Advances in Bioscience and Biotechnology
Vol.4 No.7A2(2013), Article ID:34155,11 pages DOI:10.4236/abb.2013.47A2004
Methylene blue protects mitochondrial respiration from ethanol withdrawal stress
![]()
Departments of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, USA
Email: Marianna.Jung@unthsc.edu
Copyright © 2013 Marianna Jung, Daniel Metzger. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 14 May 2013; revised 15 June 2013; accepted 30 June 2013
Keywords: Cell Viability; Ethanol Withdrawal; Methylene Blue; Mitochondrial Respiration; Cytochrome C Oxidase
ABSTRACT
Methylene blue (MB), a tricyclic phenothiazine drug, has been reported to enhance mitochondrial functions including mitochondrial respiration. By comparison, stress associated with abrupt ethanol withdrawal (EW) impedes mitochondrial functions. We investigated whether MB protects mitochondrial respiration and cell survival from EW stress through a key mitochondrial enzyme, cytochrome c oxidase (COX). We also investigated whether the MB’s protection involves the inhibition of an excitatory neurotransmitter, glutamate. Male rats were exposed to and withdrawn from ethanol-diet (7.5%, 5 weeks). MB (0.5 mg/kg, intraperitoneal) was injected for the last 5 days of ethanol-diet and on the first day of EW. Cerebellum was then harvested to measure mitochondrial respiration and COX expression using real-time XF respirometer and immunohistochemistry, respectively. Separately, HT22 cells (a murine hippocampal cell line) were exposed to and abruptly withdrawn for 4 hours from chronic ethanol (100 mM, 3 days). MB was administered during EW with or without a COX inhibitor (NaN3) or glutamate. Mitochondrial respiration, COX content, and cell viability were then assessed using real-time XF respirometer, an immunoblot method, and Calcein assay, respectively. MB attenuated the suppressing effects of EW on mitochondrial respiration, COX content, and cell survival. This protection was reduced after NaN3 or glutamate cotreatment. These results suggest that MB treatment help maintain mitochondrial respiratory and cellular integrity through COX-upregulation and glutamateinhibition upon EW stress. MB treatment may help identify mitochondrial mechanisms underlying hyperexcitatory CNS disorders.
1. INTRODUCTION
In mitochondria, a series of enzyme complexes is located in membranes to mediate the transfer of electrons to the terminal enzyme complex, cytochrome c oxidase (COX), where most of mitochondrial O2 is consumed [1]. In the process of electron transfer, a proton gradient is created across mitochondrial membranes, which drives force to generate ATP. As such, mitochondrial O2 consumption (mitochondrial respiration) is a critical event for cellular energy production and often measured as an indicator of mitochondrial functional integrity. Mitochondrial respiratory defect is found in a variety of CNS disorders. Alzheimer’s disease patients show mitochondrial dysfunction and energy hypometabolism [2]. Brains that undergo severe stress associated with abrupt ethanol withdrawal (EW) show impeded mitochondrial respiration compared to healthy brain [3]. Accordingly, mitochondrial targeting drugs have been the focus of many areas of CNS pathology.
MB (methylthionine hydrochloride), originally discovered as a synthetic dye, easily crosses the blood-brain barrier and accumulates in nervous tissue after systemic administration [4,5]. These properties of MB have attracted many researchers to this drug, revealing MB’s potential to treat methemoglobinemia, cyanide poisoning [6], Alzheimer Disease [7], and psychosis [8]. The medical safety of MB at a low dose has been shown in clinical trials to attenuate cognitive dysfunction in bipolar disorder patients [clinical trials number (NCT) 00214877], post-traumatic stress symptoms (NCT01188694), malaria (NCT00545935), and other medical conditions. In animal studies, MB improves spatial memory retention [9], delays the onset of Alzheimer’s disease [10,11], and attenuates ischemia/reperfusion injury in rats [12]. MB counteracts the damaging effect of rotenone, an inhibitor of mitochondrial electron transfer complex I on retinal neurons [13]. These neurodegenerative disorders bear common fundamental deficits in mitochondrial function, implying that MB exerts a beneficial effect on mitochondria compromised by these disorders. MB is a redox compound, meaning that it undergoes cycling between the reduced (MBH2) and oxidized (MB) form. This property of MB has been reported to facilitate electron transfer across mitochondrial electron transfer complexes, minimize electron leakage, and inhibit superoxide production [12,14].
In contrast to the protective effects of MB on mitochondria, the abrupt termination of long-term heavy ethanol consumption damages mitochondrial and neuronal integrity. Controlling EW stress is clinically important because alcoholic patients must be detoxified when they are treated for alcohol-related or -unrelated illnesses [15, 16]. We have previously demonstrated that EW stress provokes oxidative damage to mitochondrial proteins, the excessive opening of the mitochondrial membrane permeability pore, and the inhibition of COX activity [3, 17,18]. Others have also reported that liver mitochondria from ethanol-exposed and withdrawn rats are more vulnerable to lipid peroxidation than mitochondria from control-diet rats [19]. As such, abrupt EW impedes whereas MB enhances the important functions of mitochondria. We thus, sought to investigate MB’s potential to protect mitochondrial respiration and cell survival from EW stress. EW stress is initiated by the removal of the inhibitory stimulus effect of ethanol. Therefore, EW is hyperexcitatory in nature and associated with excitatory neurotransmitters such as glutamate. Based on these facts, we also tested whether MB’s protection against EW stress is mediated through glutamate inhibition. MB has been reported to have a hormetic dose-response, with neuroprotective effects at a low dose and neurotoxic effects at a high dose [20]. The current study used a low dose of MB and focused on the beneficial effects of MB on mitochondria and cellular integrity.
2. MATERIALS AND METHODS
2.1. Materials
Analytic grade reagents were purchased from Sigma Aldrich (St. Louis, MO), Cell Signaling Technology (Danvers, MA), Santa Cruz Biotechnology (Santa Cruz, CA), and Mitosciences (Eugene, OR). Diet ingredients were obtained from Research Organics (Cleveland, OH) or MP Biomedicals (Irvine, CA). HT22 cells, a murine hippocampal cell line, were the generous gift of Dr. David Schubert (Salk Institute, San Diego, CA). All reagents for mitochondrial respiration assay were purchased from Seahorse Bioscience (North Billerica, MA).
2.2. Animals
Male Sprague-Dawley rats (Charles River, Wilmington, MA) were 3 months old at the beginning of this study. They were housed individually at controlled temperature (22˚C - 25˚C) and humidity (55%), with ad libitum access to water and a 12-hour-light/dark cycle. All animal experimentation was conducted in accordance with the Guide to the Care and Use of Laboratory Animals [DHHS Publication No. (NIH) 85 - 23, revised 1996, Office of Science and Health Reports, DRR/NIH, Bethesda, MD] and was approved by the University of North Texas Health Science Center Animal Care and Use Committee.
