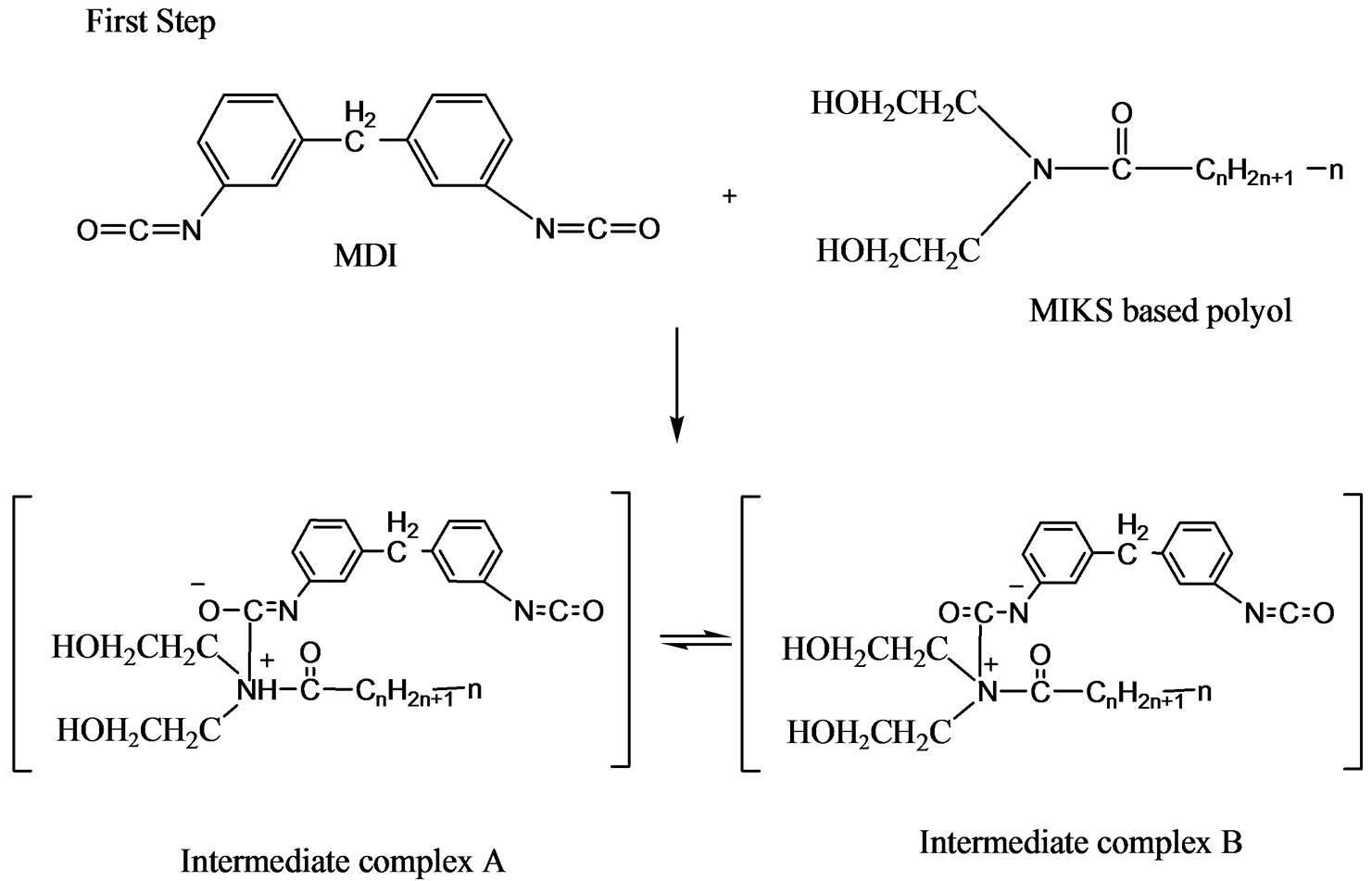 (a)
(a)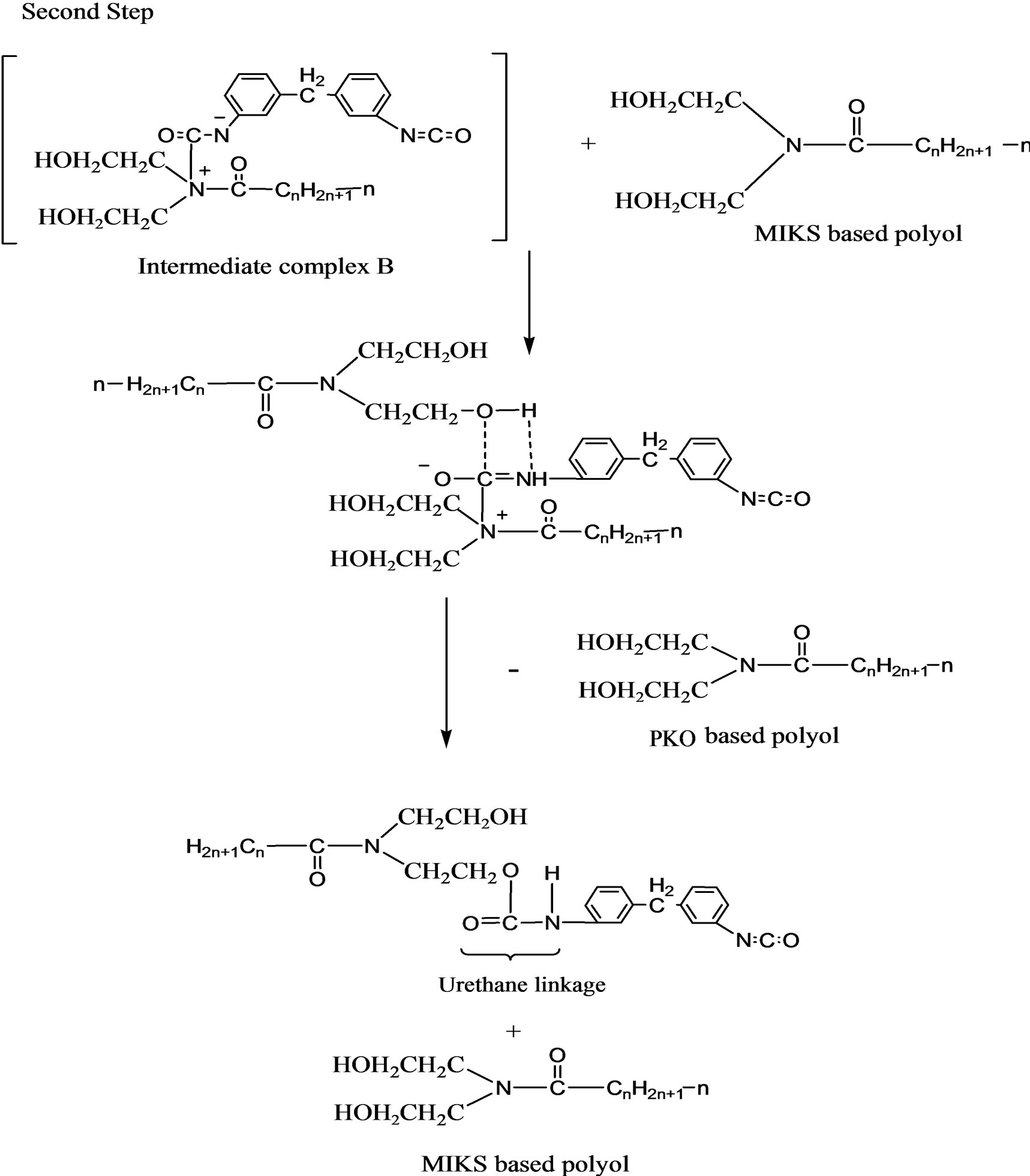 (b)
(b)
Figure 5. (a) Mechanism for the intermediate species formation via nucleophilic substitution reaction with amine of PKO-p as a nucleophile; (b) Mechanism for the urethane linkage formation via nucleophilic substitution reaction with amine of PKO-p as a nucleophile.
