Journal of Biomaterials and Nanobiotechnology
Vol.3 No.2(2012), Article ID:18929,14 pages DOI:10.4236/jbnb.2012.32020
Conjugation of Magnetite Nanoparticles with Melanogenesis Substrate, NPrCAP Provides Melanoma Targeted, in Situ Peptide Vaccine Immunotherapy through HSP Production by Chemo-Thermotherapy
![]()
1Institute of Dermatology & Cutaneous Sciences, Sapporo, Japan; 2Department of Dermatology, School of Medicine, Sapporo Medical University, Sapporo, Japan; 3Department of Pathology 1, School of Medicine, Sapporo Medical University, Sapporo, Japan; 4Department of Chemical Engineering, Faculty of Engineering, Kyushu University, Fukuoka, Japan; 5Department of Biotechnology, School of Engineering, Nagoya University, Nagoya, Japan; 6Department of Chemistry, Fujita Health University School of Health Sciences, Toyoake, Japan; 7Meito Sangyo Co., Nagoya, Japan; 8Faculty of Health and Welfare, Kawasaki University of Medical Welfare, Kurashiki, Japan; 9Department of Biological Chemistry, College of Bioscience and Biotechnology, Chubu University, Kasugai, Japan.
Email: jimbow@sapmed.ac.jp
Received January 9th, 2012; revised February 20th, 2012; accepted March 7th, 2012
Keywords: Melanogenesis; Chemo-Thermo-Immunotherapy; Nanomedicine; Melanoma; Magnetite Nanoparticles
ABSTRACT
In order to develop melanoma-targeted in situ peptide vaccine immunotherapy, magnetite nanoparticles were conjugated with a melanogenesis substrate, N-propionyl cysteaminylphenol (NPrCAP). Magnetite nanoparticles introduced thermotherapy which caused non-apoptotic cell death and generation of heat shock protein (HSP) upon exposure to alternating magnetic field (AMF). NPrCAP was expected to develop a melanoma-targeted therapeutic drug because of its selective incorporation into melanoma cells and production of highly reactive free radicals, that result in not only oxidative stress but also apoptotic cell death by reacting with tyrosinase.
1. Introduction
Management of metastatic melanoma is extremely difficult challenge for physicians and scientists. Currently only 10% with metastatic melanoma patients survive for five years because of the lack of effective therapies [1]. There is, therefore, an emerging need to develop innovative therapies for the control of advanced metastatic melanoma.
Intracellular hyperthermia using magnetite nanoparticles (10 - 100 nm-sized, Fe3O4) has been shown to be effective for treating cancers in not only primary but also metastatic lesions [2-4]. Incorporated magnetite nanoparticles generate heat within the cells after exposure to AMF due to hysteresis loss [5]. In this treatment, there is not only the heat-mediated cell death but also immune reaction due to the generation of HSPs [6-15]. HSP expression induced by hyperthermia has been shown to be involved in tumor immunity, providing the basis for developing a cancer thermo-immunotherapy.
Exploitation of biological properties unique to cancer cells may also provide a novel approach to overcome difficult challenge to the melanoma treatment. Melanogenesis is inherently cytotoxic and uniquely occurs in melanocytic cells; thus, tyrosine analogs that are tyrosinase substrates can be good candidates for developing drugs to melanoma-targeting therapies [16]. N-propionyl and N-acetyl derivatives of 4S-cysteaminylphenol (NPrand NAcCAP) were synthesized, and found to possess cytotoxic effects on in vivo and in vitro melanomas through the oxidative stress that derives from production of cytotoxic free radicals [17-21].
Based upon these rationales, we now provide evidence that melanoma-targeted in situ peptide vaccine immunotherapy has been successfully developed by conjugation of magnetite nanoparticles with a chemically modified melanogenesis substrate, and that a novel melanomatargeted chemo-thermo-immunotherapy (CTI Therapy) is provided for patients suffering from advanced metastatic melanoma.
2. Chemical Modification of Melanogenesis Substrate as a Potential Source for Development of Selective Drug Delivery System and Melanoma Toxicity
2.1. Melanogenesis Cascade in Melanoma Cells
The major advance of drug discovery for targeted therapy to cancer cells can be achieved by exploiting their unique biological property. The biological property unique to the melanocyte and melanoma cell resides the biosynthesis of melanin pigments within specific compartments, melanosomes. Melanogenesis begins with the conversion of amino acid, tyrosine to dopa and subsequently to dopaquinone in the presence of tyrosinase. This pathway is unique to all of melanocytes and melanoma cells including “amelanotic” melanoma. With the interaction of melanocyte-stimulating hormone (MSH)/ melanocortin 1 receptor (MC1R), the melanogenesis cascade begins from activation of microphthalmia transcription factor (MITF) for induction of either euor pheomelanin biosynthesis. Tyrosinase is the major player in this cascade. Tyrosinase is a glycoprotein and its glycosylation process is regulated by a number of molecular chaperons, including calnexin in the endoplasmic reticulum [22,23]. Vesicular transport then occurs to carry tyrosinase and its related proteins from trans-Golgi network to melanosomal compartments. In this process a significant number of transporters, such as small GTPbinding protein, adaptor proteins and PI3-kinase are involved, and early and late endosomes are closely associated with the production of these compartments. Once melanin biosynthesis is completed to conduct either euor pheomelanogenesis within melanosomal compartments, they will move along dendritic processes and transferred to surrounding keratinocytes in normal skin [24-26]. In metastatic melanoma cells, however, there will be no melanosome transfer inasmuch as there will be no receptor cells such as keratinocytes, and melanosomes synthesized are aggregated in autophagic vacuoles of melanoma cells. Thus a chemically modified melanogenesis substrate can be retained in melanoma cells once they are incorporated into their melanogenesis cascade, hence providing a unique drug delivery system (DDS) to melanoma cells.
2.2. Synthesis of Sulfur Analogs (Amine and Amide) of Tyrosine, Cysteaminylphenols and Their Melanocyte Toxicity
In order to utilize melanin biosynthesis pathway for developing cytocidal compounds in controlling melanoma growth, N-acetyl and N-propionyl derivatives of cysteaminylphenols (CAPs) have been synthesized [27,28] (Figures 1 and 2). These compounds are found to possess cytocidal effect on in vivo and in vitro melanocytes through the oxidative stress. For example, both NPrCAP and NAcCAP can selectively disintegrate follicular melanocytes after single or multiple ip administration to new-born or adult C57 black mice [3,29]. In the case of adult mice after repeated ip administration of NPrCAP, white follicles with 100% success can be seen at the site where hair follicles were plucked to stimulate new melanocyte growth and to activate tyrosinase synthesis. A single ip injection of NPrCAP into a new born mouse resulted in the development of silver follicles in the entire body coat. The selective disintegration of melanocytes can be seen as early as in 12 hr after a single ip administration. None of surrounding keratinocytes or fibroblasts showed such membrane degeneration and cell death.
A high, specific uptake of NAcCAP was seen by melanoma cell lines compared to non-melanoma cells. A melanoma-bearing mouse showed, on the whole body autoradiogram, the selective uptake and covalent binding of NAcCAP in melanoma tissues of lung and skin. The specific cytotoxicity of NPrCAP and NAcCAP was examined on various types of culture cells by MTT assay [30], showing that only melanocytic cells except HeLa possessed the low IC50. The cytotoxicity on DNA synthesis inhibition was time-dependent and irreversible on melanoma cells, but was transient on HeLa cells [21].
We also examined to what extent the melanoma growth can be blocked in both in vitro culture and in vivo lung metastasis assays by administration of NAcCAP combined with buthionine sulfoximine (BSO), which blocks the effect of anti-oxidants through reducing glutathione levels. There was a marked growth inhibition of cultured melanoma cells in the presence of BSO, indicating that the selective cytotoxicity by our NPrand NAc-CAP is mediated by the production of cytotoxic free radicals. The in vivo lung metastasis experiment also showed the decreased number of lung melanoma colonies [17]. The problem was, however, that a fairly large number of amelanotic melanoma lesions were seen to grow in the lung. NPrCAP has been developed and con-
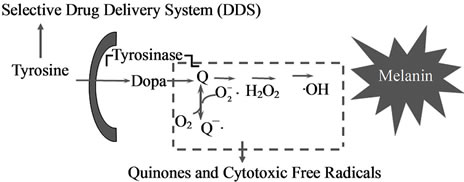
Figure 1. Exploitation of melanogenesis cascade for better management of melanoma.
