Advances in Bioscience and Biotechnology
Vol.1 No.2(2010), Article ID:2078,6 pages DOI:10.4236/abb.2010.12013
A new fibrinogenase from Echis multisquamatis venom is a perspective agent for limited proteolysis and defibrinogenation
![]()
1Kyiv National Shevchenko University, Kyiv, Ukraine;
2Palladin Institute of biochemistry, Kyiv, Ukraine.
Email: bio.cherv@gmail.com
Received 17 February 2010; revised 8 March 2010; accepted 24 March 2010.
Keywords: fibrinogenase; serine proteinase; snake venom; fibrinogen
ABSTRACT
A serine proteinase with a fibrinogenase activity was isolated from the venom of viper Echis multisquamatis. Isolation was performed by the combination of Q-sepharose and Heparine-agarose chromatography. The enzyme has apparent molecular weight 35 ± 1 kDа. It posesses strong fibrinogen β-chain, moderate α- chain proteolytic activity, arginine-amidase activity as the majority of serine fibrinogenases. The Km value was determined for β-chain fibrinogenolytic activity: Km = 8.3 µM. Kinetic parameters for amidase activity were also determined. Amino-acid composition was revealed. Limited hydrolysis of fibrinogen by the obtained fibrinogenase allowed us to detemine stable hydrolytic subproducts with definite molecular weights. The manner of the proteolytic processes suggests possible use of this fibrinogenase in probing fibrinogen structure dinamics by limited proteolysis. Applicability of the obtained fibrinogenase in therapeutic practice is speculative, but presented data about its nature are encouraging and require additional investigation.
1. INTRODUCTION
Snake venoms are mixtures of high-reactive enzymes which can influence the haemostasis components. The enzymes affect coagulation factors and proteins of endothelial cells’ and platelets’ membranes. They influence the system both non-selectively (through nonspecific hydrolysis) and selectively. The latter way is activation or inhibition of enzymes of coagulation, fibrinolysis and platelets activation. They also often are resistant to physiological inhibitors [1,2].
There is a group of fibrinogenases among snake venom enzymes. These direct-acting endopeptidases do not cleave off fibrinogen fibrinopeptides and therefore do not induce polymerization [3,4]. These enzymes are either serineor metalloproteinases.
They are classified according to their action on the α- or β-chains of fibrinogen. Fibrinogenases slow down blood coagulation, because truncated fibrinogen does not form as strong a fibrin clot as the native one. This action is more evident for α-fibrinogenases [5].
As a rule, α-fibrinogenases contain Zink and have molecular weights of 20-26 kDa on an average. Disulfide bonds are often found in their structures. Most of them are inhibited by blood serum proteins and have affinity both to fibrinogen and to stabilized fibrin [3,4].
On the other hand, serine proteinases are mostly β-fibrinogenases and cleave the α-chain but moderately (there are some α-specific ones, though) [6,7]. Apart from fibrinogenase and arginine-esterase activities common for all of serine fibrinogenases, some can degrade fibrin, too. Average molecular weights vary between 23-32 kDa. Serine fibrinogenases unlike metalloproteinases are more heat-stable and less dependent on pH changes. Most of them are glycoproteins [4,6,8].
There is extensive sequence homology between the fibrin(ogen)olytic serine proteinases and the plasminogen activators and thrombin-like venom serine proteinases. However they do not have thrombin-like activity, and β-chain hydrolysis takes part in different non-thrombin sites [4,8].
Serine proteinases, including fibrinogenolytic enzymes are very abundant in Viperidae venoms in which they may account for 20% of their total protein content [9]. The unique specifity of snake venom proteinases makes them potentially useful in research of fibrinogen-depletion [10,11] and limited proteolysis.
The aim of our work was to obtain and characterize the fibrinogenase from Echis multisquamatis venom.
2. MATERIALS AND METHODS
2.1. Materials
Q-sepharose, acrylamide, Bis, SDS, TEMED were purchased from GE Healthcare. Heparin-agarose type II was from Sigma. Chromogenic substrates were purchased from Chromogenix.