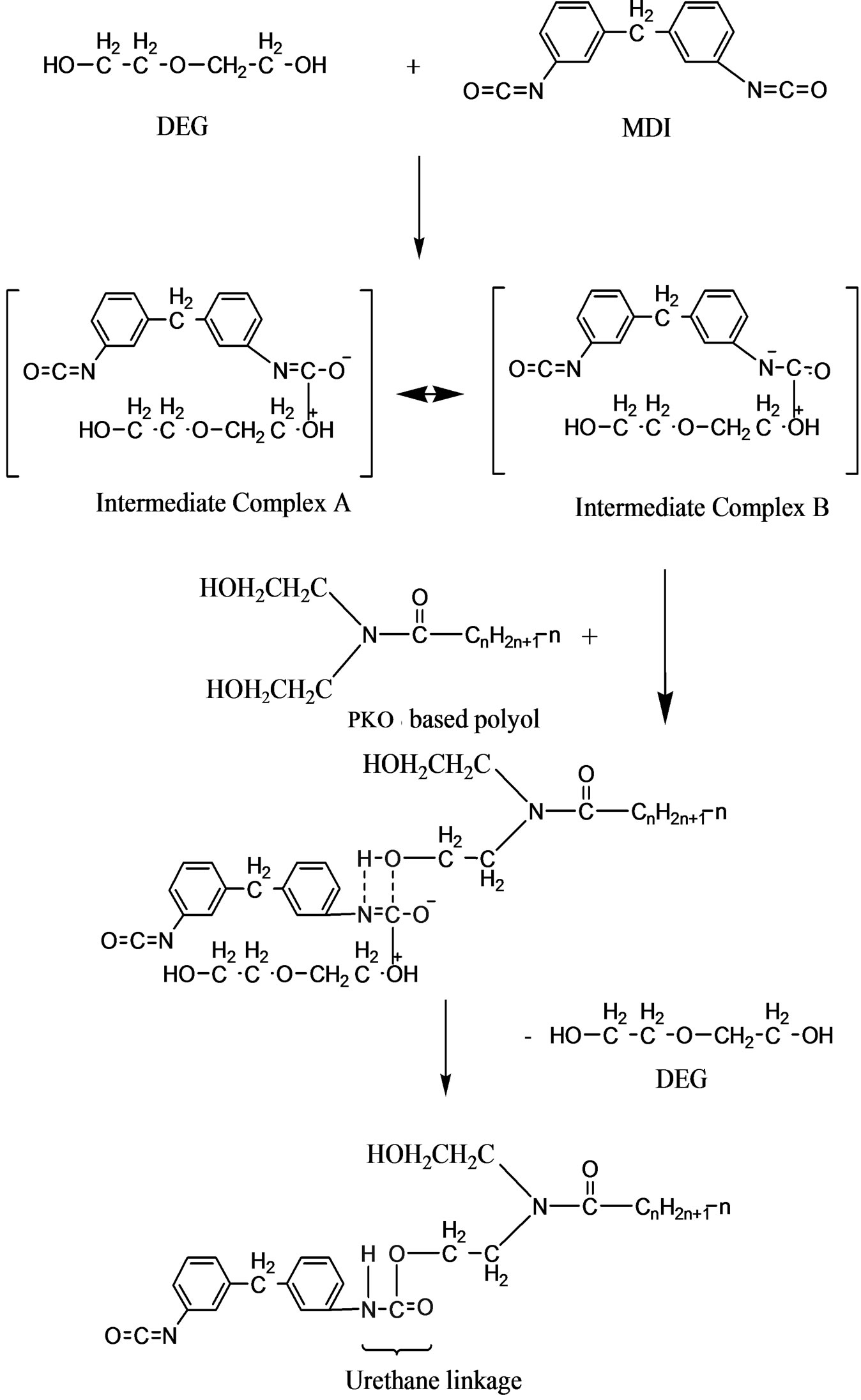
Figure 6. Mechanism for the urethane linkage formation via nucleophilic substitution reaction with DEG as a nucleophile.