2.3. Ethanol-Diet, EW, and MB Injection
The induction of ethanol dependence was accomplished by a method that has been routinely employed in our laboratory [21]. Rats were divided into 4 groups (5 - 7 rats/group) according to ethanol-diet and MB treatment. The four groups were control dextrin-diet groups with or without MB treatment and ethanol-diet groups with or without MB treatment. Rats in the ethanol-diet groups received a liquid diet containing 7.5% (wt./vol.) ethanol for 5 weeks. Control-diet rats received a liquid diet with dextrin isocalorically substituted for ethanol. MB (0.5 mg/kg, once/day, I.P.) was injected for last 5 days of ethanol-diet and on the first day of EW. This regimen of MB administration is based on the rationale that most of alcoholic patients who seek for treatments receive medication near and/or during a detoxification phase. The liquid diets are nutritionally balanced containing multiple ingredients including pulverized casein, vitamin and mineral mixtures, sucrose, and cellulose [22]. One hundred ml of fresh diet was placed in each cage daily for 5 weeks and then abruptly removed to initiate EW stress. Thereafter, animals were fed conventional chow pellets. The experience of EW stress in these rats was ensured according to a standard method of monitoring EW signs such as tremor, rigidity, and hyperactivity [21].
2.4. Isolation of Mitochondria from Brain Tissues
Rats were humanely sacrificed under anesthesia [xylazine (20 mg/kg, I.P.) and ketamine (100 mg/kg, I.P.)] 24 hours after the removal of ethanol-diet. Immediately thereafter, cerebellar mitochondria were isolated by conventional differential centrifugation with slight modifications [23]. We chose cerebellum because this brain area is one of the most vulnerable brain areas to ethanol or EW. Briefly, cerebellum was dissected, rinsed, and rapidly transferred to a homogenizer containing ice-cold isolation buffer (320 mM sucrose, 1 mM dipotassium ethylene diamine tetraacetic acid, 10 mM Tris-Hcl). The homogenate was centrifuged at 1330 g for 5 minutes at 4˚C and the supernatant was saved. The pellet was resuspended in 0.5 volume of the original isolation buffer and centrifuged again under the same conditions. The two supernatants were combined and centrifuged further at 21,200 g for 10 minutes. The resulting pellet was resuspended in 12% Percoll solution (GE Healthcare BioSciences AB, Uppsala, Sweden) and centrifuged at 6900 g for 10 minutes. The resulting soft pellet was washed once with mitochondrial isolation buffer and centrifuged at 6900 g for 10 minutes. The pellet containing mitochondria was used in this study. Bradford assay was conducted to analyze mitochondrial protein concentrations according to manufacturer’s instructions (Bio-Rad, Hercules, CA, USA) and a previous method [24].
2.5. Mitochondrial Respiration in Rats
Mitochondrial respiration was assessed by measuring mitochondrial O2 consumption rate (pmoles/minutes) according to a method provided by the XF respirometer manufacturer (Seahorse Bioscience). XF sensor cartridge was hydrated overnight in XF calibration buffer (at 37˚C, no CO2). Isolated mitochondria were diluted with mitochondrial assay solution (Seahorse Bioscience) to yield a final concentration of 200 µg/ml. Diluted mitochondria (50 µl) were transferred into each well of XF microplate and spun down at 4˚C for 10 - 20 minutes at 2000 - 3600 g. A consistent monolayer of mitochondrial adhesion to the bottom of the wells was visually ensured. A volume of 450 µl of succinate (5.5 mM) and rotenone (2.2 µM) was then added to each well. The XF microplate was warmed at 37˚C (no CO2) for 8 - 10 minutes and placed in XF respirometer. Real-time (data are obtained while mitochondria respire) mitochondrial respiration was subsequently recorded every 5 - 7 minutes.
2.6. Immunohistochemical Analysis of COX in Rats
Rats were deeply anesthetized with the mixture of ketamine (60 mg/kg, I.P.) and xylazine (10 mg/kg) at 24 hours of EW and perfused intracardially with saline, followed by 4% paraformaldehyde in phosphate buffer. Cerebellar areas containing vermis were divided into halves by a midsagittal cut and this sample was used for the immunohistochemistry. Briefly, the fixed tissues were rinsed in 70% ethanol overnight, dehydrated using different ethanol concentrations, xylene, and mixture of xylene/ paraffin, and then embedded in paraffin. For consistency from animal to animal, multi-tissue blocks from the same groups were embedded together. The sections started approximately 1.5 mm parasagital from the midline of the cerebellar vermis and ended after 250 sections at 2.5 mm. For each rat, the 9th section from the midline was used for the immunohistochemistry analysis [21]. The slides were deparaffinized in xylene, rehydrated through decreasing ethanol concentrations, and washed with phosphate-buffered solution (PBS). The slices were then moisturized at 95˚C for antigen retrieval. A primary antibody against COX subunit IV (Cell signaling, Danvers, MA) was diluted (1:1000) in blocking buffer (10% normal goat serum). COX is composed of 13 subunits. We chose subunit IV because this subunit is particularly vulnerable to EW [25]. The slices were rinsed in PBS and then incubated with broad spectrum poly-horseradish-peroxidase-conjugate for 40 minutes. The antigen-antibody bindings were visualized with a diaminobenzidine color reaction and examined with an inverted Carl Zeiss microscope and a digital camera. All photographs were taken of the cerebellar cortex containing Purkinje layers. Slides stained with non-immunoreactive serum used for a negative control had negligent diaminobenzidine staining.
2.7. HT22 Cell Culture
The HT22 cell line was originally selected from HT4 cells that were immortalized from mouse primary hippocampal neurons using a temperature-sensitive small virus- 40 T antigen. HT22 cells were maintained as described previously [26]. Briefly, HT22 cells were grown in DMEM, supplemented with 10% charcoal-stripped fetal bovine serum (HyClone, Logan, UT) and 1% penicillin/ streptomycin at 37˚C in an atmosphere containing 5% CO2 and 95% air. HT22 cells (106 cells) were plated into Petri dishes (Greiner Bio-One, Monroe, NC). The following day, the cells were exposed to vehicle solution (ethanol-free DMEM medium) or 100 mM ethanol for 3 days. At the end of the 3 day-ethanol exposure, ethanolcontaining solution was abruptly replaced with ethanolfree solution (DMEM medium) to create EW stress and then incubated for 4 hours before assays began.