2.2. Chromatography
The crude venom was dissolved in 0.05 M Tris-HCl buffer of pH 8.9 and was applied onto an Q-sepharose column pre-equilibrated with the same buffer. The column volume was 3 ml, a flow rate of 2.55 ml/min/cm2. The solution was eluted with step gradient of NaCl, the fraction with fibrinogenase activity was taken at the ionic strength of 0.125 M (Figure 1). The active fraction was further purified by Heparin-agarose type IІ, (Sigmа) equilibrated with 0.05 M Tris-HCl buffer of pH 7.4, column volume 2 ml, flow rate of 1.27 ml/min/cm2. The fraction with fibrinogenase activity was taken at the ionic strength of 0.3 M NaCl (Figure 2). Three chroma-
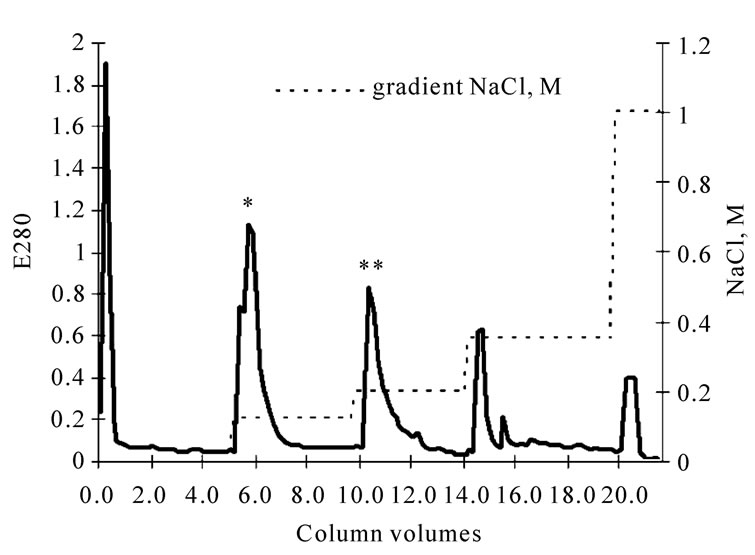
Figure 1. Chromatogram of fibrinogenase (*) and prothrombin activator (**) fraction of crude E. multisquamatis venom on Q-sepharose.
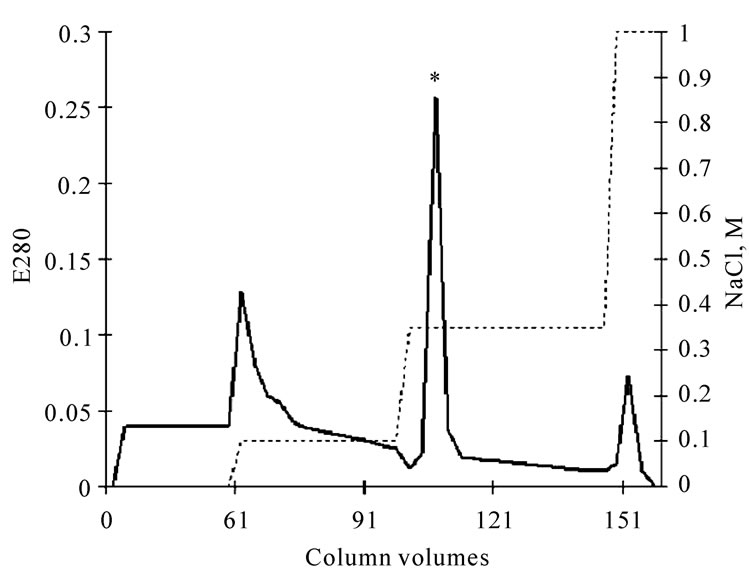
Figure 2. Chromatogram of fibrinogenase (*) of the fibrinogenase fraction of crude E.multisquamatis venom on Нeparinagarose type II.
tographycal cycles were required due to the volume obtained after Q-sepharose process.
2.3. Electrophoretic Analysis
Protein fractions were analysed in SDS-PAGE Laemli system [12], the proteins are identified using coomassie brilliant blue (R-250). Molecular weight markers “Fermentas” #0671 (170, 130, 100, 72, 55, 40, 33, 24 kDа).
2.4. Absorbance Coefficient Estimation
To determine the absorbance coefficient we estimated adsorption at 280 nm and carried on the Bradford analysis [13].
2.5. Amino-Acid Composition Analysis
Amino-acid composition was revealed using automatic amino-acid analyzer Т-339 “Mikrotekno”. Hydrolysis of protein was performed in the presence of 6 M NaCl, 120°C [14].
