Advances in Enzyme Research
Vol.1 No.3(2013), Article ID:36966,15 pages DOI:10.4236/aer.2013.13007
Crystal structure, biochemical and biophysical characterisation of NHR1 domain of E3 Ubiquitin ligase neutralized
![]()
1Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom; *Corresponding Author: vbolanos-garcia@brookes.ac.uk
2UMR-CNRS 6251, IPR, Université de Rennes, Campus de Beaulieu, France
3Institut Pasteur, CNRS URA2578, Paris, France
4Department of Biological and Medical Sciences, Oxford Brookes University, Oxford, England
Copyright © 2013 Deepti Gupta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 30 May 2013; revised 15 August 2013; accepted 22 August 2013
Keywords: Notch; Signaling; Neuralized; NHR1; Bearded; Tom; Endocytosis; Neurogenesis
ABSTRACT
Notch signaling controls diverse developmental decisions of central importance to cell activity. One of the conserved positive regulators of Notch signaling is Neuralized, the E3 Ubiquitin ligase enzyme that regulates signaling activity by endocytosis. Neuralized has two novel repeats, NHR1 and NHR2, with a RING finger motif at the C-terminus. Both endocytosis of the Notch ligand, Delta, and inhibition of Notch signaling by Tom, a bearded family member, require the NHR1 domain. Here we describe the first crystal structure of NHR1 domain from Drosophila melanogaster, solved to 2.1 Å resolution by X-ray analysis. Using NMR and other biophysical techniques we define a minimal binding region of Tom, consisting of 12 residues, which interacts with NHR1 and show by interfacial analysis of protein monolayers that NHR1 binds PI4P. Taken together, the studies provide insight into molecular interactions that are important for Notch signaling.
1. INTRODUCTION
The neurogenic genes of Drosophila melanogaster encode elements responsible for the cell-cell communication process that determines choice between epidermal and neuronal cell fates. One such gene, neuralized (neur), encodes a 753 amino acid peripheral membrane protein consisting of two Neur homology repeats (NHR1 and NHR2) and a C-terminal RING finger motif. Genetic analysis suggests that this gene is required for the activity of the Notch ligand Delta for Neuralized-mediated endocytosis and Notch-dependent signaling [1,2]. The activity of Delta depends on the endocytic events for normal trafficking which, in the absence of Neuralized, are greatly compromised by accumulation of inactive protein at the cell surface [1,3]. Drosophila Neuralized exists as a complex with Delta, which it ubiquitinates in a RING-finger-dependent manner to promote Delta endocytosis and signaling. Over-expression or suppression of Neuralized has been shown to result in increased or decreased Delta endocytosis and Notch-dependent signaling, respectively, suggesting the obligatory role of Neuralized in Notch signaling [4,5].
Recently, discovered members of the Bearded (Brd) family of proteins have been shown to directly regulate the Notch signaling pathway. These proteins are expressed in areas of active Notch signaling and have at least one high affinity binding site for Notch activated Suppressor of Hairless protein [6]. These proteins antagonise the Notch signaling pathway and inhibit Neuralized mediated endocytosis of Delta [7]. This inhibition of Neuralized by Bearded proteins is important for the spatial regulation of Delta signaling [1]. Genetic studies and yeast two hybrid experiments have shown that the interaction of Neuralized with Delta requires the NHR1 domain and the interaction of Tom (Twin of m4, a member of Bearded protein family) also occurs through the same domain [7]. While the work presented in this study was undergoing, He and collaborators (2011) published the solution structure of Neuralized NHR1 domain [8]. In their study, they also analysed the complex with a 20 residue peptide of Tom by NMR and ITC. Their study showed that the Neuralized NHR1 domain is structurally similar to B30.2/SPRY domain with a similar binding interface but higher target specificity [8].
In addition to interactions with Delta and Bearded proteins, Neuralized also interacts with phosphoinositides, phosphorylated derivatives of phosphatidylinositol (PtdIns), which are phospholipids present in minor amounts in the eukaryotic cell membrane. This interacttion localises the Neuralized peripheral membrane protein at the plasma membrane in the absence of Delta [9,10]. Phosphoinositides are involved in classical signal transduction at the cell surface, including regulation of membrane trafficking, mitogenesis and apoptosis as well as endocytosis. Phosphatidyl inositol phosphates (PIP) exist in multiple forms, phosphorylated at positions 3 - 5 of the inositol ring. The interaction of phosphoinositides has been localised to a lysine-rich region in the N-terminus of Neuralized, while the NHR1 domain is suggested to be dispensable for the interaction [9].
Here we describe the crystal structure of NHR1 at 2.1 Å resolution, define the stoichiometry and affinity of the interaction of NHR1 with Tom and show that NHR1 binds to phosphatidyl inositol-4-phosphate (PI4P) in solution and when adsorbed below lipid monolayers. Taken together, the studies provide insight into the structural features of NHR1 in the crystal state and the interactions mediated by this domain that are important for Notch signaling.