2.8. Mitochondrial Respiration of HT22 Cells
HT22 cells (1000 cells/well) were seeded into XF microplates and exposed to ethanol (100 mM) for 3 days, and abruptly withdrawn from ethanol to create EW stress [18, 27]. The cell plate was processed according to a manufacture’s (Seahorse Bioscience) instruction, placed on an O2 sensor cartridge, and inserted into the XF respirometer. This step was done immediately after 4 hours of EW. MB’s protection against EW-induced mitochondrial respiratory suppression was tested by treating cells with MB (250 nM) during the 4-hour-EW period. The role of COX in mitochondrial respiration was tested by treating cells with a COX inhibitor (NaN3, 0.1 µM). NaN3 was preloaded in the reagent delivery chambers of the O2 sensor cartridge and injected into the wells while the XF respirometer was reading O2 consumption rate. The effects of antioxidants on mitochondrial respiration were tested by preloading [50 µM of CoQ10 (Coenzyme Q10) or 1 µM of BHT (2,6-di-tert-butyl-4-methylphenol)] in this way. In addition, these antioxidants were chronically administered to a separate set of cells during the 4 hours of EW. Control cells were exposed to only dimethyl sulfoxide. O2 consumption rates (pmoles/minutes) were obtained approximately every 5 - 7 minutes.
2.9. Immunoblotting of COX in HT22 Cells
Since glutamate is known to mediate hyperexcitatory EW stress, we tested the effect of glutamate in the presence or absence of MB on COX protein content in HT22 cells. HT22 cells were treated with 3 mM glutamate for 24 hours with or without MB treatment (250 nM) for last 4 hours. After cells were allowed to grow overnight, a protease inhibitor and 1 ml of radioimmunoprecipitation assay buffer were added to lyse cells. Cells were then quickly collected and placed on ice. Bradford protein assay was conducted to assess total protein amount. Samples containing 30 µg of protein were resolved by SDS-PAGE on 10% cross-linked gels and then electrophoretically transferred onto polyvinylidene fluoride membranes. Nonspecific binding was blocked with 5% fat-free milk. Blots were washed in PBS containing 0.05% Tween 20 and then probed overnight with mouse monoclonal antibodies (Mitosciences, Eugene, OR) against COX subunit IV (1:1000 dilution). Blots were then incubated with horseradish peroxidase-conjugated secondary antibodies (1:10,000 dilution) for one hour at room temperature. Bands were detected using immunoblotting luminescence (UVP, Upland, CA) and quantified by an image densitometer. Immunoblottings for β-actin were carried out as a positive and a loading control.
2.10. Calcein-Acetoxymethylester Viability Assay
The effect of MB on cell death induced by EW and/or glutamate was tested using Calcein assay. HT22 cells were exposed to ethanol (100 mM) for 3 days and withdrawn for 4 hours as described earlier. MB (250 nM) and/or glutamate (3 mM) was treated during the 4-hour of EW period. A separate set of ethanol-free cells was exposed to glutamate (3 mM) for 24 hours with or without MB treatment for last 4 hours to test MB’s protection against glutamate cytotoxicity. The role of COX in the effect of MB or glutamate was tested by NaN3 (0.1 µM) cotreatment during EW. Following the removal of the medium from the 96-well plates, the cells were incubated with fluorogenic Calcein-AM (2.5 µM, Molecular Probes, Eugene, OR) in PBS. Twenty minutes later, fluorescence intensity was determined using a Bio-Tek FL600 microplate reader (Winooski, VT) with an excitation/emission filter set at 485/530 nm. Cell culture wells treated with methanol served as a blank condition.
2.11. Statistical Analysis
All numerical data are expressed as means ± standard error of mean (SEM) and were analyzed by a singlefactor-ANOVA (EW with or without drug treatment) or two-factor-ANOVA (EW × drug treatment). When ANOVA detected statistically significant treatment effects, post hoc Tukey’s tests were performed to identify specific differences between groups. p values of < 0.05 were taken to indicate statistical significance.
3. RESULTS
3.1. MB Protects Mitochondrial Respiration from EW in Rats
We have previously demonstrated that mitochondria from ethanol withdrawn cells are less able to respire than mitochondria from healthy cells [3]. Using brain tissues obtained from ethanol withdrawn rats, the current study extended this phenomenon to MB’s protection. Figure 1 illustrates that brain mitochondria from ethanol withdrawn rats show a smaller degree of respiration than those from control-diet rats or MB-treated ethanol withdrawn rats. MB-treatment to control-diet rats did not significantly alter mitochondrial respiration (data not shown). These data indicate that EW suppresses mitochondrial respiration in a manner that is protected by MB.
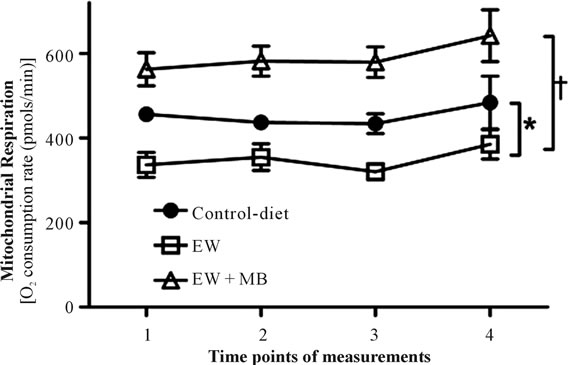
Figure 1. MB protects mitochondrial respiration from EW in rats. Male rats received ethanol-diet (7.5% wt./vol.) or controldextrin diet for 5 weeks and then abruptly withdrawn from the diet. MB (0.5 mg/kg, once/day, I.P.) was injected for last 5 days of ethanol-diet and on the first day of withdrawal. Rats were sacrificed 24 hours after the removal of the diet. Immediately thereafter, mitochondria were isolated from cerebellum to measure mitochondrial respiration at 4 time points with every 5 - 7 minutes per time points using XF respirometer. *p < 0.01 vs. control-dextrin diet, †p < 0.001 vs. EW + MB. Values are means ± standard error of mean (SEM) for 5 - 7 rats/group.
3.2. MB Protects COX from EW in Rats
Our previous study has shown that EW suppresses COX expression in rats. We tested whether MB attenuates such effect of EW on COX. Ethanol withdrawn rats show COX positive immunostaining (green fluorescence) to a lesser degree than control-diet rats (Figure 2). MB-treated ethanol withdrawn rats show much more COX positive staining than vehicle-treated ethanol withdrawn rats. These results indicate that MB treatment preserves COX expression upon EW stress.
3.3. COX Mediates MB’s Protection for Mitochondrial Respiration in Ethanol Withdrawn Cells
We next investigated how MB improves mitochondrial respiration compromised by EW stress (Figure 3). We hypothesized that MB’s up-regulation of COX mediates protection against EW-induced mitochondrial respiratory suppression. To test this hypothesis, we coadministered MB with a COX inhibitor (NaN3) during the 4 hours of EW in cells. Separately, MB followed by NaN3 was acutely administered while XF respirometer recorded basal mitochondrial respiration. Both chronic and acute treatments of MB attenuated the inhibiting effect of EW on mitochondrial respiration. However, this effect of MB was diminished in the presence of NaN3. These results indicate that COX mediates the MB’s protection against EW-induced mitochondrial respiratory suppression.