2.6. Amidase Activity Assay
Hydrolysis of chromogenic substrates (S2238 – H-D-PhePip-Arg-pNA, S2251 – D-Val-Leu-Lys-pNA, S2765 – Z-D-Arg-Gly-Arg-pNA, S236 – pyro-Glu-L-Pro-L-ArgpNA, S2302 – H-D-Pro-Phe-Arg-pNa) under the influence of fibrinogenase was studied using the reader (Titerteck-multiscan), E405 – E492. The analysis was done in 0.05M Tris-HCl buffer of pH 7.4 solution, 37°C. Chromogenic substrates concentration – 30 mM, that of fibrinogenase – 0.010 mg/ml.
2.7. Inhibitory Assay
The fibrinogenase was verified for inhibition by serine proteinases (DFF, PMSF, benzamidine), metalloproteinases (EDTA) and cystein proteinases (PCMB) inhibitors. PMSF – phenylmethylsulphonyl fluoride; DFP – Diisopropyl-fluorophosphate; PCMB – p-chloromercuribenzoate; EDTA – ethylenediaminetetraacetic acid. For the assay we used the thrombin-specific chromogenic substrate S2238 (H-D-Phe-Pip-Arg-pNA) using the reader (Titerteck-multiscan), E405 – E492. The analysis was done in 0.05M Tris-HCl buffer of pH 7.4 solution, 37°C. S2238 concentration was 40-120 mM, that of fibrinogenase – 0.010 mg/ml, and that of an inhibitor 0.8 mM.
2.8. Fibrinogenase Activity
1.5 mg/ml fibrinogen had been incubated with 0.050 mg/ml protein fractions in 0.05M Tris-HCl buffer of pH 7.4, 37°C for 10 minutes before being treated with 1 NIH/ml thrombin. The prolongation of clot formation time comparing to the control (native fibrinogen) was then determined.
2.9. Kinetic Parameters
To evaluate the Michaelis constants we studied hydrolysis of chromogenic substrates and native fibrinogen.
Hydrolysis of chromogenic substrates was studied using the reader (Titerteck-multiscan), E405 – E492. The analysis was done in 0.05 M Tris-HCl buffer of pH 7.4, 25°C. Chromogenic substrates concentration – 40-120 mM, that of fibrinogenase – 0.010 mg/ml.
Plasminogen-free fibrinogen (0.5-2.5 mg/ml) was incubated with the fibrinogenase (0.008 mg/ml) under similar conditions. The hydrolyzates were analyzed using SDS-PAGE Laemmli electrophoresis in the presence of 0.2% β-mercaptoethanol and quantitatively evaluated by Totallab TL100.
2.10 Fibrin(ogen) Fragments Obtaining
Obtaining of fibrin(ogen) degradation products was performed according to the method [15].
2.11. Fibrinogenase Treatment of Fibrin(ogen) Fragments
Fibrinogen fragments (1.3 mg/ml) were incubated with the fibrinogenase (0.08 mg/ml) during 60 minutes in 0.05 M Tris-HCl buffer of pH 7.4 solution, 25°C. Analyzed with electrophoresis as described above.
3. RESULTS
To achieve determined goal we fractionated crude venom, purified the fibrinogenase and analyzed it electrophoretically, determined substrate specificity and conducted an inhibitory assay.
Fractioning the Echis multisquamatis venom on Q-sepharose revealed two fractions of enzymes able to act on haemostasis components. The one eluted at ionic strength 0.2 M NaCl contained a prothrombin activator (Figures 1 and 2).
Therefore, 50 mg of crude venom yielded 2.5 mg of electrophoretically pure fibrinogenase. Its molecular weight is 35 ± 1 kDa. According to densitometry assay using Totallab TL100 (Figure 3) the content of the enzyme in crude venom was 5.83%, so the yield was 85.6%.
Amino-acid composition was revealed (Table 1). It was comparable with amino-acid compositions of some snake venom serine proteinases: bothrops protease A (β-fibrinogenase), shedaoenase (α-fibrinogenase) and afaасytin (αβ-fibrinogenase).
The enzyme was characterized under pH 7.4 as consistent with its evaluated pH optimum. It degraded chromogenic substrates. To qualitative determine the amidase activity of fibrinogenase we evaluated Michaelis constant for degradation of every substrate (Table 2). The affinities to kallikrein, plasmin, and thrombin chromogenic substrates highest exceeded affinities to others.