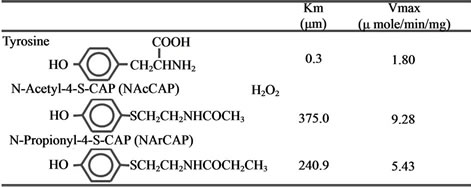
Figure 2. Synthesis and chemical structures of NAcCAP and NPrCAP and their tyrosinase kinetics.
jugated with magnetite nanoparticles in the hope of increasing the cytotoxicity and overcoming the problem.
3. Conjugation of Magnetite Nanoparticles with N-Propionyl 4S-Cysteaminylphenol, NPrCAP for Melanoma Targeted ChemoThermo-Immunotherapy
3.1. Synthesis for Conjugates of Magnetite Nanoparticles and NPrCAP, and Their Selective Aggregation in Melanosomal Compartments
Magnetite nanoparticles have been employed for thermotherapy in a number of cancer treatments including human gliomas and prostate cancers [31-33]. They consist of 10 - 100 nm-sized iron oxide (Fe3O4) with a surrounding polymer coating and become magnetized when placed in AMF [3]. We expected the combination of NPrCAP and magnetite nanoparticles to be a potential source for developing not only anti-melanoma pharmacologic but also immunogenic agent. It was expected that NPrCAP/magnetite nanoparticles complex could be selectively incorporated into melanoma cells because NPrCAP possesses a specific effect on DDS. The degradation of melanoma tissues occurs from oxidative and heat stresses by exposure of NPrCAP to tyrosinase and by exposure of magnetite nanoparticles to AMF. These two stress processes produce the synergistic or additive effect for generating tumor-infiltrating lymphocytes (TIL) that will kill melanoma cells in distant metastases (Figure 3).
We synthesized, in our initial study, the conjugates of NPrCAP with magnetite nanoparticles, NPrCAP/ML and NPrCAP/CML, in which NPrCAP was embedded in neutral and cationic liposomes respectively (Figure 4). There were, however, non-specific electrostatic interacttion between cationic magneto-liposomes (NPrCAP/ CML) and various non-target cells [33] as well as nonspecific aggregations in neutral magneto-liposomes (NPrCAP/ML). A promising technique is the use of tumor-targeted magnetite nanoparticles, and this approach was extended by synthesizing another type of magnetite nanoparticles, NPrCAP/M and NPrCAP/PEG/M, on which NPrCAP is superficially and directly bound on the surface of magnetite nanoparticles without using liposomes. They are chemically stable, can be produced in
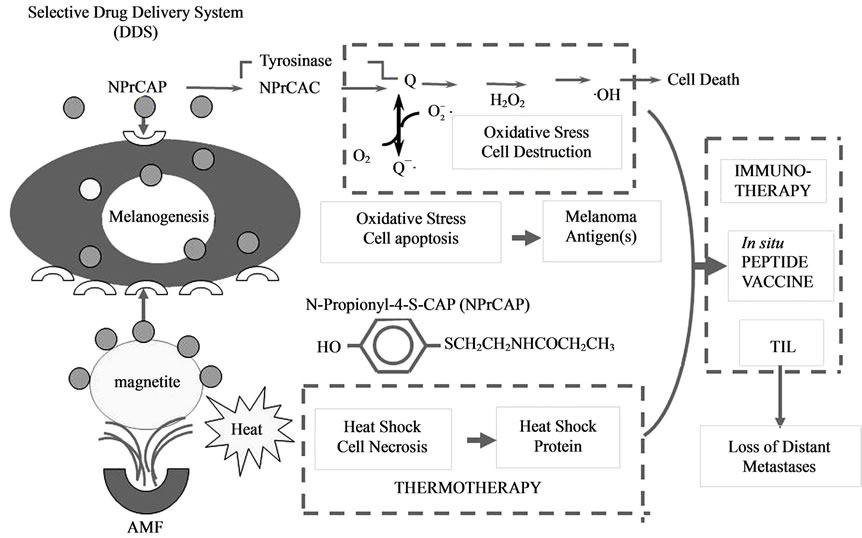
Figure 3. Strategy for melanogenesis-targeted CTI therapy by conjugates of NPrCAP and magnetite nanoparticles with AMF exposure.
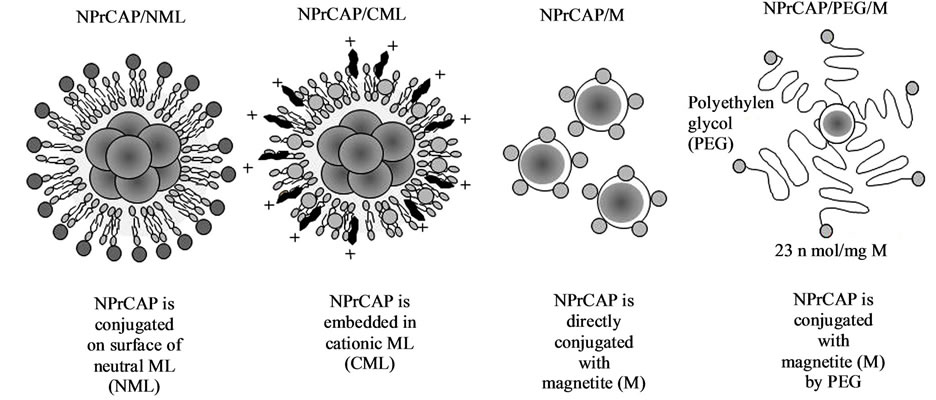
Figure 4. Conjugates of NPrCAP/magnetite nanoparticles for developing melanogenesis-targeted melanoma nanomedicine.
large quantities and may develop effective mela-nomaargeted chemotherapy (by NPrCAP) and thermo-immunotherapy (by magnetite nanoparticles with HSP), hence providing a basis for a novel CTI therapy. Most of the experiments described below were carried out by employing NPrCAP/M. A preliminary clinical trial, however, used NPrCAP/PEG/M to which polyethylene glycol (PEG) was employed to conjugate NPrCAP and magnetite nanoparticles.
In our studies, we could see that NPrCAP/M nanoparticle conjugates were selectively aggregated in melanoma cells compared to non-melanoma cells (Figure 5(a)). Specifically NPrCAP/M nanoparticles were found to be incorporated into and aggregated in melanosomal compartments after ip administration by electron microscopy (Figure 5(b)). After AMF exposure, there will be selective disintegration of melanoma tissues as can be seen by Berlin Blue staining [34,35] (Figure 5(c)). The conjugates of NPrCAP and magnetite nanoparticles will be selectively aggregated on the cell surface of melanoma cells through still unknown surface receptor and then incorporated into melanoma cells by early and late endosomes. The conjugates will then be incorporated into melanosomal compartment as the stage I melanosomes and late endosomes share common compartments, to which tyrosinase will be transported from trans-Golgi network by vesicular transport (Figure 6).
3.2. In Vivo Chemo-Thermo-Immunotherapy in Mouse Melanoma by Conjugates of Magnetite Nanoparticles and Melanogenesis Substrate, NPrCAP
In this study, we employed three cell lines of B16 melanoma, i.e., B16F1, B16F10 and B16-ovalbmine (OVA)
cells and compared the thermotherapeutic protocols in detail by evaluating the growth of the re-challenge melanoma as well as the duration and rates of survival of melanoma bearing mice (Figure 7).
By employing B16F1 and F10 cells, we first evaluated the chemotherapeutic effect of NPrCAP/M with or without AMF exposure which generates heat. NPrCAP/ M without heat inhibited growth of primary transplants to the same degree as did NPrCAP/M with heat, indicating that NPrCAP/M alone has a chemotherapeutic effect. However, there was a significant difference in the melanoma growth inhibition of re-challenge transplants between the groups of NPrCAP/M with and without heat. NPrCAP/M with AMF exposure showed the most significant growth inhibition in re-challenge melanoma and increased life span of the host animals, i.e., almost complete rejection of re-challenge melanoma growth whereas NPrCAP/M without heat was much less, i.e., 30% - 50%, indicating that NPrCAP/M with heat possesses a thermoimmunotherapeutic effect (Figures 8(a)-(c)).