3.4. Antioxidants Fail to Protect Mitochondrial Respiration from EW
MB has been reported to exert an antioxidant activity such that it minimizes electron leakage from the electron transfer complexes and the formation of reactive O2 species (ROS). In contrast, EW increases ROS produc-

Figure 2. MB protects COX from EW in rats Male rats received ethanol-diet (7.5% wt./vol.) or control-dextrin diet for 5 weeks and then abruptly withdrawn from the diet. MB (0.5 mg/ kg, once/d, I.P.) was injected for last 5 days of ethanol-diet and on the first day of withdrawal. At 24 hours of EW, rats were deeply anesthetized and perfused intracardially with saline followed by paraformaldehyde. Brain (cerebellum) slices were immunostained with COX subunit IV antibody (green fluorescence) and imaged using a fluorescence microscope at a magnification of 40×. Arrows indicate COX positive cerebellar neuronal cells. A horizontal bar indicates an actual length of 20 µm.
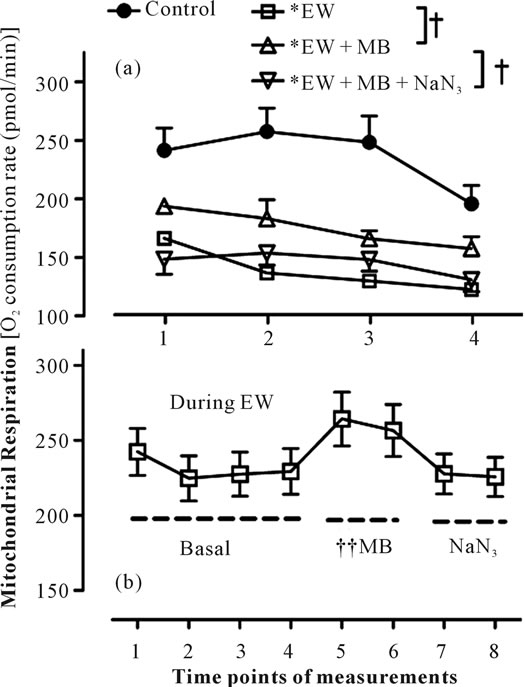
Figure 3. COX mediates MB’s protection for mitochondrial respiration in ethanol withdrawn cells. HT22 cells were exposed to ethanol (100 mM, 3 days) and withdrawn for 4 hours. MB (250 nM) with or without NaN3 (0.1 µM) was administered during the 4 hours of EW period. Mitochondrial respiration was then measured using XF respirometer (Figure 3(a)). For a separate set of ethanol withdrawn cells, MB followed by NaN3 was acutely injected while basal mitochondrial respiration was recorded. *p < 0.001 vs. vehicle-treated control-cells, †p < 0.01 vs. EW + MB (Figure 3(a)). ††p < 0.05 vs. basal or NaN3 condition (Figure 3(b)). Values are means ± SEM for 6 wells/group.
tion and protein oxidation [17,28]. If MB’s antioxidant activity mediates its mitochondrial respiratory protection, treating cells directly with antioxidants would increase mitochondrial respiration upon EW stress. We administered antioxidants BHT and CoQ10 during the 4 hours of EW (Figure 4(a)) or acutely injected (Figure 4(b)) them into cells while basal respiration was recorded. However, neither compound was able to increase mitochondrial respiration of ethanol withdrawn or control-cells. The failure of these antioxidants to increase mitochondrial respiration of ethanol withdrawn cells suggests that an antioxidant activity may not mediate or not directly mediate MB’s protection against EW-induced mitochondrial respiratory suppression.
3.5. MB Protects Cell Viability from EW through Glutamate Inhibition
Considering that mitochondria provide cellular energy for cell survival, it is reasonable to speculate that mitoprotective MB protects cell viability. We have previously demonstrated that EW provokes cell death [27] and neuronal loss [21]. Since glutamate is known to mediate hyperexcitatory EW stress [29,30], we hypothesized that MB protects cell survival from EW through glutamate inhibition. Figure 5 illustrates that EW decreases cell viability and this effect of EW is substantially attenuated

Figure 4. Antioxidants fail to protect mitochondrial respiration from EW. HT22 cells were exposed to ethanol (100 mM, 3 days) and withdrawn for 4 hours. Antioxidants, BHT (1 µM) or CoQ10 (50 µM) was applied to cells during the 4 hours of EW (Figure 4(a)). For a separate set of ethanol withdrawn cells, antioxidants were acutely injected (Figure 4(b)) while XF respirometer was reading basal mitochondrial respiration. *p < 0.001 vs. vehicle-treated control-cells. Values are means ± SEM for 6 wells/group.
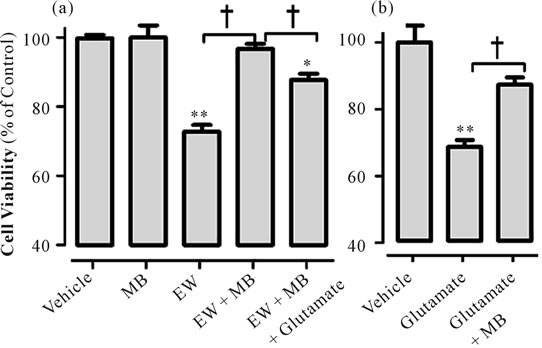
Figure 5. MB protects cell viability from EW through glutamate inhibition. HT22 cells were exposed to ethanol (100 mM, 3 days) and withdrawn for 4 hours. MB (250 nM) with or without glutamate (3 mM) were applied during EW (Figure 5(a)). Separately, HT22 cells were exposed to glutamate for 24 hours with or without MB (250 nM) treatment during the last 4 hours of glutamate-exposure (Figure 5(b)). Cell viability was then measured using Calcein assay. *p < 0.01 or **p < 0.001 vs. vehicle alone, †p < 0.01 vs. EW + MB or glutamate + MB. N = 6 wells/group. Values are means ± SEM for 6 wells/group.
by MB treatment. However, MB was less cytoprotective after glutamate cotreatment (Figure 5(a)). Reciprocally, glutamate treatment acted like EW stimulus by inhibiting cell viability and this effect of glutamate was attenuated by MB treatment (Figure 5(b)). These results indicate that there is a counteracting relationship between MB and glutamate at the level of cell viability and that MB protects cells from EW stress by inhibiting glutamate insults.