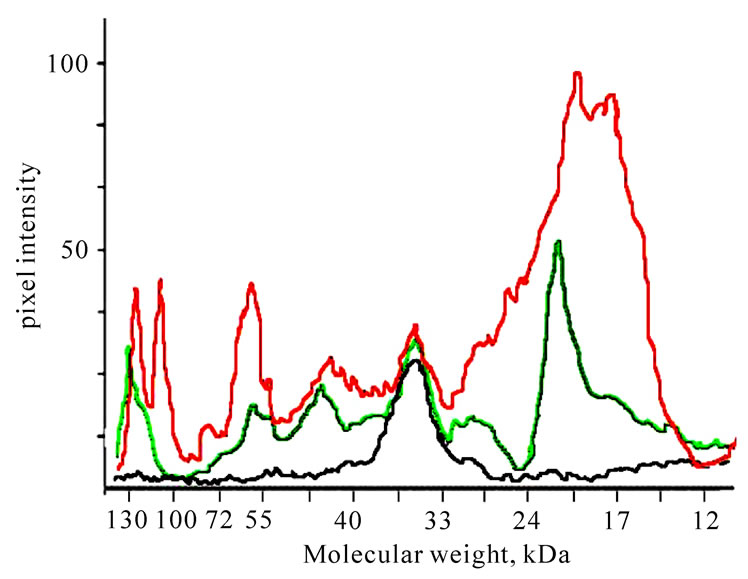
Figure 3. Densitogram of crude E. multisquamatis venom (red), fibrinogenase fraction after Q-sepharose (green) and purified fibrinogenase after Heparin-Sepharose (black), calculated in Totallab TL100.
Inhibitory assay was performed. Of the used inhibitors of different types of proteinases, PMSF, benzamidine and DFР had inhibitory effect on amidase activity of the enzyme. It means that it belongs to the serine proteinases. Degree of inhibitory effect was evaluated by Michaelis constant calculation (Table 3).
The target of proteolytic action of the fibrinogenase on fibrinogen was found to be the β-chain of the molecule that loses 3.3 ± 0.2 kDa of its weight at the early stages of hydrolysis (Figure 4) already, while the α-chain begins degradation after 40 minutes of incubation losing 9 kDa. Further hydrolysis results in more profound degradation.
To evaluate Michaelis constant we analysed the serie of electrophoregrams with densitometry soft Totallab TL100. Evaluated Michaelis constants for β-chain is 8.3 μM.
To localize the initial sites of β-chains cleavage we compared the abilities of fibrinogenase to hydrolyze fibrinogen, fibrin DD-fragment, fibrinogen E2- and D-fragments [18]. Studied fragments are representing different sequences of fibrinogen. D and DD – distal C-terminal parts of molecule, E2-fragment – central N terminal parts, except of Aα 1-17 Bβ 1-54 sequences [19]. The concentration of fibrinogenase was enough for profound Bβ-chain and initial α-chain degradation. Hydrolysis was not observed for all fragments (Figures 5(a), (b) and (c)) but was evident for fibrinogen (Figure 5(d)). Thus we concluded that target of initial fibrinogen proteolysis by fibrinogenase is contained in N-terminal part of fibrinogen Bβ-chain.