In this research, nucleophilic substitution reaction with PKO-p has greater tendency to take place compared to DEG due to the presence of the nitrogen atom in PKO-p which is more electropositive than oxygen atom in DEG. Amine compound is more intend to react with isocyanate compound compared to hydroxyl compound [11]. Ashida and co-workers identified that amine compound with stronger alkalinity is more likely to react with carbon atom of the isocyanate [12]. Alkalinity of amine compound increased with increasing carbon number of alkyl group because the electron in carbon atom is transferred to stabilize the alkylaminium ion. The alkalinity of the amine compound is much depending on the stability of the alkylaminium ion [13]. PKO-p contains a very long carbon chain (fatty acid) that it can easily stabilize the alkylaminium ion when an intermediate complex is formed. Thus, the PKO-p is more reactive than DEG to react with MDI.
DEG will attach to the urethane prepolymer by nucleophilic substition reaction to form intermediate complex (Figure 6). The extended chain is formed by urethane bond. Adding DEG to the urethane prepolymer will increase the chain length of PU (Figure 7) and inhibit side reaction to occur such as the formation of urea between the reaction of NCO group in urethane prepolymer and water molecules from the surrounding.
3.3. NMR Spectroscopy Analysis
NMR spectra are used to study the structure of the polymeric chains. The 13C NMR sepctrum of the polyurethane
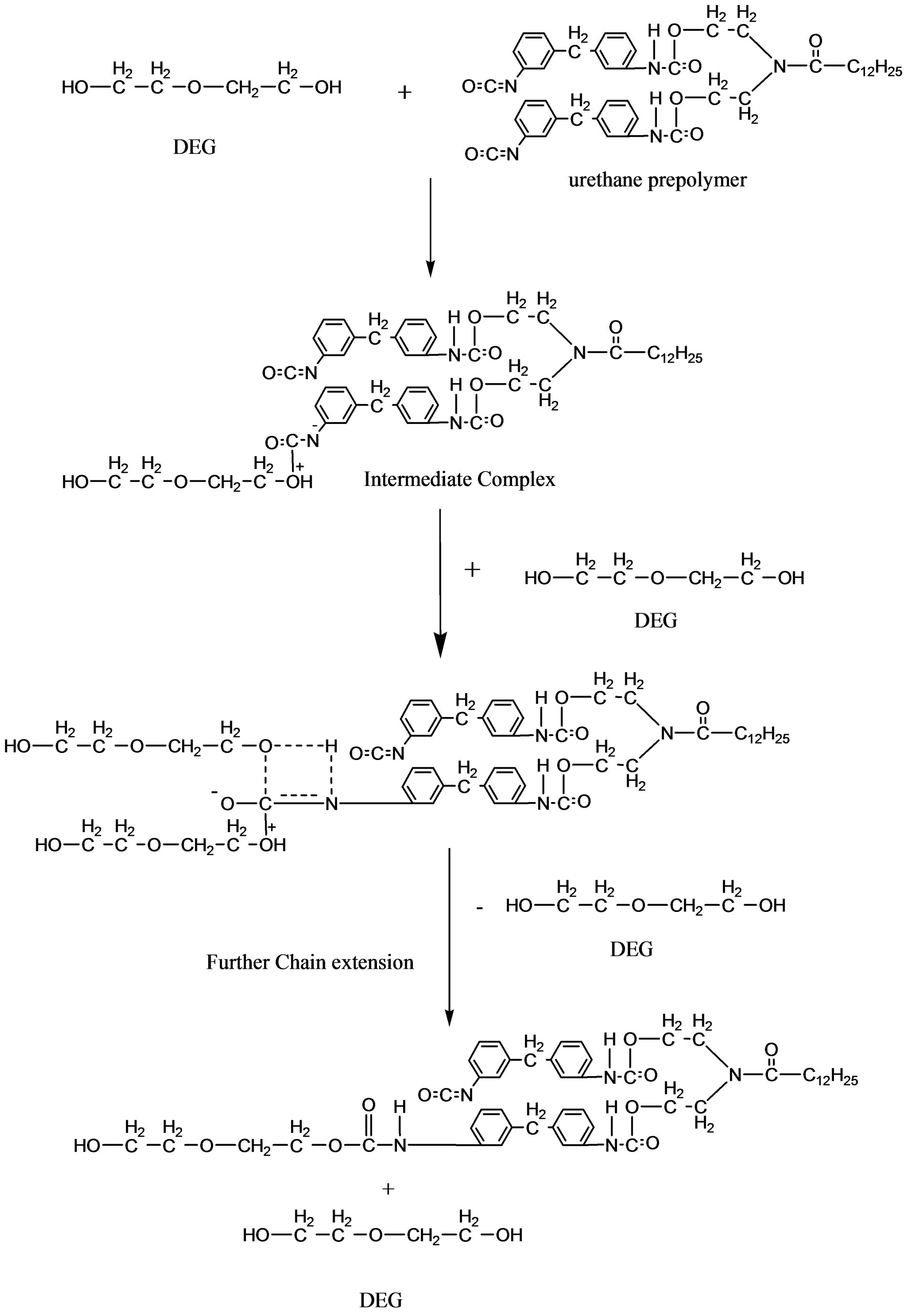
Figure 7. Addition of DEG increased the chain length of the urethane prepolymer.
is as shown in Figure 8 and depicted as follows:
13C NMR (DMSO-d6) for PU1: δ (ppm) = 154.0 - 154.2 (C(O)ONH), 129.3 - 137.2 (C=C), 119.0 (C=CN(H)), 72.5 (CH2O), 69.0 (CH2C(O)OC), 60.6 - 64.1 (C(O)OCNH), 22.5 - 33.9 (CH2), 14.3 (CH3).
13C NMR (DMSO-d6) for PU2: δ (ppm) = 153.4 - 154.5 (C(O)ONH), 128.8 - 137.0 (C=C), 118.4 (C=CN(H)), 72.3 (CH2O), 68.6 (CH2C(O)OC), 60.2 - 63.5 (C(O)OCNH), 22.1 - 33.5 (CH2), 14.0 (CH3).
13C NMR (DMSO-d6) for PU3: δ (ppm) = 153.7 (C(O)- ONH), 129.0 - 137.1 (C=C), 118.7 (C=CN(H)), 72.4 (CH2O), 68.8 (CH2C(O)OC), 60.4 - 63.7 (C(O)OCNH), 22.3 - 33.6 (CH2), 14.2 (CH3).
13C NMR (DMSO-d6) for PU4: δ (ppm) = 154.7 (C(O)- ONH), 129.7 - 137.3 (C=C), 119.4 (C=CN(H)), 72.7 (CH2O), 69.3 (CH2C(O)OC), 60.9 - 64.4 (C(O)OCNH), 22.8 - 34.2 (CH2), 14.2 (CH3).