3.6. COX Mediates MB’s Cytoprotection against EW or Glutamate
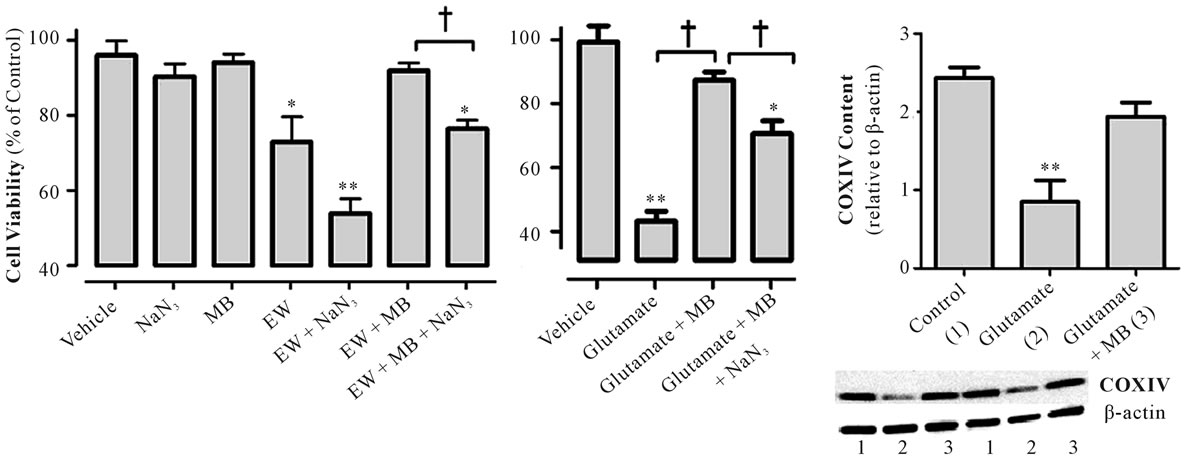
We investigated whether MB protects cell survival from EW or glutamate through COX upregulation. If this was the case, a COX inhibitor treatment would blunt the MB’s protection. We indeed observed that MB was less protective against EW (Figure 7(a)) or glutamate (Figure 7(b)) cytotoxicity in the presence of NaN3. NaN3 treatment alone (without MB) during EW exacerbated EW-induced cell death. In addition, glutamate at the dose that inhibited cell viability decreased the protein content of COX and this effect of glutamate was attenuated by MB treatment (Figure 7(c)). These results support the idea that COX mediates MB’s protection against EWor glutamate-induced cytotoxicity.
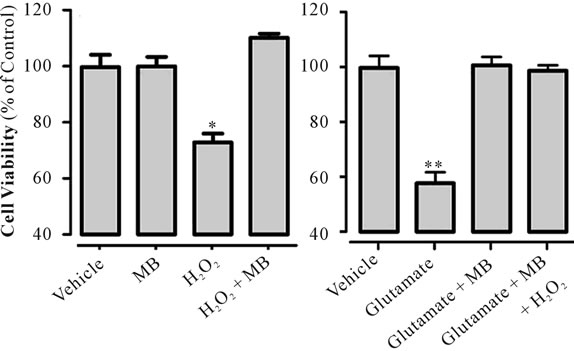
3.7. A Prooxidant Drug Does Not Abolish MB’s Protection against Glutamate Cytotoxicity
If MB’s protection against glutamate cytotoxicity is
 (a) (b)
(a) (b)
Figure 6. A prooxidant drug does not abolish MB’s protection against glutamate cytotoxicity. HT22 cells were treated with H2O2 (30 µM, 24 hours) with or without MB (250 nM) during the last 4 hours of H2O2-exposure (Figure 6(a)). Separate cells were treated with glutamate (3 mM, 24 hours) with MB (250 nM) with or without H2O2 (30 µM) during the last 4 hours of glutamate treatment (Figure 6(b)). Cell viability was then measured using Calcein assay. *p < 0.01 (7A), **p < 0.001 (Figure 6(b)) vs. all other 3 groups. N = 6 wells/group. Data (%) are relative to vehicle-treated control-cells (at 100%). Values are means ± SEM for 6 wells/group.
 (a) (b) (c)
(a) (b) (c)
Figure 7. COX mediates MB’s cytoprotection against EW or glutamate. HT22 cells were exposed to ethanol (100 mM, 3 days) and withdrawn for 4 hours. NaN3 (0.1 µM) with or without MB (250 nM) was applied during EW (Figure 7(a)). Separately, HT22 cells were exposed to glutamate (3 mM) for 24 hours (Figure 7(b)). MB (250 nM) with or without NaN3 (0.1 µM) was applied during the last 4 hours of glutamate-exposure. Cell viability and COX protein content (Figure 7(c)) were then measured using Calcein assay and immunoblot, respectively. *p < 0.01 or **p < 0.001 vs. vehicle alone, †p < 0.01 vs. EW + MB, or glutamate + MB. N = 6 wells/group. Values are means ± SEM for 4 or 5 rats/group.
mediated through its antioxidant activity, a prooxidant drug (H2O2) would blunt the MB’s protection. H2O2 alone inhibited cell viability and this effect was blunted in the presence of MB, indicating MB’s antioxidant protection for cell survival (Figure 6(a)). However, the cell viability of glutamate-exposed cells did not differ after treatment with MB alone or in combination with H2O2 (MB + H2O2) (Figure 6(b)). These data indicate that a prooxidant stimulus does not alter MB’s protection against glutamate cytotoxicity.
4. DISCUSSION
In the current study, we have demonstrated that MB protects against mitochondrial respiratory suppression and cell death induced by stress associated with the abrupt termination of chronic ethanol. The MB’s protection is at least partly attributed to COX upregulation and glutamate inhibition. These findings are the first report that MB protects mitochondrial and cellular integrity from EW insults.
Mitochondria appear to be one of the major targets for EW. Mitochondria isolated from ethanol withdrawn rats or cells show an increased level of free radical production, protein oxidation, mitochondria membrane swelling, and membrane potential collapse [18,28]. This abnormal biology of mitochondria may have a cause-effect relationship with mitochondrial respiratory dysfunction. However, little information is available about how to mitigate the damaging effect of EW on mitochondria. MB has been shown to accumulate within mitochondria [31], improve mitochondrial respiration [32,33], and attenuate neurodegeneration associated with mitochondrial aging [10,34]. The mitoprotective effects of MB opposite to the mitotoxic effects of EW led us to the idea that MB may mitigate EW-induced mitochondrial respiratory dysfunction. Indeed, both in vivo and in vitro treatments of MB showed a robust increase in mitochondrial respiration compared to vehicle-treated ethanol withdrawn mitochondria. In contrast to these beneficial effects of MB on mitochondria, Lu et al. [35] have demonstrated that MB exerts mitochondria-dependent apoptosis and free radical generation. However, Lu et al. have used a much higher dose of MB (20 M) compared to 250 nM of MB used in the current study. As mentioned earlier, MB has a biphasic effect such that it is protective at a low dose but toxic at a high dose [20]. The toxic effect of MB is readily avoidable by using a low dose. In fact, the medical safety of MB at a low dose has been shown in various clinical trial studies.