4. DISCUSSION
We purified a new fibrinogenase of the E. multisquamatis venom by the two-step chromatographical protocol. It is
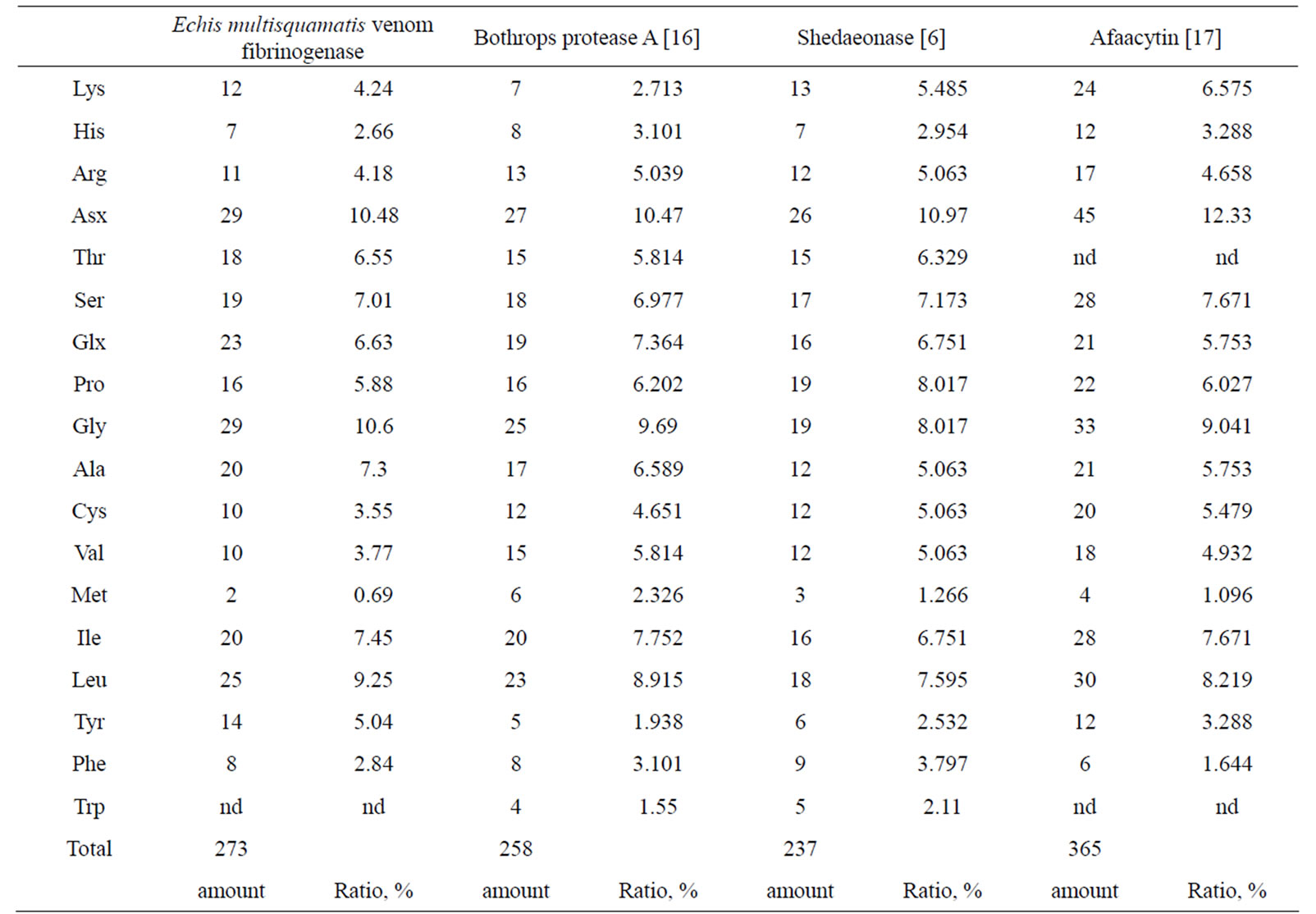
Table 1. Amino acid composition of fibrinogenase from the venom of Echis multisquamatis compared with that of other fibrinogenases.
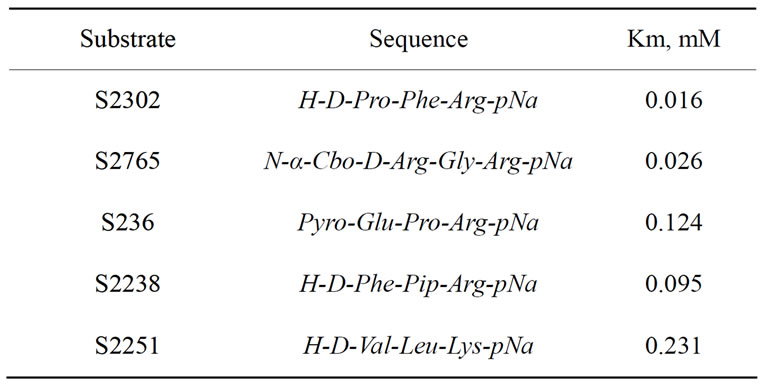
Table 2. Cinetics of amidase activity of E. multisquamatis venom fibrinogenase.