2. EXPERIMENTAL
2.1. Cloning, Expression and Purification
PCR-amplified DNA coding sequence of Drosophila melanogaster Neuralized gene, residues (106 - 260) were ligated into the BamH1 and Xho1 sites of pRSFDuet1 vector (Novagen). Protein expression was carried in BL21 (DE3) pLysS cells at 15˚C in LB. The protein expressed as a N-terminal 6XHis-tag was purified from the bacterial lysate using affinity, and size exclusion chromatography. Protein containing fractions were analysed on SDS PAGE, and then concentrated in buffer containing 50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 1 mM DTT.
2.2. Biochemical Analysis
A protein calibration curve was prepared using protein standards of low molecular weight (calibration kit, GE Healthcare) to estimate the molecular mass of NHR1. These included Apotropin (6.5 kDa), Ribonuclease A (13.7 kDa), Carbonic Anhydrase (29 kDa), Ovalbumin (43 kDa) and Conalbumin (75 kDa). 1 mg/ml concentration of each standard was prepared in 50 mM Tris/HCl, 300 mM NaCl and 1 mM DTT and analysed on Superdex75 16/60 column (GE Healthcare). The NHR1 containing peaks were also analysed by Mass spectroscopy.
2.3. Crystallization and Data Collection
Crystallization was performed using the hanging-drop vapour diffusion method at 20˚C with protein concentrated to 5 - 10 mg/ml. Needle shaped crystals of NHR1 were grown in 0.4 M magnesium nitrate and 10% PEG3350 in 4 days. NHR1 crystals were placed in 30% (v/v) ethylene glycol solution with 20% PEG 3350 and 0.4 M magnesium nitrate prior to flash freezing in liquid nitrogen. Diffraction data was collected at 100 K at ESRF (European Synchrotron Radiation Facility) beamline ID23-1. 180 diffraction images, with a 1˚ oscillation per frame, were recorded, leading to a complete data set with some redundancy. The crystal to detector distance was 296.34 mm and wavelength of 0.95370 Å.
2.4. X-ray Structure Determination and Refinement
The images were auto-indexed, then scaled and integrated using the programs DENZO and SCALEPACK [11]. The intensities obtained from SCALEPACK were converted to structure factor amplitudes using TRUNCATE (CCP4 package) [12]. Matthews coefficient, VM, indicated the probable presence of one NHR1 molecule (VM = 2.2 Å3 Da−1) with 44.14% solvent content in the asymmetric unit, designated chain A. The structure was solved by molecular replacement using the program AMoRe [13] with the co-ordinates of the solution structure of NHR1 as a probe [PDB code: 2YUE]. Seven rounds of refinement were performed using REFMAC5 [14], as shown in Table 1. The σA-weighted 2Fo-Fc and Fo-Fc electron-density maps were visually inspected to allow rebuilding and refitting using Coot [15]. Water molecules were introduced progressively throughout the refinement process. No further refinement was performed when the Rcryst and Rfree values showed no additional improvement (Table 1). The stereochemistry was validated using PROCHECK [16] and quality of the final NHR1 model was assessed by Ramachandran plot. Surface potentials were calculated using Adaptive Poisson-Boltzmann Solver (APBS) [17].