Specifically our study indicated that the most effective thermo-immunotherapy for re-challenge B16 F1 and F10 melanoma cells can be obtained at a temperature of 43˚C for 30 min with the treatment repeated three times on every other day intervals without complete degradation of the primary melanoma (Figure 8(b)). Our therapeutic conditions and their effects differ from those of magnetically mediated hyperthermia on the transplanted melanomas reported previously by Suzuki et al. [36]. Cationic magneto-liposomes-mediated hyperthermia for B16 melanoma showed that hyperthermia at 46˚C once or twice led to regression of 40% - 90% of primary tumors and to 30% - 60% survival of mice, whereas their hyperthermia at 43˚C failed to induce regression of the secondary tumors with 0% survival of mice [36].
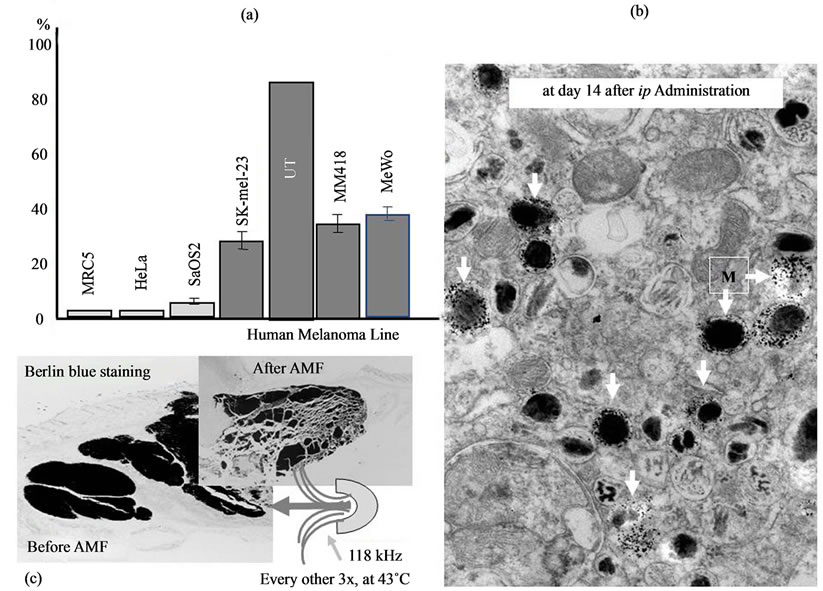
Figure 5. Selective aggregation of NPrCAP/M nanoparticles in human melanoma cell lines (a), their selective accumulation in melanosomal compartments of melanoma cells as can be seen in electron micrograph (see arrows) (b) and selective disintegration of melanoma tissues after exposure to AMF as can be seen in Berlin Blue iron staining (c).
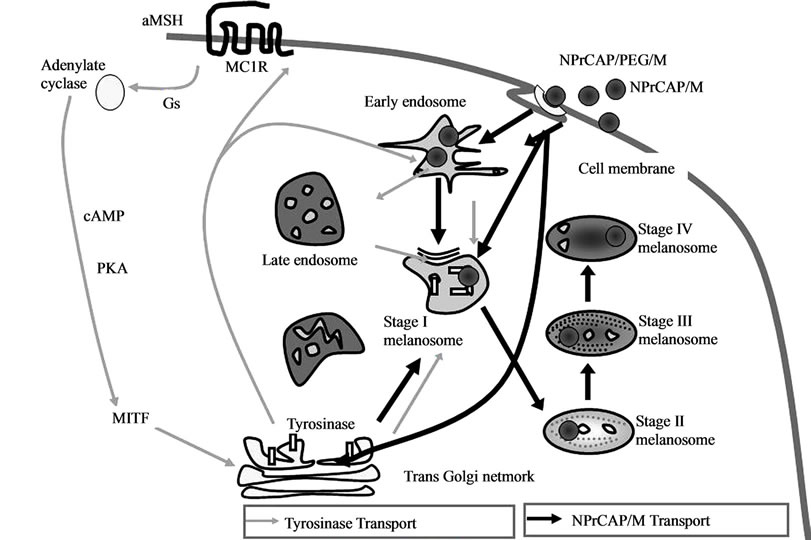
Figure 6. Selective accumulation of NPrCAP/M in melanoma cells. Conjugates of NPrCAP and magnetite nanoparticles (NPrCAP/M and NPrCAP/PEG/M) are aggregated on the cell surface of melanoma cells and incorporated into melanosomal compartments through early and late endosomes inasmuch as the stage I melanosomes derive from the specific late endosomes, to which tyrosinase will be transported from trans-Golgi network by vesicular transport.
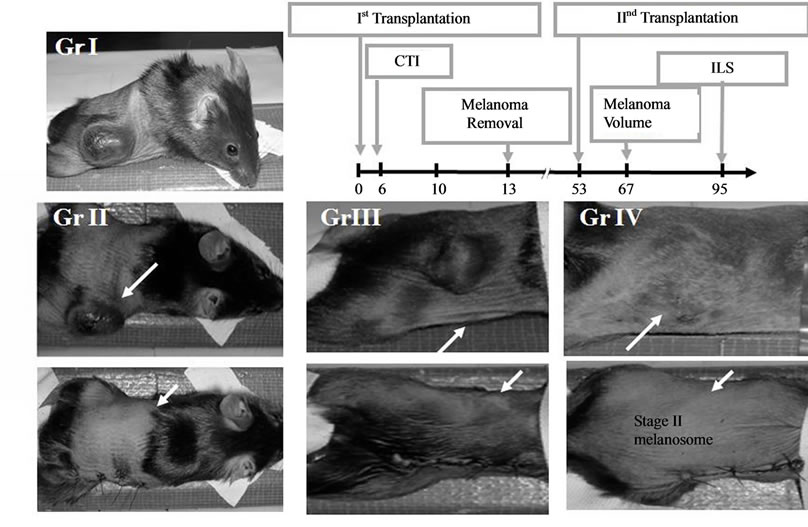
Figure 7. Growth inhibition of melanoma cells by primary first transplant and secondary re-challenge transplant in C57 black mice. Gr.I; control without NPrCAP/M treatment and second re-challnege melanoma transplant. Gr.II: control without NPrCAP/M treatment, but received second re-challenge of melanoma; Gr.III: control with NPrCAP/M treatment without AMF exposure, but received second re-challenge melanoma transplant. Gr.IV: mice with NPrCAP/M and AMF exposure as well as second rechallenge melanoma transplant. After removal of the first melanoma transplant, all mice (except Gr.I) received the second re-challenge melanoma transplant on the opposite site of trunk.
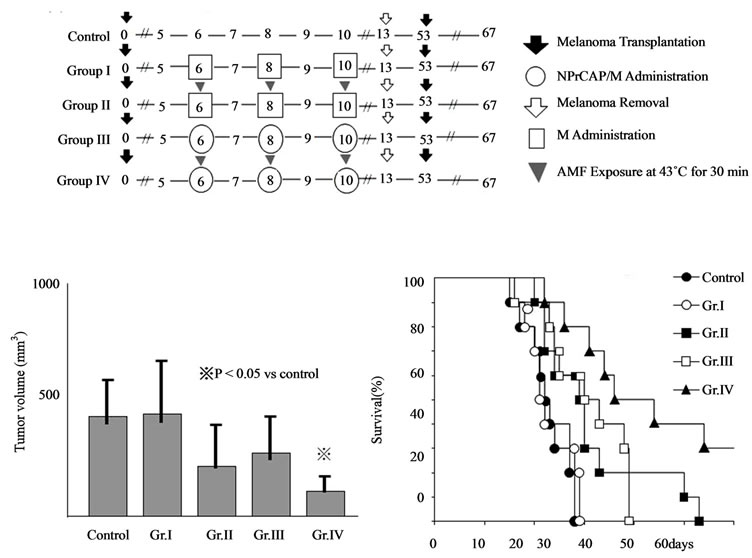
Figure 8. Melanoma growth and survival of melanomabearing mice by CTI therapy using NPrCAP/M with and without AMF exposure. (a) Protocols of Group I, II, III and IV of experimental mice; (b) Tumor volumes of re-challenge B16F1 melanoma on day 14; (c) Kaplan-Meier survival after tumor re-challenge.