Mitochondria contain four electron transfer complexes (I-IV) in the membranes with the terminal complex (IV), cytochrome c oxidase (COX). COX is unique in a sense that it consumes most of O2 entering cells to create proton gradients across mitochondrial membranes, thereby helping ATP production. As such, COX is directly involved in mitochondrial O2 consumption (mitochondrial respiration) and has been studied in conjunction with MB. For instance, MB increases the activity of COX in rats with concurrent memory improvement [9]. The COX enhancing effect of MB concurs with cellular antiaging effects in primary rat retinal ganglion cells [36] and in normal human lung fibroblasts cells [10]. If MB promotes the activities of electron transfer complexes, it is reasonable to speculate that MB would counteract the effects of electron transfer complex inhibitors. MB indeed attenuates retinal neurodegeneration, neurotoxic effects, and hypometabolism induced by an inhibitor of electron transfer complex I (rotenone) [13], II (methylmalonate) [37], and IV (NaN3) [38], respectively. Our current findings reveal that EW stress acts as a COX inhibitor whereas MB acts as a COX promoter, thereby protecting mitochondrial respiration from EW. These results are the first report that MB helps maintain mitochondrial integrity challenged by EW stress.
Abrupt EW typically triggers hyperexcitatory stress that perturbs neuronal and cellular homeostasis. One of the major events during EW is the up-regulation of excitatory glutamatergic neurotransmission [29,30]. The excessive level of glutamate causes cell death (glutamate excitotoxicity) [39] accompanied by mitochondrial dysfunction. Cerebral ischemia induces abnormally high levels of glutamate release that correlates with the impairment of mitochondrial energy-generation [40]. An impaired electron transfer complex is often found in CNS disorders, in which glutamate excitotoxicity plays an important role, such as Parkinson’s [41,42] or Huntington’s [43] disease. Based on these reports, we asked a question whether mitoprotective MB in turn protects against EWinduced cell death through glutamate inhibition. MB’s protection against hyperexcitatory stress is shown in a previous study done by de-Oliveira and Guimaraes [44]. They injected MB into the dorsal periaqueductal gray, a brain area that is responsible for defensive or hyperexcitatory reaction. They observed that compared to vehicle-injected rats, MB-injected rats spent more time in the open arms of the elevated plus-maze, an indicator of an anxiolytic behavior. Furian et al. [37] injected MB into the rat brain area of the intrastriatum and found that MB attenuated electrographically recorded seizure activity induced by methylmalonate. Directly relevant to the effect of MB on excitatory neurotransmitters, MB attenuates the damaging effect of N-methyl-D-aspartic acid on the dorsal motor neurons [45] or the inhibiting effect of glutamate on cell viability [46]. Our findings (Figure 5) agree with these studies in that MB attenuates EWinduced cell death and this MB’s protection is reduced by glutamate treatment. Collectively, our current findings along with others’ studies suggest that MB has a potential to mitigate hyperexcitatory brain insults. They also strengthen the idea that MB’s cytoprotection against EW is at least partly attributed to the inhibition of glutamate insults.
We further investigated a potential link between MB’s protection for mitochondria and cell survival. We hypothesized that the effect of MB on COX helps protect cell survival from EW or glutamate cytotoxicity. To test this hypothesis, we coadministered a COX inhibitor (NaN3) with MB and measured mitochondrial respiration and cell survival. Results revealed that the COX inhibitor reduced MB’s protection against EW-induced mitochondrial respiratory suppression and cell death. The COX inhibitor also reduced MB’s ability to protect cell survival from glutamate. These results suggest that MB counteracts the cytotoxic effects of EW or glutamate through COX upregulation. This view is further supported by an immunoblot experiment, showing that glutamate at a dose that inhibits cell survival also suppresses the expression of COX protein in a manner that is attenuated by MB treatment (Figure 7). A counteracting relationship between COX and glutamate is shown in an early study in which COX inhibition provokes abnormal glutamate release in rat brain accompanied by a fall in ATP/ ADP ratio [47]. By comparison, a promoting relationship exists between COX (complex IV) and MB, such that MB restores the activities of electron transfer complex II-IV compromised by ischaemia/reperfusion injury in rat liver [48]. Our current study is in line with these studies and provides empirical evidence that there is crosstalk between MB’s mitochondrial and cellular protection against EW or glutamate at the level of COX.
One of the remarkable effects of MB is its ability to modulate redox status. MB reduces electron leakage to mitochondrial matrix from electron transfer complexes and inhibits superoxide generation [12,14]. MB inhibits inducible NO synthase [49] that increases ROS production [50]. Mitochondria produce most of intracellular ROS and thus, it is not surprising that the antioxidant activities of MB occur largely in mitochondria. Aksu et al. [51] have reported that MB injection for 14 days attenuated hepatic damage in rats by reducing oxidative stress. MB decreased plasma membrane lipid peroxidation in equine sperm [52]. Based on these antioxidant effects of MB, we anticipated that MB’s antioxidant activity might be involved in its mitochondrial and cellular protection against EW. Not surprisingly, MB treatment protected against cell death induced by prooxidant H2O2 treatment (Figure 6), demonstrating MB’s antioxidant effects. If the antioxidant effect of MB mediates protection against EW or glutamate excitotoxicity, prooxidant H2O2 treatment would blunt the MB’s protection. Our results were somewhat unexpected because MB maintained its cytoprotection against glutamate in the presence of H2O2 (Figure 6). Moreover, antioxidant treatments failed to improve mitochondrial respiration of ethanol withdrawn cells (Figure 4). These results raise a possibility that MB’s antioxidant activities are sufficient for cellular or mitochondrial protection when oxidative stress is a sole or a direct cause for damage. However, MB’s antioxidant activities may not be sufficient for the protection when cellular or mitochondrial deficit is attributed to multiple factors such as COX inhibition in addition to oxidative stress.
In conclusion, our studies extend MB’s mitochondrial and cellular protection to the case of EW.
The regulation of COX-glutamate relationship appears to contribute to the MB’s protection, perhaps independently on or secondary to an antioxidant activity. Our findings may provide a new insight into MB’s effect on EW and support the idea that MB is a useful tool to study mitochondrial disorders associated with hyperexcitatory disorders.
5. ACKNOWLEDGEMENTS
This work has been supported by funding from NIH/NIAAA (AA 018747). We wish to thank Xiaohua Ju for her technical assistance.
![]()
![]()
REFERENCES
- Gonzalez-Lima, F. and Cada, A. (1998) Quantitative histochemistry of cytochrome oxidase activity: Theory, methods, and regional brain vulnerability cytochrom oxidase in neuronal metabolism and Alzheimer’s disease. Plenum Press, New York. doi:10.1007/978-1-4757-9936-1
- Atamna, H. and Kumar, R. (2010) Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. Journal of Alzheimer’s Disease, 20, S439-S452.
- Jung, M.E., Ju, X., Metzger, D.B. and Simpkins, J.W. (2011) Ethanol withdrawal hastens the aging of cytochrome c oxidase. Neurobiology of Aging, 33, 618.e621- 632.