The presence of peak at 153.0 ppm - 154.0 ppm along
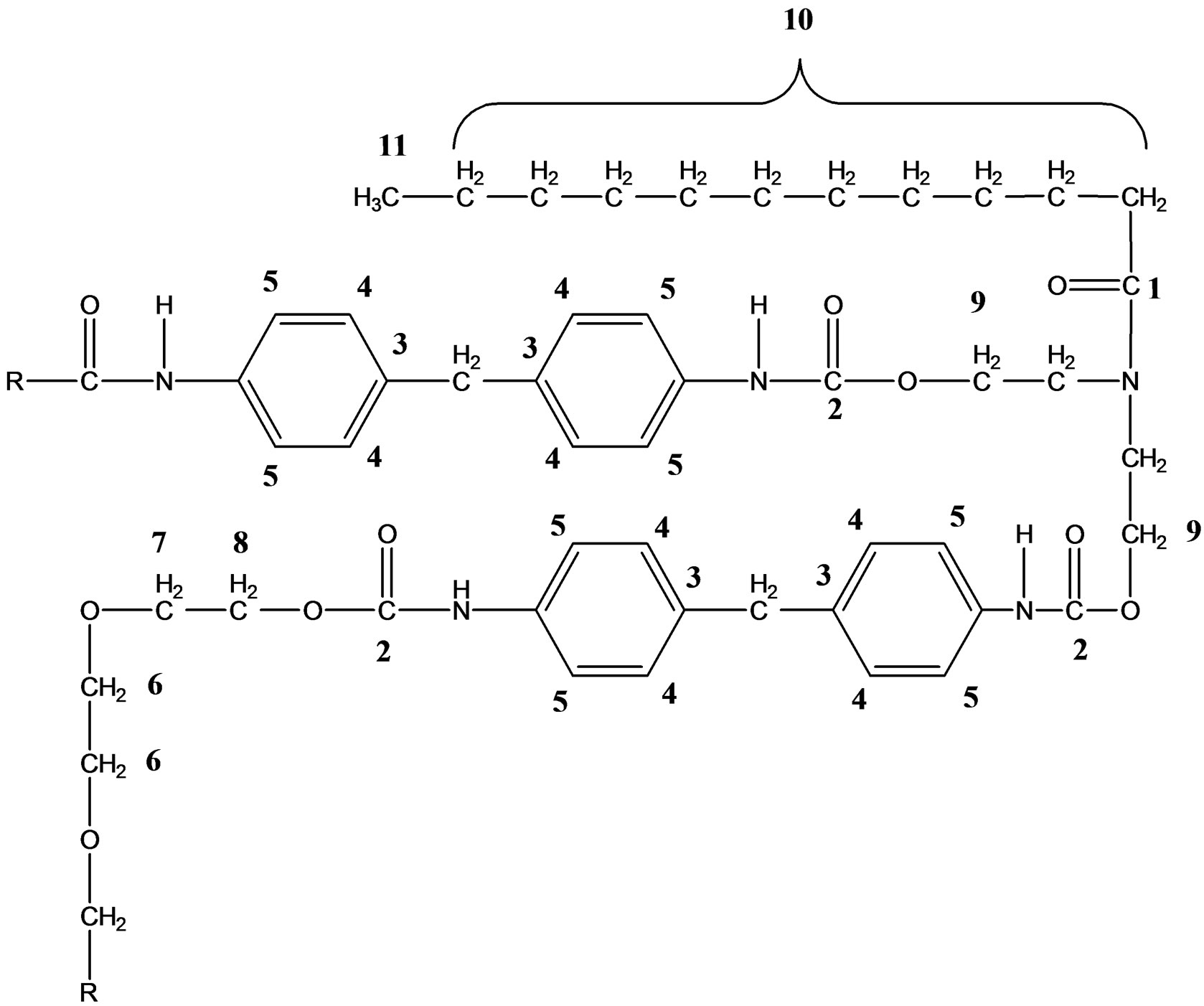
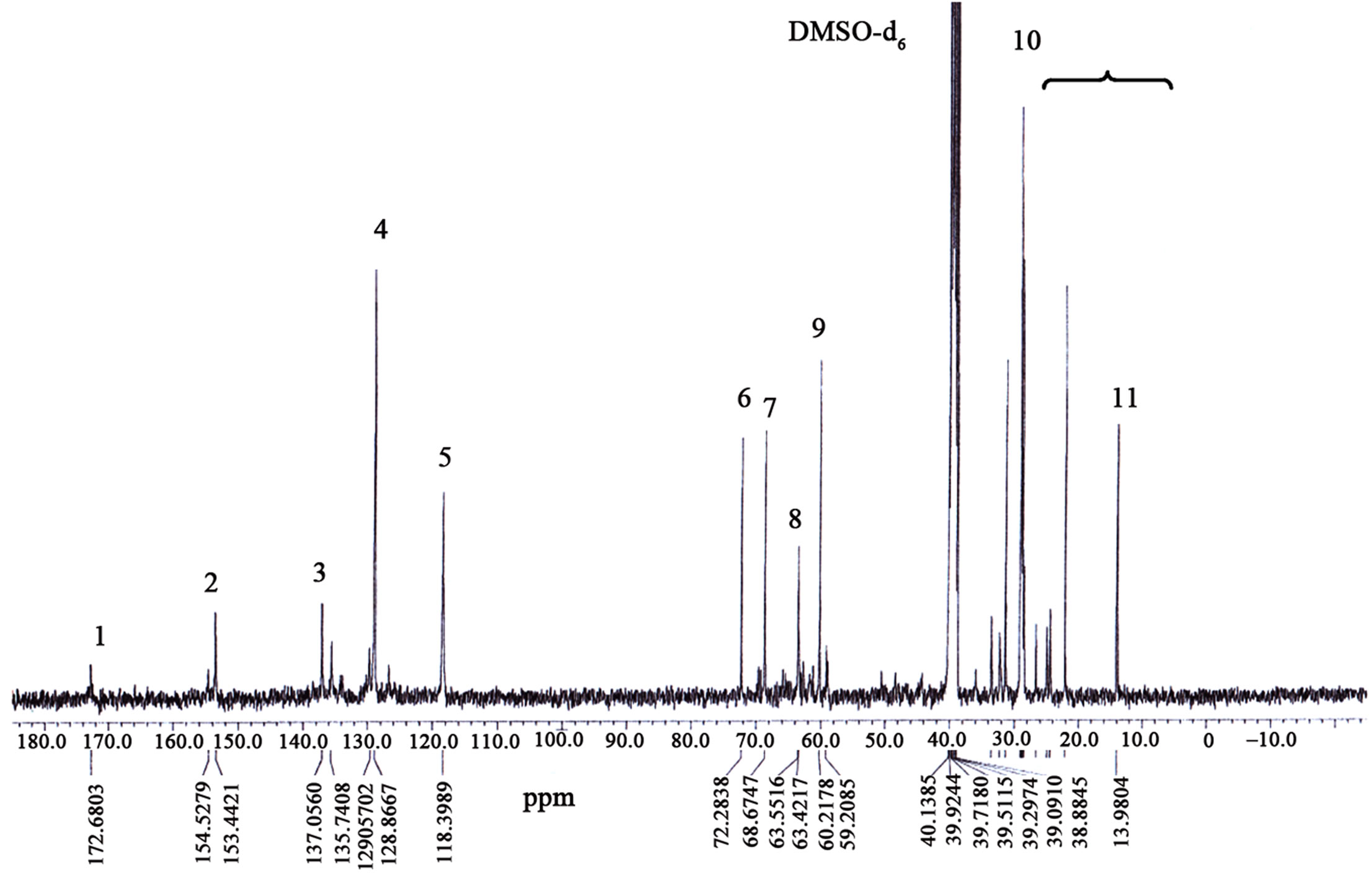
Figure 8. 13C NMR spectrum of the PKO-p prepolymerised PU.
with the disappearance of NCO peak of MDI at 125.7 ppm concluded that the diisocyanate of MDI reacted completely to -OH of PKO-p to form urethane polymeric chains [12]. Besides, the C-OH peak in PU2, PU3 and PU4 were detected around 59.2 ppm - 59.8 ppm indicating an excess of PKO-p and DEG. The CH2 peaks varies from 22.3 - 34.2 ppm due to numerous fatty acid contents in PKO such as lauric acid, palmitic acid, linoleic acid, linolenic acid and so on. In the 13C NMR analysis, lauric acid is assumed as the main carbon chain in the polyurethane since its composition is the highest at 50% - 52% in PKO. Urea linkage is also untraceable in the FT-NMR spectra.
4. Conclusions
The FTIR spectra indicated the progress of the prepolymerization of PU based on palm kernel oil-based polyol. The disappearance of NCO peak of MDI in 13C NMR and FTIR spectra justified that the MDI has completely reacted with polyol to form urethane linkage. Besides, the C=O and NH peaks in the FTIR spectra indicated the formation of hydrogen bonding in the polyurethane chain.