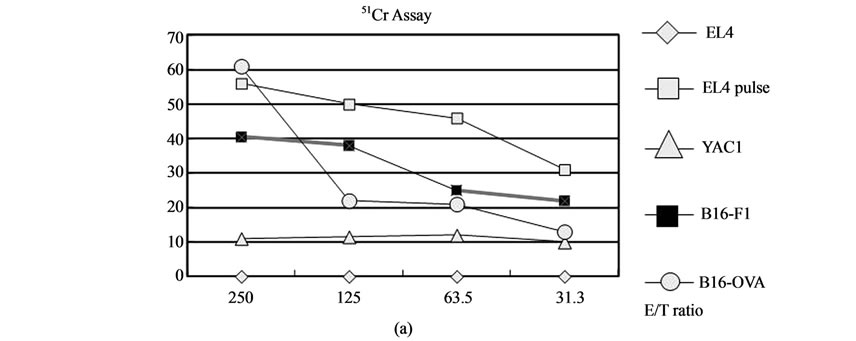
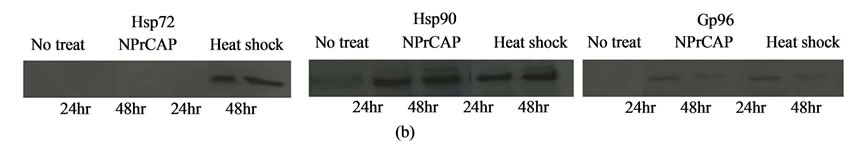
Figure 9. Hyperthermia of melanoma cells using B16-OVA cells for induction of CTL (a) and generation of HSPs (b) in CTI therapy. (a) Cytotoxic activity of spleen cells after CTI therapy against B16-OVA cells, B16F1 cells, EL4 cells, EL4 cells pulsed with SL8 peptide or YAC-1 cells was determined by standard 51Cr-release assay. B16-OVA cells were subjected to hyperthermia using NPrCAP/M with AMF exposure in vitro. (b)The expression of Hsp72, Hsp90 and Gp96 was determined by western blotting with an antiHsp72 mAb, anti-Hsp90 mAb or anti KDEL mAb.
3.3. Production of Heat Shock Protein by Chemothermo-Immunotherapy Using NPrCAP/Magnetite Nanoparticle Conjugates
It has been reported that the intracellular hyperthermia using magnetic nanoparticles is effective for treating certain types of cancer in not only primary but also metastatic lesions [37-43]. Incorporated magnetic nanoparticles generate heat within the cells after exposure to the AMF due to hysteresis loss or relaxational loss [44,45]. Hyperthermic treatment using CMLs, which are cationic liposomes containing 10 nm magnetite nanoparticles, induced antitumor immunity by enhancement of HSP expression [38,46-48].
In our animal study, those animals bearing B16F1 and B16F10 melanoma cells showed, to certain degree, rejecttion of second re-challenge melanoma transplantation by administration of both NPrCAP alone and NPrCAP/M minus AMF exposure [49]. Our working hypothesis for this finding is that there is a difference in the cytotoxic mechanism and immunogenic property of NPrCAP/M between experimental groups with and without AMF exposure. The animals with NPrCAP/M without AMF exposure resulted in non-necrotic, apoptotic cell death. The animals with NPrCAP/M plus AMF exposure, on the other hand, resulted in non-apoptotic, necrotic cell death with immune complex production of melanoma peptide as well as Hsp70 and a small amount of Hsp 90. The latter group of NPrCAP/M plus AMF exposure showed the most significant growth inhibition of the re-challenged melanoma growth which resulted in the almost complete survival of the host animals as long as for 3 months that we have conducted our experimental protocol [34]. In the latter study [34], we also found that repeated hyperthermia (3 cycles of NPrCAP/M administration and AMF irradiation) was required to induce the maximal antitumor immune response.
In the study using B16-OVA cells, the hyperthermia of melanoma cells using NPrCAP/M with AMF exposure further showed antitumor immune responses via crosspresentation of HSP-chaperoned antigen (Figures 9(a), 9(b), 10(a)-(d)). Moreover, the HSPs-antigen peptide complex released from melanoma cells treated with this intracellular hyperthermia was taken-up by dendritic cells (DCs) and cross-presented HSP-chaperoned peptide in the context of MHC class I molecules [49]. As stated above, our CTI therapy with AMF exposure induced NPrCAPas well as heat-mediated melanoma cell necrosis to NPrCAP/M incorporated cells. If melanoma cells escaped from necrotic cell death, repeated hyperthermia should produce necrotic cell death of previously heat shockedmelanoma cells in which HSPs were induced. In addition, our data of CTI therapy with AMF exposure using B16- OVA cells suggested that Hsp72/Hsc73, Hsp90, and ERresident HSPs participated in the induction of CD8+ T
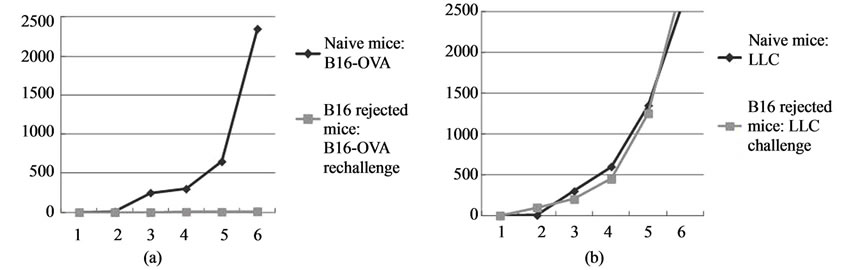
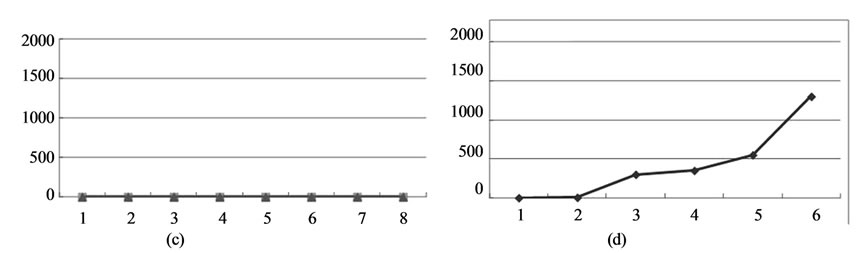
Figure 10. Tumor-specific immunity induced by CTI therapy. Naïve mice or mice cured by CTI therapy were rechallenged with B16-OVA melanoma cells (a) or lung carcinoma LLC (b). Failure of the melanoma growth by secondary re-challenge after CTI therapy to the primary melanoma transplant (a). There was no growth inhibition of LLC cells in mice which received transplantation of B16-OVA melanoma cells and received CTI therapy (b). Antitumor immunity is dependent on CD8+ T cells. Mice cured by CTI therapy were depleted of CD4+ or CD8+ T cells by an intraperitoneal injection of rat IgG (c) or anti-CD4 mAb (c) or anti-CD8 mAb (d). The growth inhibition of secondary re-challenge melanoma transplant failed after pretreatment with anti-CD8 mAb, but not by anti-CD4 mAb.
cell response. In particular, among HSPs, Hsp72 was largely responsible for the augmented antigen presentation to CD8+ T cells. As Hsp72 is known to up-regulate in response to hyperthermia or heat shock treatment [46], newly synthesized Hsp72 has a chance to bind to the heat-denatured melanoma-associated antigen.
3.4. Melanocytotoxic and Immunogenic Properties of NPrCAP Compared to Hydroquinone
Monobenzyl ether of hydroquinone has long been known to produce the skin depigmentation at both drug-applied area by direct chemical reaction with tyrosinase and non-applied distant area by immune reaction with still unknown mechanism [50-53]. The melanogenesis-related cytotoxicity primarily derives from tyrosinase-mediated formation of dopaquinone and other quinone intermediates, which produce reactive oxygen species such as superoxide and H2O2 [19,54-56]. This unique biological property of melanin intermediates not only causes cell death, but also may produce immunogenic properties. We postulated that the cytotoxic action of NAcCAP and NPrCAP appears to involve two major biological processes. One is cytostatic process which derives from the DNA synthesis inhibition through the interaction of quinone and free radicals with SH-enzymes and thymidine synthase. Another is the cytocidal process by damage of DNA and mitochondrial ATP through oxidative stress and interaction with SH-enzyme [21]. They bind protein disulphide isomerise [57]. Although we have not yet studied which one of these two processes is responsible for the immune reaction, it is likely that the cyotocidal process of NPrCAP is involved in the induction of immune reaction.