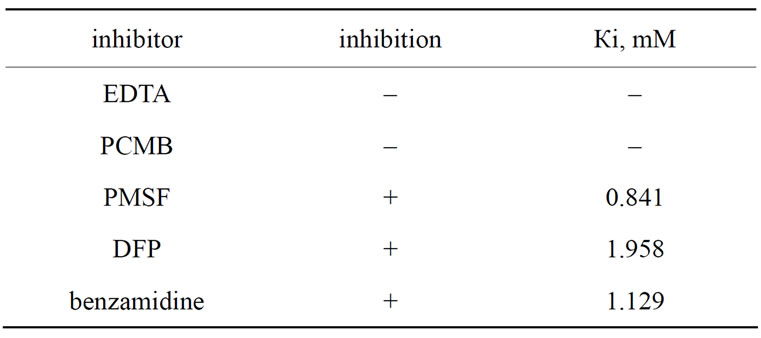
Table 3. The influence of some inhibitors on amidase activity of E. multisquamatis venom fibrinogenase.
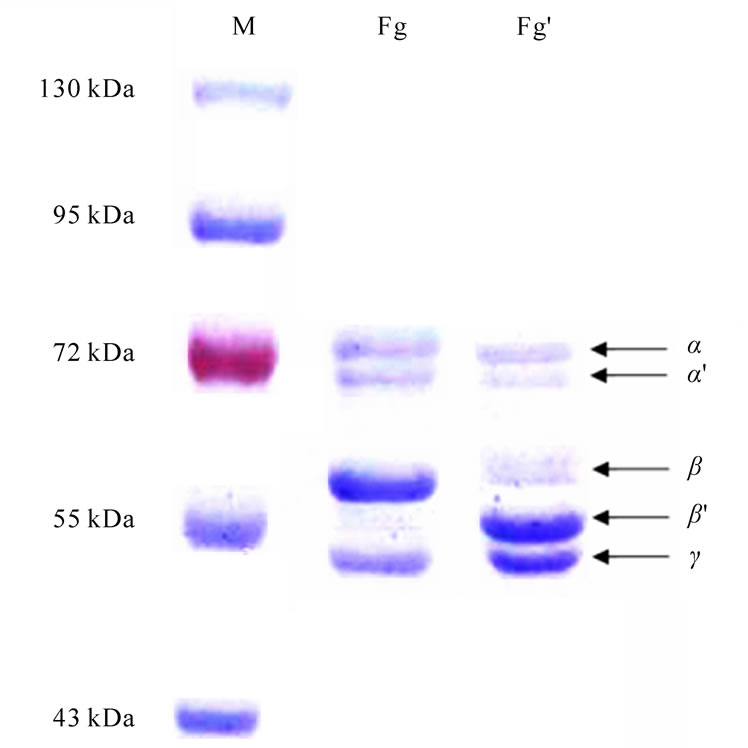
Figure 4. Electrophoregram of fibrinogen hydrolyzed by fibrinogenase from the venom of E.multisquamatis. Fg – native fibrinogen; Fg’ – early stages hydrolytic product (incubation 7,5 min). Samples were prepared in the presence of 0.2% β-mercaptoethanol. M – molecular weight markers.
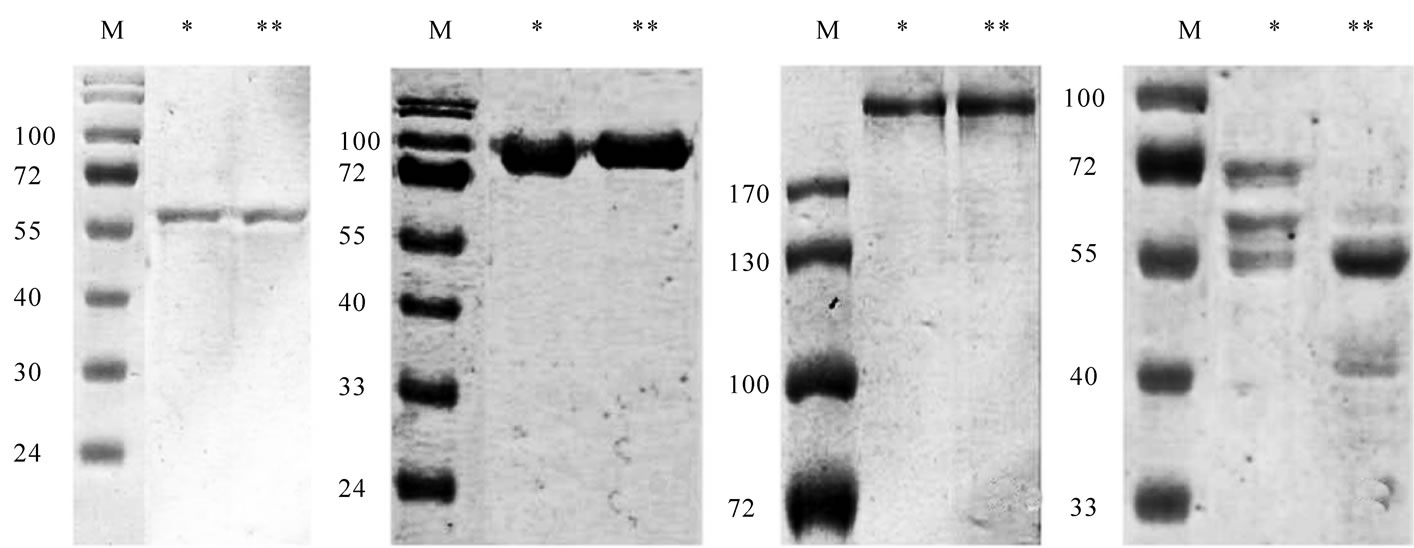 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 5. Electrophoregram of fibrinogen E2-fragment (a), D-fragment (b), fibrin DD-fragment (c) and fibrinogen (d) treated by fibrinogenase. * - before treatment, ** - after treatment. Samples D were prepared in the presence of 0,2% β-mercaptoethanol.
N-terminal specific serine β-fibrinogenase with molecular weight of 35 ± 1 kDa. It has amidase activity of a wide range and no fibrinolytic one (data not shown).
The course of fibrinogenolysis suggests its possible usefulness in limited proteolysis fibrinogen study. The limited proteolysis technique gives information about structure, conformation and irregular sections of proteins consistent with spectroscopic data. It also may be applicable to obtain fragments of proteins that would preserve to an extent both tertial structure and functional active sites of the native molecules [20-22].
According to the dynamics of hydrolysis, one can assume that fibrinogenolysis leads to conformational changes in the molecule which in turn reveal sites of further degradation [23]. The structure-functional properties of truncated fibrinogen are of interest for studies of fibrin polymerization and of all types protein-protein and proteinblood cells interaction concerning fibrin(ogen). The partly degraded form of fibrinogen can be a useful tool for study of fibrinogen structure and functions [24].