Several advantages are foreseen from this study. Some important advantages are being identified through this method of polyurethane production. Firstly, the source of polyurethane polyol is truly renewable, where it does not lead to permanent depletion of resources which has a limited global availability namely the palm kernel oil. Secondly, the polyurethane film made from this PKO-p is a clear and semi flexible film. It is intended for coating application especially onto the fiberboard due to hydrophobic nature of the polyurethane.
5. Acknowledgements
The authors wish to thanks Ministry of Science, Technology and Innovation for the funding of this project under project Grant No. 03-01-02-SF0509. A highly gratitude to Ministry of Higher Education for the financial support to the main researcher of this project under Graduate Scheme Program of MyBrain, Grant No. UKMST-02-FRGS0137-2009 and UKM-GUP-2011-0158.
REFERENCES
- I. Clemitson, “Castable Polyurethane Elastomers,” Taylor & Francis Group, New York, 2008. doi:10.1201/9781420065770
- L. Jin, M. D. Zhu and L. Zhen, “FTIR Studies on the Compatibility of Hard-Soft Segments for Polyurethaneimide Copolymers with Different Soft Segments,” European Polymer Journal, Vol. 38, No. 4, 2002, pp. 661-665. doi:10.1016/S0014-3057(01)00247-6
- K. H. Badri, S. H. Ahmad and S. Zakaria, “Production of a High-Functionality RBD Palm Kernel Oil-Based Polyester Polyol,” Journal of Applied Polymer Science, Vol. 81, No. 2, 2000, pp. 384-389. doi:10.1002/app.1449
- O. George, “Principles of Polymerization,” 2nd Edition, John Wiley & Sons Inc., New York, 1981.
- T. S. Velayutham, A. W. H. Majid, A. B. Ahmad, Y. K. Gan and S. N. Gan, “Synthesis and Characterization of Polyurethane
 Coatings Derived from Polyols Synthesized with Glycerol, Phthalic Anhydride and Oleic Acid,” Progress in Organic Coatings, Vol. 66, No. 4, 2009, pp. 367- 371. doi:10.1016/j.porgcoat.2009.08.013
Coatings Derived from Polyols Synthesized with Glycerol, Phthalic Anhydride and Oleic Acid,” Progress in Organic Coatings, Vol. 66, No. 4, 2009, pp. 367- 371. doi:10.1016/j.porgcoat.2009.08.013 - S. Li, R. Vatanparast and H. Lemmetyinen, “Cross-Linking Kinetics and Swelling Behavior of Aliphatic Polyurethane,” Polymer, Vol. 41, No. 15, 2000, pp. 5571-5576. doi:10.1016/S0032-3861(99)00785-5
- F. Mutsuhisa, K. Ken and N. Shohei, “Microphase Separated Structure and Mechanical Properties of Norbornane Diisocyanate-Based Polyurethane,” Polymer, Vol. 48, No. 4, 2007, pp. 997-1004. doi:10.1016/j.polymer.2006.12.057
- M. Boyd, “Organic Chemistry,” 4th Edition, Allyn and Bacon, Boston, 1983.
- H. Yong, Z. M. Bo, W. Bo, J. Z. lin and N. Jun, “Synthesis and Properties of Novel Polyurethane
 Acrylate Containing 3-(2-Hydroxyethyl) Isocyanurate Segment,” Progress in Organic Coatings, Vol. 67, 2009, pp. 264-268.
Acrylate Containing 3-(2-Hydroxyethyl) Isocyanurate Segment,” Progress in Organic Coatings, Vol. 67, 2009, pp. 264-268. - G. M. Loudon, “Organic Chemistry,” The Benjamin/Cummings Publishing Company Inc., San Francisco, 1988.
- R. Herrington and K. Hock, “Flexible Polyurethane Foams,” 2nd Edition, Dow Chemical Company, Midland, 1997.
- H. Sei, “Catalysts for Polyurethanes and Polyisocyanurates,” International Progress in Urethanes, Techmonic Publishing Co. Inc., Westport, 1981.
- S. T. W. Graham, “Organic Chemistry,” John Wiley & Sons Inc., New York, 1992.
NOTES
*Corresponding author.