- Peter, C., Hongwan, D., Kupfer, A. and Lauterburg, B.H. (2000) Pharmacokinetics and organ distribution of intravenous and oral methylene blue. European Journal of Clinical Pharmacology, 56, 247-250. doi:10.1007/s002280000124
- O’Leary 3rd, J.C., Li, Q., Marinec, P., Blair, L.J., Congdon, E.E., Johnson, A.G., Jinwal, U.K., Koren 3rd, J., Jones, J.R., Kraft, C., Peters, M., Abisambra, J.F., Duff, K.E., Weeber, E.J., Gestwicki, J.E. and Dickey, C.A. (2010) Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Molecular Neurodegeneration, 5, 45. doi:10.1186/1750-1326-5-45
- Sills, M.R. and Zinkham, W.H. (1994) Methylene blueinduced Heinz body hemolytic anemia. Archives of Pediatrics and Adolescent Medicine, 148, 306-310. doi:10.1001/archpedi.1994.02170030076017
- Wischik, C.M., Edwards, P.C., Lai, R.Y., Roth, M. and Harrington, C.R. (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proceedings of the National Academy of Sciences of the United States of America, 93, 11213-11218. doi:10.1073/pnas.93.20.11213
- Narsapur, S.L. and Naylor, G.J. (1983) Methylene blue. A possible treatment for manic depressive psychosis. Journal of Affective Disorders, 5, 155-161. doi:10.1016/0165-0327(83)90008-3
- Callaway, N.L., Riha, P.D., Bruchey, A.K., Munshi, Z. and Gonzalez-Lima, F. (2004) Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacology Biochemistry and Behavior, 77, 175-181. doi:10.1016/j.pbb.2003.10.007
- Atamna, H., Nguyen, A., Schultz, C., Boyle, K., Newberry, J., Kato, H. and Ames, B.N. (2008) Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB Journal, 22, 703- 712. doi:10.1096/fj.07-9610com
- Oz, M., Lorke, D.E. and Petroianu, G.A. (2009) Methylene blue and Alzheimer’s disease. Biochemical Pharmacology, 78, 927-932. doi:10.1016/j.bcp.2009.04.034
- Salaris, S.C., Babbs, C.F. and Voorhees 3rd, W.D. (1991) Methylene blue as an inhibitor of superoxide generation by xanthine oxidase. A potential new drug for the attenuation of ischemia/reperfusion injury. Biochemical Pharmacology, 42, 499-506. doi:10.1016/0006-2952(91)90311-R
- Zhang, X., Rojas, J.C. and Gonzalez-Lima, F. (2006) Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotoxicity Research, 9, 47-57. doi:10.1007/BF03033307
- Kelner, M.J., Bagnell, R., Hale, B. and Alexander, N.M. (1988) Methylene blue competes with paraquat for reduction by flavo-enzymes resulting in decreased superoxide production in the presence of heme proteins. Archives of Biochemistry and Biophysics, 262, 422-426. doi:10.1016/0003-9861(88)90393-1
- Buchsbaum, D.G., Buchanan, R.G., Poses, R.M., Schnoll, S.H. and Lawton, M.J. (1992) Physician detection of drinking problems in patients attending a general medicine practice. Journal of General Internal Medicine, 7, 517-521. doi:10.1007/BF02599456
- Mayo-Smith, M.F. (1997) Pharmacological management of alcohol withdrawal. A meta-analysis and evidencebased practice guideline. American Society of Addiction Medicine Working Group on Pharmacological Management of Alcohol Withdrawal. Journal of the American Medical Association, 278, 144-151. doi:10.1001/jama.1997.03550020076042
- Jung, M.E., Simpkins, J.W., Wilson, A.M., Downey, H.F. and Mallet, R.T. (2008) Intermittent hypoxia conditioning prevents behavioral deficit and brain oxidative stress in ethanol-withdrawn rats. Journal of Applied Physiology, 105, 510-517. doi:10.1152/japplphysiol.90317.2008
- Jung, M.E., Wilson, A.M., Ju, X., Wen, Y., Metzger, D.B. and Simpkins, J.W. (2009) Ethanol withdrawal provokes opening of the mitochondrial membrane permeability transition pore in an estrogen-preventable manner. Journal of Pharmacology and Experimental Therapeutics, 328, 692- 698. doi:10.1124/jpet.108.146829
- Rouach, H., Clement, M., Orfanelli, M.T., Janvier, B., Nordmann, J. and Nordmann, R. (1983) Hepatic lipid peroxidation and mitochondrial susceptibility to peroxidative attacks during ethanol inhalation and withdrawal. Biochimica et Biophysica Acta, 753, 439-444. doi:10.1016/0005-2760(83)90068-1
- Rojas, J.C., Bruchey, A.K. and Gonzalez-Lima, F. (2011) Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Progress in Neurobiology, 96, 32-45. doi:10.1016/j.pneurobio.2011.10.007
- Jung, M.E., Yang, S.H., Brun-Zinkernagel, A.M. and Simpkins, J.W. (2002) Estradiol protects against cerebellar damage and motor deficit in ethanol-withdrawn rats. Alcohol, 26, 83-93. doi:10.1016/S0741-8329(01)00199-9
- Ju, X., Mallet, R.T., Downey, H.F., Metzger, D.B. and Jung, M.E. (2012) Intermittent hypoxia conditioning protects mitochondrial cytochrome c oxidase of rat cerebellum from ethanol withdrawal stress. Journal of Applied Physiology, 112, 1706-1714. doi:10.1152/japplphysiol.01428.2011
- Yan, L.J., Yang, S.H., Shu, H., Prokai, L. and Forster, M.J. (2007) Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis, 28, 1036-1045. doi:10.1002/elps.200600574
- Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Annals of Biochemistry, 72, 248-254. doi:10.1016/0003-2697(76)90527-3
- Jung, M.E., Agarwal, R. and Simpkins, J.W. (2007) Ethanol withdrawal posttranslationally decreases the activity of cytochrome c oxidase in an estrogen reversible manner. Neuroscience Letters, 416, 160-164. doi:10.1016/j.neulet.2007.01.065
- Perez, E., Liu, R., Yang, S.H., Cai, Z.Y., Covey, D.F. and Simpkins, J.W. (2005) Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Research, 1038, 216-222. doi:10.1016/j.brainres.2005.01.026
- Jung, M.E., Wilson, A.M. and Simpkins, J.W. (2006) A nonfeminizing estrogen analog protects against ethanol withdrawal toxicity in immortalized hippocampal cells. Journal of Pharmacology and Experimental Therapeutics, 319, 543-550. doi:10.1124/jpet.106.103630
- Jung, M.E., Yan, L.J., Forster, M.J. and Simpkins, J.W. (2008) Ethanol withdrawal provokes mitochondrial injury in an estrogen preventable manner. Journal of Bioenergetics and Biomembranes, 40, 35-44. doi:10.1007/s10863-008-9129-y
- Prendergast, M.A., Harris, B.R., Mullholland, P.J., Blanchard 2nd, J.A., Gibson, D.A., Holley, R.C. and Littleton, J.M. (2004) Hippocampal CA1 region neurodegeneration produced by ethanol withdrawal requires activation of intrinsic polysynaptic hippocampal pathways and function of N-methyl-D-aspartate receptors. Neuroscience, 124, 869-877. doi:10.1016/j.neuroscience.2003.12.013
- Tsai, G.E., Ragan, P., Chang, R., Chen, S., Linnoila, V.M. and Coyle, J.T. (1998) Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. American Journal of Psychiatry, 155, 726-732.