The properties of the fibrinogenase from the venom of E. multisquamatis suggest that it should reasonably be studied as a potential fibrinogen-depleting agent [25]. Hyperfibrinogenemia is known to be not only a symptom but also a cause of thrombotic complications in case of inflammatory processes and cardiovascular diseases [26]. Fibrinogen depletion through hydrolysis by a fibrinogenase may be promising for intravascular coagulation prevention [27].
It is known that serine fibrinogenases have no hemorrhagic activity, do not influence protein C and fibrinolytic systems and do not hydrolyze PAR-receptors. It is therefore possible that they can provide fibrinogen depletion without at least the known side effects [11].
A new fibrinogenase was characterized and peculiarities of its functional activity determined. The obtained results encourage further investigations that may contribute both to science and medicine.
REFERENCES
- Braud, S., Bon, C. and Wisner, A. (2000) Snake venom proteins acting on hemostasis. Biochimie, 82(9-10), 851- 859.
- Segers, W.H. and Ouyang, C. (1991) Snake venoms and blood coagulation. Snake venoms. In: Lee, C.-Y., Ed., Handbook of Experimental Pathology, 52, 703-711.
- Markland, F.S. (1988) Fibrin(ogen)olytic enzymes from snake venoms. In: Pirkle, H. and Markland, F.S., Jr., Ed., Haemostasis and Animal Venoms, 149-165.
- Swenson, S. and Markland, F.S. (2005) Snake venom fibrin(ogen)olytic enzymes. Toxicon, 45(8), 1021-1039.
- Kini, R.M. (2006) Anticoagulant proteins from snake venoms: Structure, function and mechanism. Biochemical Journal, 397(3), 377-387.
- Jiao, H.M., Yang, L.X., Lu, B., et al. (2005) Shedaoenase, a novel fibrinogenase from the venom of Agkistrodon shedaoenthesis Zhao. Acta Biochimica et Biophysica Sinica, 37(12), 835-842.
- Samel, M., Subban, J., Siigur, J. and Siigur, E. (2002) Biochemical characterization of fibrinogenolytic serine proteinases from vipera lebetina snake venom. Toxicon, 40(1), 51-54.