Monobenzy ether of hydroquinone was shown to produce a reactive ortho-quinone generated by tyrosinasecatalyzed oxidation and self-coupling and thiol conjugation reactions [58]. It was also shown to induce cell death without activating the caspase cascade or DNA fragmentation, indicating that the death pathway is nonapoptotic [58,59]. It was further reported that monobenzyl ether of hydroquinone induced the immunogenicity to melanocytes and melanoma cells by forming quinonehaptens to tyrosinase protein and by inducing the release of tyrosinaseand melanoma antigen recognized by T cells-1 (MART-1) containing CD63+ exosomes following melanosome oxidative stress induction. The drug further augmented the processing and shedding of melanocyte differentiation antigens by inducing melanosome autophagy and enhanced tyrosinase ubiquitination, ultimately activating DCs, which induced cytotoxic human melanoma-reactive cells. These T cells eradicated melanoma in vivo [59]. It is necessary to examine if NPrCAP will also take an immune-biological process similar to that reported in the case of monobenzyl ether of hydroquinone.
4. Discussion
The immune system can respond to cancer cells in two ways: by reacting against tumor-specific antigens (molecules that are unique to cancer cells) or against tumorassociated antigens (molecules that are expressed differentially by cancer cells and normal cells) [60]. The immunotherapy for cancer cells is further subdivided into immunotherapy with antibodies and T cells, therapeutic cancer vaccines, therapeutic vaccines combined with chemotherapy and immunoprevention of cancer cells. Several clinical trials using melanoma peptides or an antibody that blocks cytotoxic T-lymphocyte-associated antigen on lymphocytes have been shown to improve overall melanoma survival [61-63]. Exploitation of a specific biological property to cancer cells may, however, be another approach for developing novel cancer-targeted drugs. Promising oncogene-targeted melanoma therapy has successfully introduced recently [64].
Hyperthermia increases the expression of intracellular HSPs which is important in and necessary for the induction of antitumor immunity [46,65]. Over expression of HSPs, such as Hsp 70, increases tumor immunogenicity by augmenting the chaperoning ability of antigenic peptides and presentation of antigenic peptides in MHC class I molecules [66,67]. In this process professional antigen presenting DCs play unique and important roles in taking up, processing and presenting exogenous antigens in association with MHC class I molecules. Our working hypothesis for induction of in situ vaccination immunotherapy is that CTI therapy causes degradation of melanoma tissues which results in the release of HSP/melanoma antigen complex. This complex is taken up by professional antigen-presenting DCs through HSP receptor (Figure 11).
Based upon these animal experiments, a preliminary human clinical trial has been carried out by employing NPrCAP/PEG/M plus AMF after we obtained the approval of our human clinical trial for a limited number of stage III and IV melanoma patients (Clinical Trial Research No. 18-67, Sapporo Medical University). The therapeutic protocol followed the basically identical experimental schedule as that of animal experiments. In the clinical trials, however, we utilized NPrCAP/PEG/M which was made by conjugating polyethylene glycol between NPrCAP and magnetite nanoparticles. Among
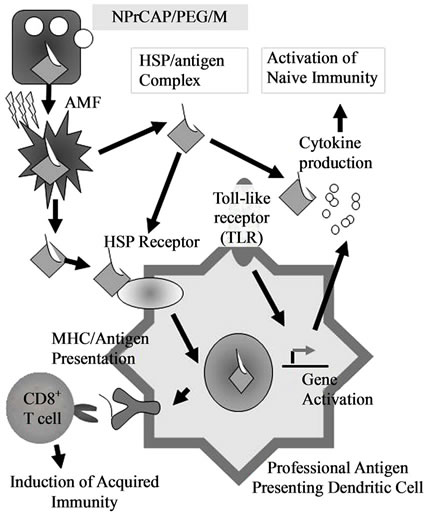
Figure 11. Scheme of intracellular hyperthermia using NPrCAP/PEG/M with AMF exposure. NPrCAP/PEG/M nanoparticles are selectively incorporated into melanoma cells. Intracellular hyperthermia can induce necrotic cell death and adjacent live melanoma cells suffer heat shock, resulting in increased level of intracellular HSP-peptide complexes. Repeated hyperthermia turns heat-shocked cells to necrotic cells, leading to the release of HSPs-peptide complexes into extracellular milieu. The released HSPspeptide complexes are taken-up by DCs. Then, DCs migrate into regional lymph nodes and cross-present HSP chaperoned antigenic peptides to CD8+ T cells in the context of MHC class I molecules, thereby inducing anti-melanoma cytotoxic CD8+ T cells.
two of four patients showed complete and partial responses to our treatment and have been able to carry out normal daily activities after CTI therapy. In one patient, for example, four distant cutaneous metastasis sites were evaluated and either significant regression or shrinkage of all of these four melanoma lesions was seen. The patient was able to survive 30 months after several trials of CTI therapy. The pathological and immunological specimens revealed dense aggregation of lymphocytes and macrophages at the site of CTI therapy. Importantly there was a trend to have an almost identical distribution of CD8+ T cells and MHC class 1 positive cells. Another patient had many lymph node metastases, but still has been surviving more than 32 months. In order to evaluate the overall therapeutic effect to advanced melanoma, it is important to have larger-scaled clinical trials and define concisely the molecular interaction between chemotherapeutic and thermo-immunotherapeutic effect in our CTI therapy.
5. Summary and Conclusion
In this study, we examined to what extent the conjugates of magnetite nanoparticles and melanogenesis substrate can generate melanoma-targeted vaccines through the chemotherapeutic and thermotherapeutic effects on primary transplant of B16 mouse melanoma cells with and without AMF exposure (heat generation). Specifically we evaluated the immunotherapeutic effect on the second, re-challenge transplant of the same melanoma cells to see if the growth of distant metastatic melanomas can be inhibited. We also investigated the possible association of HSP production, CD8+ T cell activation and MHC expression along with rejection of the re-challenge melanoma.
Our approach using melanogenesis substrate and magnetite nanoparticles is based upon the expectation that the combination of 1) direct killing of melanoma cells by chemotherapeutic and thermo-therapeutic effect of melanogenesis-targeted drug (NPrCAP/M) and 2) indirect killing by immune reaction (in situ peptide vaccine) after exposure to AMF. It is hoped from these rationales that a tumor-specific DDS is developed by NPrCAP and selective cell death can be achieved by exposure of conjugates of NPrCAP/M nanoparticles to AMF, which then can induce HSP expression through either necrotic or nonnecrotic process or combination of the two, without damaging non-cancerous tissues. Finally a novel immunotherapy targeted to metastatic melanoma lesions is achieved through “in situ peptide vaccine”.
6. Acknowledgements
This work was supported by a Health and Labor Sciences Research Grant-in-Aid (H21-Nano-006) for Research on Advanced Medical Technology from the Ministry of Health, Labor and Welfare of Japan.
We also express our sincere appreciation to Drs. Akiko Sato, Makito Sato, Yasue Ishii-Osai and Tomoaki Takada who devoted to carry out basic experiments and clinical trials of this study.
REFERENCES
- C. M. Balch, A. C. Buzaid, S. J. Soong, M. B. Atkins, N. Cascinelli, D. G. Coit, I. D. Fleming, J. E. Gershenwald, A. Houghton Jr., J. M. Kirkwood, K. M. McMasters, M. F. Mihm, D. L. Morton, D. S. Reintgen, M. I. Ross, A. Sober, J. A. Thompson and J. F. Thompson, “Final version of the American Joint Committee on Cancer staging system for Cutaneous Melanoma,” Journal of Clinical Oncology, Vol. 19, No. 16, 2001, pp. 3635-3648.