- Hassan, H.M. and Fridovich, I. (1979) Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Archives of Biochemistry and Biophysics, 196, 385-395. doi:10.1016/0003-9861(79)90289-3
- Lindahl, P.E. and Oberg, K.E. (1961) The effect of rotenone on respiration and its point of attack. Experimental Cell Research, 23, 228-237. doi:10.1016/0014-4827(61)90033-7
- Visarius, T.M., Stucki, J.W. and Lauterburg, B.H. (1997) Stimulation of respiration by methylene blue in rat liver mitochondria. FEBS Letters, 412, 157-160. doi:10.1016/S0014-5793(97)00767-9
- Callaway, N.L., Riha, P.D., Wrubel, K.M., McCollum, D. and Gonzalez-Lima, F. (2002) Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neuroscience Letters, 332, 83- 86. doi:10.1016/S0304-3940(02)00827-3
- Lu, Y., Jiao, R., Chen, X., Zhong, J., Ji, J. and Shen, P. (2008) Methylene blue-mediated photodynamic therapy induces mitochondria-dependent apoptosis in HeLa cell. Journal of Cellular Biochemistry, 105, 1451-1460. doi:10.1002/jcb.21965
- Daudt 3rd, D.R., Mueller, B., Park, Y.H., Wen, Y. and Yorio, T. (2012) Methylene blue protects primary rat retinal ganglion cells from cellular senescence. Investigative Ophthalmology & Visual Science, 53, 4657-4667. doi:10.1167/iovs.12-9734
- Furian, A.F., Fighera, M.R., Oliveira, M.S., Ferreira, A.P., Fiorenza, N.G., de Carvalho Myskiw, J., Petry, J.C., Coelho, R.C., Mello, C.F. and Royes, L.F. (2007) Methylene blue prevents methylmalonate-induced seizures and oxidative damage in rat striatum. Neurochemistry International, 50, 164-171. doi:10.1016/j.neuint.2006.07.012
- Riha, P.D., Rojas, J.C. and Gonzalez-Lima, F. (2010) Beneficial network effects of methylene blue in an amnestic model. Neuroimage, 54, 2623-2634. doi:10.1016/j.neuroimage.2010.11.023
- Jung, M.E., Watson, D.G. and Simpkins, J.W. (2005) Suppression of protein kinase Cepsilon mediates 17betaestradiol-induced neuroprotection in an immortalized hippocampal cell line. Journal of Neurochemistry, 95, 745- 755. doi:10.1111/j.1471-4159.2005.03424.x
- Santos, M.S., Moreno, A.J. and Carvalho, A.P. (1996) Relationships between ATP depletion, membrane potential, and the release of neurotransmitters in rat nerve terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and ischemia. Stroke, 27, 941-950. doi:10.1161/01.STR.27.5.941
- Adam-Vizi, V. (2005) Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxidants & Redox Signaling, 7, 1140-1149. doi:10.1089/ars.2005.7.1140
- Dauer, W. and Przedborski, S. (2003) Parkinson’s disease: Mechanisms and models. Neuron, 39, 889-909. doi:10.1016/S0896-6273(03)00568-3
- Tabrizi, S.J., Cleeter, M.W., Xuereb, J., Taanman, J.W., Cooper, J.M. and Schapira, A.H. (1999) Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Annals of Neurology, 45, 25-32. doi:10.1002/1531-8249(199901)45:1<25::AID-ART6>3.0.CO;2-E
- de-Oliveira, R.W. and Guimaraes, F.S. (1999) Anxiolytic effect of methylene blue microinjected into the dorsal periaqueductal gray matter. Brazilian Journal of Medical and Biological Research, 32, 1529-1532. doi:10.1590/S0100-879X1999001200012
- Liu, C.Y., Xie, D.P. and Liu, J.Z. (2004) Microinjection of glutamate into dorsal motor nucleus of the vagus excites gallbladder motility through NMDA receptor—nitric oxide—cGMP pathway. Neurogastroenterology and Motility, 16, 347-353. doi:10.1111/j.1365-2982.2004.00525.x
- Poteet, E., Winters, A., Yan, L.J., Shufelt, K., Green, K.N., Simpkins, J.W., Wen, Y. and Yang, S.H. (2012) Neuroprotective actions of methylene blue and its derivatives. PLoS One, 7, e48279. doi:10.1371/journal.pone.0048279
- Sanchez-Prieto, J. and Gonzalez, P. (1988) Occurrence of a large Ca2+-independent release of glutamate during anoxia in isolated nerve terminals (synaptosomes). Journal of Neurochemistry, 50, 1322-1324. doi:10.1111/j.1471-4159.1988.tb10611.x
- Collange, O., Charles, A.L., Bouitbir, J., Chenard, M.P., Zoll, J., Diemunsch, P., Thaveau, F., Chakfe, N., Piquard, F. and Geny, B. (2012) Methylene blue protects liver oxidative capacity after gut ischaemia-reperfusion in the rat. European Journal of Vascular & Endovascular Surgery, 45, 168-175. doi:10.1016/j.ejvs.2012.11.011
- Mayer, B., Brunner, F. and Schmidt, K. (1993) Inhibition of nitric oxide synthesis by methylene blue. Biochemical Pharmacology, 45, 367-374. doi:10.1016/0006-2952(93)90072-5
- Patel, J.D., Krupka, T. and Anderson, J.M. (2007) iNOSmediated generation of reactive oxygen and nitrogen species by biomaterial-adherent neutrophils. Journal of Biomedical Materials Research Part A, 80, 381-390. doi:10.1002/jbm.a.30907
- Aksu, B., Umit, H., Kanter, M., Guzel, A., Aktas, C., Civelek, S. and Uzun, H. (2009) Effects of methylene blue in reducing cholestatic oxidative stress and hepatic damage after bile-duct ligation in rats. Acta Histochemica, 112, 259-269. doi:10.1016/j.acthis.2008.12.002
- Silva, D.F., Selfridge, J.E., Lu, J.E.L., Cardoso, S.M. and Swerdlow, R.H. (2012) Mitochondrial abnormalities in Alzheimer’s disease: Possible targets for therapeutic intervention. Advances in Pharmacology, 64, 83-126. doi:10.1016/B978-0-12-394816-8.00003-9