- Matsui, T., Sakurai, Y., Fujimura, Y., et al. (1998) Purification and amino acid sequence of halystase from snake venom of Agkistrodon halys blomhoffii, a serine protease that cleaves specifically fibrinogen and kininogen. European Journal of Biochemistry, 252(3), 569-575.
- Wisner, A., Braud, S. and Bon, C. (2001) Snake venom proteinases as tools in hemostasis studies: structure-function relationship of a plasminogen activator purified from Trimeresurus stejnegeri venom. Haemostasis, 31(3-6), 133- 140.
- Koh, D.C.I., Armugam, A. and Jeyaseelan, K. (2006) Snake venom components and their applications in biomedicine. Cellular and Molecular Life Sciences, 63(24), 3030-3041.
- Gardiner, E.E. and Andrews, R.K. (2008) The cut of the clot(h): Snake venom fibrinogenases as therapeutic agents. Journal of Thrombosis and Haemostasis, 6(8), 1360-1362.
- Laemli, R.V. (1970) Cleavage of structural poteteins during the assembly of the head of bacteriophage T4. Nature, 227(5259), 680-685.
- Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248-254.
- Speckmann, D., Stein, W. and Moore, S. (1958) Automatic recording apparatus for use in the chromatography of amino acids. Analytical Chemistry, 30, 1190-1196.
- Platonova, T.M., Chernyshenko, Т.М. and Sokolovska, L.I. (2006) Effect peculiarities of D-domain-containing specific inhibitors of polymerization on the fibrin self-assembly process. Ukrainian Biochemical Journal, 78(3), 124-129.
- Murayama, N., Saguchi, K. and Mentele, R. (2003) The unusual high molecular mass of Bothrops protease A, a trypsin-like serine peptidase from the venom of Bothrops jararaca, is due to its high carbohydrate content. Biochimica et Biophysica Acta (BBA) – Proteins & Proteomics, 1652(1), 1-6.
- Lababa-Djebari, F., Martin-Eauclaire, M.-F.I., Mauco, G. and Marchot, P. (1995) Afaасytin, an αβ-fibrinogenase from Cerastes cerastes (Horned Viper) venom, activates purified factor X and induces serotonin release from human blood platelets. European Journal of Biochemistry, 233, 756-765.
- Collen, D., Kudryk, B., Hessel, B. and Blomback, B. (1975) Primary structure of human fibrinogen and fibrin. Isolation and partial characterization of chains of fragment D. Journal of Biological Chemistry, 250(15), 5808-5817.
- Gãrdlund, B. (1977) Human fibrinogen amino acid sequence of fragment E and of adjacent structures in the Aα- and Bβ-chains. Thrombosis Research, 10(5), 689-702.
- Fontana, A., Fassina, G., Vita, C., et al. (1986) Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry, 25(8), 1847-1851.
- Fontana, A., de Laureto, P.P., Spolaore, B., et al. (2004) Probing protein structure by limited proteolysis. Acta Biochimica Polonica, 51(2), 299-321.
- Zappacosta, F., Pessi, A., Bianchi, E., et al. (1996) Probing the tertiary structure of proteins by limited proteolysis and mass spectrometry: the case of minibody. Protein Science, 5(5), 802-813.
- Chernyshenko, V.O. and Korolova D.S. (2008) Echis multi- squamatis venom enzymes acting on haemostasis. Bridges in Life Sciences Annual Scientific Review Meeting, Zagreb, 4 October 2008.
- Cooley, B.C. and Isermann, B.H. (2004) Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood, 103(5), 1728-1734.
- Chernyshenko, V.O. and Maksymovych, I.S. (2009) Echis multisquamatis venom fibrinogenase is a prospective fibrinogen-depletive agent. 1st World Conference on Physico-Chemical Methods in Drug Discovery and Development, Rovinj, 27 September-1 October 2009, 40.
- Siebenlist, K.R., DiOrio, J.P., Budzynski, A.Z. and Mosesson, M.W. (1990) The polymerization and thrombinbinding properties of des-(Bbeta 1-42)-fibrin. Journal of Biological Chemistry, 265(30), 18650-18655.
- Liu, M., Counsell, C., Zhao, X.L. and Wardlaw, J. (2005) Fibrinogen-depleting agents for acute ischemic stroke. Stroke, 36(1), 173-174.