- A. Ito, M. Shinkai, H. Honda and T. Kobayashi, “Medical application of Functionalized Magnetic Nanoparticles,” Journal of Bioscience and Bioengineering, Vol. 100, No. 1, 2005, pp. 1-11. doi:10.1263/jbb.100.1
- N. Kawai, A. Ito, Y. Nakahara, M. Futakuchi, T. Shirai, H. Honda, T. Kobayashi and K. Kohri, “Anticancer effect of hyperthermia on Prostate Cancer Mediated by Magnetite Cationic Liposomes and Immune-Response Induction in Transplanted Syngeneic Rats,” The Prostate, Vol. 64, No. 4, 2005, pp. 373-381. doi:10.1002/pros.20253
- M. Yanase, M. Shinkai, H. Honda, T. Wakabayashi and J. Yoshida, T Kobayashi, “Antitumor Immunity Induction by Intracellular Hyperthermia Using Magnetite Cationic Liposomes,” Cancer Science, Vol. 89, No. 7, 1998, pp. 775-782. doi:10.1111/j.1349-7006.1998.tb03283.x
- M. Shinkai, M. Yanase, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Intracellular hyperthermia for Cancer Using Magnetite Cationic Liposomes: in vitro study,” Cancer Science, Vol. 87, No. 11, 1996, pp. 1179- 1183. doi:10.1111/j.1349-7006.1996.tb03129.x
- A. Ménoret and R. Chandawarkar, “Heat-shock Protein-Based Anticancer Immunotherapy: An Idea Whose Time Has Come,” Seminars in Oncology, Vol. 25, No. 6, 1998, pp. 654-660.
- P. K. Srivastava, A. Ménoret, S. Basu, R. Binder and K. Quade, “Heat Shock Proteins Come of age: Primitive Functions Acquired New Roles in an Adaptive World,” Immunity, Vol. 8, No. 6, 1998, pp. 657-665. doi:10.1016/S1074-7613(00)80570-1
- Y. Tamura, N. Tsuboi, N. Sato and K. Kikuchi, “70 kDa Heat Shock Cognate Protein Is a Transformation-Associated Antigen and a Possible Target for the host’s AntiTumor Immunity,” The Journal of Immunology, Vol. 151, No. 10, 1993, pp. 5516-5524.
- Y. Tamura, P. Peng, K. Liu, M. Daou and P. K. Srivastava, “Immunotherapy of tumors with Autologous TumorDerived Heat Shock Protein Preparations,” Science, Vol. 278, No. 5335, 1997, pp. 117-120. doi:10.1126/science.278.5335.117
- Y. Tamura and N. Sato, “Heat Shock Proteins: Chaperoning of innate and Adaptive Immunities,” Japanese Journal of Hyperthermic Oncology, Vol. 19, No. 3, 2003, pp. 131-139.
- P. K. Srivastava, “Immunotherapy for Human Cancer Using Heat Shock Protein-Peptide Complexes,” Current Oncology Reports, Vol. 7, No. 2, 2005, pp. 104-108. doi:10.1007/s11912-005-0035-8
- Y. Tamura, S. Takashima, J. M. Cho, W. Qi, K. Kamiguchi, T. Torigoe, S. Takahashi, I. Hirai, N. Sato and K. Kikuchi, “Inhibition of Natural Killer Cell Cytotoxicity by Cell Growth-Related Molecules,” Cancer Science, Vol. 87, No. 6, 1996, pp. 623-630. doi:10.1111/j.1349-7006.1996.tb00269.x
- G. Ueda, Y. Tamura, I. Hirai, K. Kamiguchi, S. Ichimiya, T. Torigoe, H. Hiratsuka, H. Sunakawa and N. Sato, “Tumor-Derived Heat Shock Protein 70-Pulsed Dendritic Cells Elicit Tumor-Specific Cytotoxic T Lymphocytes (Ctls) and Tumor Immunity,” Cancer Science, Vol. 95, No. 1, 2004, pp. 248-253. doi:10.1111/j.1349-7006.2004.tb02211.x
- A. Ito, H. Honda and T. Kobayashi, “Cancer immunotherapy based on Intracellular Hyperthermia Using Magnetite Nanoparticles: A Novel Concept of ‘Heat-ConTrolled Necrosis’ with Heat Shock Protein Expression,” Cancer Immunology, Immunotherapy, Vol. 55, No. 3, 2006, pp. 320-328. doi:10.1007/s00262-005-0049-y
- H. Shi, T. Cao, J. E. Connolly, L. Monnet, L. Bennett, S. Chapel, C. Bagnis, P. Mannoni, J. Davoust, A. K. Palucka and J. Banchereau, “Hyperthermia enhances CTL CrossPriming,” The Journal of Immunology, Vol. 176, No. 4, 2006, pp. 2134-2141.
- K. Jimbow, T. Iwashina, F. Alena, K. Yamada, J. Pankovich and T. Umemura, “Exploitation of Pigment Biosynthesis Pathway as a Selective Chemotherapeutic Approach for Malignant Melanoma,” Journal of Investigative Dermatology, Vol. 100, No. S2, 1993, pp. s231- S238.
- F. Alena, T. Ishikawa, A. Gili and K. Jimbow, “Selective in Vivo Accumulation of N-acetyl-4-S-cysteaminylphenol in B16F10 Murine Melanoma and enhancement of its in vitro and in Vivo Antimelanoma Effect by combination of Buthionine Sulfoximine,” Cancer Research, Vol. 54, No. 10, 1994, pp. 2661-2666.
- J. M. Pankovich and K. Jimbow, “Tyrosine transport in a Human Melanoma Cell Line as a basis for selective transport of Cytotoxic Analogues,” Biochemical Journal, Vol. 280, 1991, pp. 721-725.
- K. Reszka and K. Jimbow, “Electron donor and acceptor properties of Melanin Pigments in the skin,” In: J. Fuchs and L. Packer, Eds., Oxidative Stress in Dermatology, Marcel Dekker Inc., New York, 1993. pp. 287-320.
- M. Tandon, P. D. Thomas, M. Shokravi, S. Singh, S. Samra, D. Chang and K. Jimbow, “Synthesis and antitumour effect of the Melanogenesis-Based Anti-Melanoma Agent N-Propionyl-4-S-Cysteaminylphenol,” Biochemical Pharmacology, Vol. 15, No. 12, 1998, pp. 2023- 2029. doi:10.1016/S0006-2952(98)00090-2
- P. D. Thomas, H. Kishi, H. Cao, M. Ota, T. Yamashita, S. Singh and K. Jimbow, “Selective incorporation and specific Cytocidal Effect as the Cellular Basis for the antimelanoma action of Sulphur Containing Tyrosine Analogs,” Journal of Investigative Dermatology, Vol. 113, 1999, pp. 928-934. doi:10.1046/j.1523-1747.1999.00781.x
- J. Dakour, M. Vinayagamoorthy, H. Chen, D. Luo, W. Dixon and K. Jimbow, “Identification of A cDNA for Ca2+-binding Calnexin-like phosphoprotein (p90) on melanosomes in normal and Malignant Human Melanocytes,” Experimental Cell Research, Vol. 209, No. 2, 1993, pp. 288-300. doi:10.1006/excr.1993.1313
- K. Toyofuku, I. Wada, K. Hirosaki, J. S. Park, Y. Hori and K. Jimbow, “Involvement of calnexin in maturation of tyrosinase as a Molecular Chaperone,” Journal of Biochemistry, Vol. 125, 1999, pp. 82-89.
- K. Jimbow, P. F. Gomez, K. Toyofuku, D. Chang, S. Miura, H. Tsujiya and J. S. Park, “Biological role of tyrosinase Related Protein and Its Biosynthesis and transport from TGN to stage I melanosome, Late Endosome, through Gene Transfection Study,” Pigment Cell Research, Vol. 10, No. 4, 1997, pp. 206-213. doi:10.1111/j.1600-0749.1997.tb00486.x
- K. Jimbow, J. S. Park, F. Kato, K. Hirosaki, K. Toyofuku, C. Hua and T. Yamashita, “Assembly, Target Signal and Intracellular Transport of Tyrosinase Gene Family Protein in the Initial Stage of Melanosome Biogenesis,” Pigment Cell Research, Vol. 13, No. 4, 2000, pp. 222-229. doi:10.1034/j.1600-0749.2000.130403.x
- K. Jimbow, C. Hua, P. F. Gomez, K. Hirosaki, K. Shinoda, T. G. Salopek, H. Matsusaka, H. Y. Jin and T. Yamashita, “Intracellular Vesicular Trafficking of Tyrosinase Gene Family Protein in euand Pheomelanosome Biogenesis,” Pigment Cell Research, Vol. 13, No. S8, 2000, pp. S110-S117. doi:10.1034/j.1600-0749.13.s8.20.x
- T. Miura, K. Jimbow and S. Ito, “The in vivo antimelanoma effect of 4-S-cysteaminylphenol and its N-acetyl derivative,” International Journal of Cancer, Vol. 46, No. 5, 1990, pp. 931-934. doi:10.1002/ijc.2910460530
- M. Tandon, P. D. Thomas, M. Shokravi, S. Singh, S. Samra, D. Chang and K. Jimbow, “Synthesis and Antitumour Effect of the Melanogenesis-based Antimelanoma Agent N-Propionyl-4-S-cysteaminylphenol,” Biochemical Pharmacology, Vol. 55, No. 12, 1998, pp. 2023- 2029. doi:10.1016/S0006-2952(98)00090-2
- S. Ito, T. Kato, K. Ishikawa, T. Kasuga and K. Jimbow, “Mechanism of Selective Toxicity of 4-S-cysteinylphenol and 4-S-cysteaminylphenol to melanocytes,” Biochemical Pharmacology, Vol. 36, No. 12, 1987, pp. 2007- 2011. doi:10.1016/0006-2952(87)90501-6
- A. Gili, P. D. Thomas, M. Ota and K. Jimbow, “Comparison of in Vitro Cytotoxicity of N-acetyl and Npropionyl derivatives of phenolic thioether amines in melanoma and Neuroblastoma Cells and the relationship to tyrosinase and Tyrosine Hydroxylase Enzyme Activity,” Melanoma Research, Vol. 10, No. 1, 2000, pp. 9-15.
- B. Thiesen and A. Jordan, “Clinical applications of magnetic nanoparticles for hyperthermia,” International Journal of Hyperthermia, Vol. 24, No. 6, 2008, pp. 467-474.
- M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldöfner, R. Scholz, S. Deger, P. Wust, S. A. Loening and A. Jordan, “Clinical hyperthermia of Prostate Cancer Using Magnetic Nanoparticles: Presentation of a New Interstitial Technique,” International Journal of Hyperthermia, Vol. 21, No. 7, 2005, pp. 637-647.
- A. Ito, M. Fujioka, T. Yoshida, K. Wakamatsu, S. Ito, T. Yamashita, K. Jimbow and H. Honda, “4-S-cysteaminylphenol-Loaded Magnetite Cationic Liposomes for Combination Therapy of hyperthermia with chemotherapy against Malignant Melanoma,” Cancer Science, Vol. 98, No. 3, 2007, pp. 424-430. doi:10.1111/j.1349-7006.2006.00382.x
- T. Takada, T. Yamashita, M. Sato, A. Sato, I. Ono, Y. Tamura, N. Sato, A. Miyamoto, A. Ito, H. Honda, K. Wakamatsu, S. Ito and K. Jimbow, “Growth inhibition of Re-Challenge B16 Melanoma Transplant by conjugates of Melanogenesis Substrate and Magnetite Nanoparticles as the basis for Developing Melanoma-Targeted ChemoThermo-Immunotherapy,” Journal of Biomedicine and Biotechnology, 2009, Article ID: 457936. doi:10.1155/2009/457936
- M. Sato, T. Yamashita, M. Ohkura, Y. Osai, A. Sato, T. Takada, H. Matsusaka, I. Ono, Y. Tamura, N. Sato, Y. Sasaki, A. Ito, H. Honda, K. Wakamatsu, S. Ito and K. Jimbow, “N-Propionyl-Cysteaminyl-Phenol-Magnetite Conjugate (NPrCAP/M) is a nanoparticle for the Targeted Growth Suppression of Melanoma Cells,” Journal of Investigative Dermatology, Vol. 129, 2009, pp. 2233-2241. doi:10.1038/jid.2009.39
- M. Suzuki, M. Shinkai, H. Honda and T. Kobayashi, “Anticancer effect and Immune Induction by hyperthermia of Malignant Melanoma Using Magnetite Cationic Liposomes,” Melanoma Research, Vol. 13, No. 2, 2003, pp. 129-135. doi:10.1097/00008390-200304000-00004
- M. Yanase, M. Shinkai, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Intracellular hyperthermia for Cancer Using Magnetite Cationic Liposomes: An in Vivo Study,” Cancer Science, Vol. 89, No. 4, 1998, pp. 463-469. doi:10.1111/j.1349-7006.1998.tb00586.x
- N. Kawai, A. Ito, Y. Nakahara, M. Futakuchi, T. Shirai, H. Honda, T. Kobayashi and K. Kohri, “Anticancer effect of hyperthermia on Prostate Cancer Mediated by Magnetite Cationic Liposomes and Immunere-Sponse Induction in Transplanted Syngeneic Rats,” Prostate, Vol. 64, No. 4, 2005, pp. 373-381. doi:10.1002/pros.20253
- A. Ito, M. Shinkai, H. Honda and T. Kobayashi, “Medical application off unctionalized Magnetic Nanoparticles,” Journal of Bioscience and Bioengineering, Vol. 100, No. 1, 2005, pp. 1-11. doi:10.1263/jbb.100.1
- M. Yanase, M. Shinkai, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Antitumor immunity induction by Intracellular Hyperthermia Using Magnetite Cationic Liposomes,” Cancer Science, Vol. 89, No. 7, 1998, pp. 775-782. doi:10.1111/j.1349-7006.1998.tb03283.x
- A. Ito, M. Shinkai, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Augmentation of MHC class I Antigen Presentation via Heat Shock Protein Expression by hyperthermia,” Cancer Immunology, Immunotherapy, Vol. 50, No. 10, 2001, pp. 515-522. doi:10.1007/s00262-001-0233-7
- A. Ito, T. Kobayashi and H. Honda, “A mechanism of Antitumor Immunity Induced by hyperthermia,” Japanese Journal of Hyperthermic Oncology, Vol. 21, No. 1, 2005, pp. 1-19.
- A. Ito and K. Takeshi, “Intracellular Hyperthermia Using Magnetite Nanoparticles: A Novel Method for Hyperthermia Clinical Applications,” Thermal Medicine, Vol. 24, No. 4, 2008, pp. 113-129. doi:10.3191/thermalmed.24.113
- M. Shinkai, M. Yanase, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Intracellular hyperthermia for Cancer Using Magnetite Cationic Liposomes: In Vitro Study,” Cancer Science, Vol. 87, No. 11, 1996, pp. 1179- 1183. doi:10.1111/j.1349-7006.1996.tb03129.x
- R. Hergt, S. Dutz, R. Mueller and M. Zeisberger, “Magnetite Particle Hyperthermia: Nanoparticle Magnetism and Materials Development for Cancer Therapy,” Journal of Physics: Condensed Matter, Vol. 18, No. 38, 2006, pp. s2919-S2934. doi:10.1088/0953-8984/18/38/S26
- A. Ito, M. Shinkai, H. Honda, K. Yoshikawa, S. Saga, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Heat Shock Protein 70 Expression Induces Antitumor Immunity during Intracellular Hyperthermia Using Magnetite Nanoparticles,” Cancer Immunology, Immunotherapy, Vol. 52, No. 2, 2003, pp. 80-88.
- A. Ito, F. Matsuoka, H. Honda and T. Kobayashi, “Antitumor effects of combined therapy of Recombinant Heat Shock Protein 70 and Hyperthermia Using Magnetic Nanoparticles in an Experimental Subcutaneous Murine Melanoma,” Cancer Immunology, Immunotherapy, Vol. 53, No. 1, 2004, pp. 26-32. doi:10.1007/s00262-003-0416-5
- A. Ito, H. Honda and T. Kobayashi, “Cancer immunotherapy based on Intracellular Hyperthermia Using Magnetite Nanoparticles: A Novel Concept of ‘Heat Controlled Necrosis’ with Heat Shock Protein Expression,” Cancer Immunology, Immunotherapy, Vol. 55, No. 3, 2006, pp. 320-328. doi:10.1007/s00262-005-0049-y
- P. Manini, A. Napolitano, W. Westerhof, P. A. Riley and M. d’Ischia, “A Reactive Ortho-Quinone Generated by Tyrosinase-Catalyzed Oxidation of the Skin Depigmenting Agent Monobenzone: Self-Coupling and Thiol-Conjugation Reactions and Possible Implications for Melanocyte Toxicity,” Chemical Research in Toxicology, Vol. 22, No. 8, 2009, pp. 1398-1405. doi:10.1021/tx900018q
- A. Sato, Y. Tamura, N. Sato, T. Yamashita, T. Takada, M. Sato, Y. Osai, M. Okura, I. Ono, A. Ito, H. Honda, K. Wakamatsu, S. Ito and K. Jimbow, “Melanoma-targeted Chemo-Thermo-Immuno (CTI)-Therapy Using N-Propionyl-4S-Cysteaminylphenol-Magnetite Nanoparticles Elicits CTL Response via Heat Shock Protein-Peptide Complex Release,” Cancer Science, Vol. 101, No. 9, 2010, pp. 1939-1946. doi:10.1111/j.1349-7006.2010.01623.x
- K. Jimbow, H. Obata, M. A. Pathak and T. B. Fitzpatrick, “Mechanism of depigmentation by hydroquinone,” Journal of Investigative Dermatology, Vol. 62, 1974, pp. 436- 449. doi:10.1111/1523-1747.ep12701679
- J. J. Nordlund, B. Forget, J. Kirkwood and A. B. Lerner, “Dermatitis produced by applications of monobenzone in patients with Active Vitiligo,” Archives of Dermatology, Vol. 121, No. 9, 1985, pp. 1141-1144. doi:10.1001/archderm.1985.01660090055013
- R. E. Boissy and P. Manga, “On the etiology of contact/ Occupational Vitiligo,” Pigment Cell Research, Vol. 17, No. 3, 2004, pp. 208-214. doi:10.1111/j.1600-0749.2004.00130.x
- C. J. Cooksey, K. Jimbow, E. J. Land and P. A. Riley, “Reactivity of Orthoquinones Involved in tyrosi-naseDependent Cytotoxicity: Differences between alkylthioand Alkoxy-Substituents,” Melanoma Research, Vol. 2, No. 5-6, 1992, pp. 283-293. doi:10.1097/00008390-199212000-00001
- Y. Minamitsuji, K. Toyofuku, S. Sugiyama and K. Jimbow, “Sulphur Containing Tyrosinase Analogs Can Cause Selective Melanocytoxicity Involving Tyrosinase-Mediated Apoptosis,” Journal of Investigative Dermatology, Vol. 4, 1999, pp. s130-S136.
- K. Jimbow, Y. Miyake, K. Homma, K. Yasuda, Y. Izumi, A. Tsutsumi and S. Ito, “Characterization of melanogenesis and morphogenesis of melanosomes by Physicochemical Properties of melanin and melanosomes in Malignant Melanoma,” Cancer Research, Vol. 44, No. 3, 1984, pp. 1128-1134.
- P. G. Parsons, F. Favier, M. McEwan, T. Takahashi, K. Jimbow and S. Ito, “Action of cysteaminylphenols on Human Melanoma Cells in vivo and in vitro: 4-S-cysteaminylphenol Binds Protein Disulphide Isomerise,” Melanoma Research, Vol. 1, No. 2, 1992, pp. 97-104. doi:10.1097/00008390-199106000-00004
- V. Hariharan, J. Klarquist, M. J. Reust, A. Koshoffer, M. D. McKee, R.E. Boissy and I. C. Le Poole, “Monobenzyl ether of hydroquinone and 4-tertiary butyl Phenol Activate Markedly Different Physiological Responses in Melanocytes: Relevance to Skin Depigmentation,” Journal of Investigative Dermatology, Vol. 130, 2010, pp. 211-220. doi:10.1038/jid.2009.214
- J. G. van den Boorn, D. I. Picavet, P. F. van Swieten, H. A. van Veen, D. Konijnenberg, P. A. van Veelen, T. van Capel, E. C. Jong, E. A. Reits, J. W. Drijfhout, J. D. Bos, C. J. Melief and R. M. Luiten, “Skin-Depigmenting Agent Monobenzone Induces Potent T-Cell Autoimmunity toward Pigmented Cells by Tyrosinase Haptenation and Melanosome Autophagy,” Journal of Investigative Dermatology, Vol. 131, No. 6, 2011, pp. 1240-1251.
- O. J. Finn, “Molecular Origins of Cancer,” New England Journal of Medicine, Vol. 358, 2008, pp. 2704-2711.
- F. S. Hodi, F. Stephen, S. J. O’Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf, J. C. Hassel, W. Akerley, A. J. M. van den Eertwegh, J. Lutzky, P. Lorigan, J. M. Vaubel, G. P. Linette, D. Hogg, C. H. Ottensmeier, C. Lebbé, C. Peschel, I. Quirt, J. I. Clark, J. D. Wolchok, J. S. Weber, J. Tian, M. J. Yellin, G. M. Nichol, A. Hoos and W. J. Urba, “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” New England Journal of Medicine, Vol. 363, No. 8, 2010, pp. 711-723. doi:10.1056/NEJMoa1003466
- D. J. Schwartzentruber, D. H. Lawson, J. M. Richards, R. M. Conry, D. M. Miller, J. Treisman, F. Gailani, L. Riley, K. Conlon, B. Pockaj, K. L. Kendra, R. L. White, R. Gonzalez, T. M. Kuzel, B. Curti, P. D. Leming, E. D. Whitman, J. Balkissoon, D. S. Reintgen, H. Kaufman, F. M. Marincola, M. J. Merino, S. A. Rosenberg, P. Choyke, D. Vena and P. Hwu, “gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma,” New England Journal of Medicine, Vol. 364, No. 22, 2011, pp. 2119-2127. doi:10.1056/NEJMoa1012863
- C. Robert, L. Thomas, I. Bondarenko, S. O’Day, J. W. C. Garbe, C. Lebbe, J. F. Baurain, A. Testori, J. J. Grob, N. Davidson, J. Richards, M. Maio, A. Hauschild, W. H. Miller Jr., P. Gascon, M. Lotem, K. Harmankaya, R. Ibrahim, S. Francis, T. Chen, R. Humphrey, A. Hoos and J. D. Wolchok, “Ipilimumab plus dacarbazine for Previously Untreated Metastatic Melanoma,” New England Journal of Medicine, Vol. 364, No. 26, 2011, pp. 2517-2526. doi:10.1056/NEJMoa1104621
- P. B. Chapman, A. Hauschild, C. Robert, J. B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S. J. O’Day, J. A. Sosman, J. M. Kirkwood, A. M. M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R. J. Lee, K. T. Flaherty and G. A. McArthur, “Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation,” New England Journal of Medicine, Vol. 364, No. 26, 2011, pp. 2507- 2516. doi:10.1056/NEJMoa1103782
- K. Mise, N. Kan, T. Okino, M. Nakanishi, K. Satoh, Y. Teramura, S. Yamasaki, K. Ohgaki and T. Tobe, “Effect of Heat Treatment on Tumor Cells and Antitumor Effector Cells,” Cancer Research, Vol. 50, No. 19, 1990, pp. 6199-6202.
- A. Ito, F. Matsuoka, H. Honda and T. Kobayashi, “Heat Shock Protein 70 Gene Therapy Combined with hyperthermia Using Magnetic Nanoparticles,” Cancer Gene Therapy, Vol. 10, No. 12, 2003, pp. 918-925. doi:10.1038/sj.cgt.7700648
- M. Yanase, M. Shinkai, H. Honda, T. Wakabayashi, J. Yoshida and T. Kobayashi, “Intracellular hyperthermia for Cancer Using Magnetite Cationic Liposomes: An in vivo study,” Cancer Science, Vol. 89, No. 4, 1998, pp. 463-469. doi:10.1111/j.1349-7006.1998.tb00586.x
Abbreviations
AMF = alternating magnetic field
BSO = buthionine sulfoximine
CML = cationic magneto-liposome
CTI therapy = chemo-thermo-immunotherapy
DC = dendritic cell
DDS = drug delivery system
HSP/Hsp = heat shock protein
mAb = monoclonal antibody
MC1R = melanocortin 1 receptor
MITF = microphthalmia transcription factor
ML = non-cationic magneto-liposome
MSH = melanocyte stimulating hormone
NAcCAP = N-acetyl 4S-cysteaminylphenol
NPrCAP = N-propionyl 4S cysteaminylphenol
NPrCAP/M = N-propionyl 4S-cysteaminylphenol/ magnetite nanoparticle
NPrCAP/PEG/M = N-propionyl 4S-cysteaminylphenol/ polyethylene glycol/ magnetite nanoparticle
OVA = ovalbumin
PEG = polyethylene glycol
TIL = tumor infiltrating lymphocytes