Protein Data Bank accession code: The refined coordinates of the model and the structure factors have been deposited with the Protein Data Bank under the accession code 4KG0. The solution structure [PDB code: 2YUE] was compared with the crystal structure of NHR1 to search for structural differences and similarities between the two. A comparative model of NHR2 was cre-
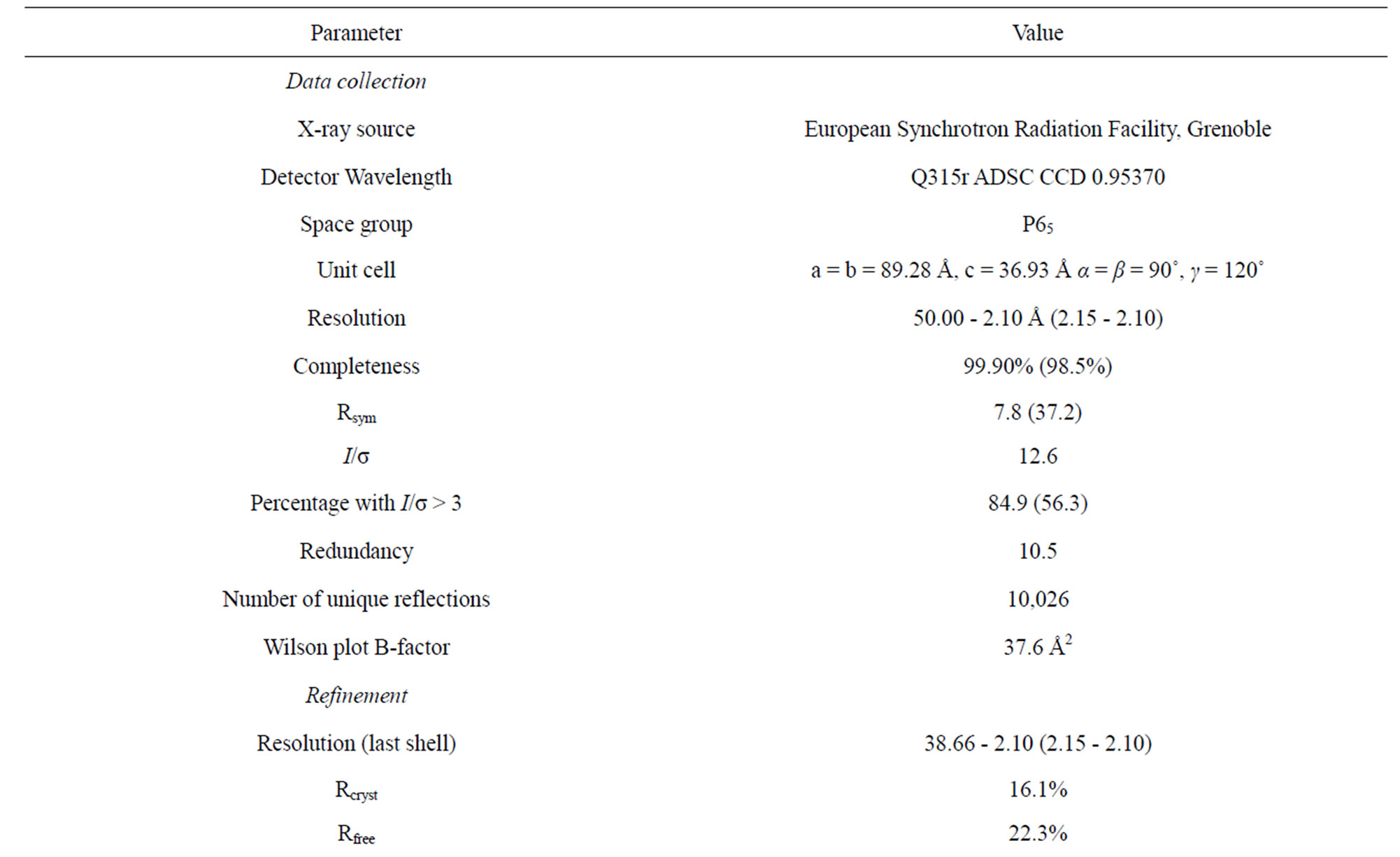

Table 1. Data collection and refinement statistics of NHR1 domain.
ated using Modeller [18].
2.5. Biophysical Analysis
Fluorescence-based thermal shift assay: A fluorescence-based thermal shift assay [19] was performed to monitor the protein unfolding and also assess ligand (12 residues Tom peptide) binding. The iCycler iQ real time detection system (Bio-Rad) was used and a temperature range of 25˚C - 80˚C was selected. 100 μl samples were prepared in 12 well strips containing protein alone (10 μM) or protein with peptide (5 μM NHR1 and 5 μM/7 μM Tom peptide) in 50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 1 mM DTT, and 25 μl of 1X Sypro orange dye (Invitrogen). A control reaction with NHR1 and a nonspecific peptide was also set. The change in fluorescence in the wells was detected by the CCD detector and the fluorescence intensity was measured at excitation at 490 nm/emission at 530 nm wavelength.
Isothermal titration calorimetry: A MicroCal VP-ITC titration microcalorimeter was used to perform isothermal titration calorimetry (ITC) in order to determine binding parameters between NHR1 and Tom. 1.4 ml of 4.5 μM Tom peptide was placed in the cell and 500 μl of 670 μM NHR1 protein was filled in the titrating syringe. Both the samples were prepared in 50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 1 mM DTT and the experiment performed at 25˚C. A total of 25 injections of 10 μl each were applied into the cell, except that the first 5 μl was discarded. The stirring speed during the titration was 300 rpm. Titrations of peptide to buffer were performed to allow base-line corrections. The Origin software was used to analyse the binding kinetics. The best-fit values of the heat of binding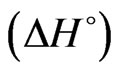 , the stoichiometry of binding
, the stoichiometry of binding 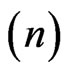 and the dissociation constant
and the dissociation constant 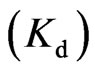 were determined from the plots of heat evolved per mol of NHR1 injected versus the NHR1-Tom peptide molar ratio using this software.
were determined from the plots of heat evolved per mol of NHR1 injected versus the NHR1-Tom peptide molar ratio using this software.
2.6. Lipid Binding Study
Lipid binding strips: 10 μg/ml of the purified NHR1 protein was incubated with the membrane lipid strips (Echelon Biosciences) at room temperature. Following the manufacture’s protocol, the strip was first incubated for 1 hour at room temperature with anti-His antibody (GE Healthcare), washed and then incubated with secondary anti-mouse alkaline phosphatase conjugate antibody (Promega) for 1 hour at room temperature. Prior to protein detection, the strip was washed as in the previous step to remove excess antibody and then detected by BCIP/NBT (5-Bromo 4-Chloro 3-Indolyl Phosphate/ Nitro Blue Tetrazolium) alkaline phosphatase tablet (Sigma) after 5 minutes of incubation at room temperature.
Interfacial Studies: Monolayers, Null Ellipsometry and Surface Rheology
A circular trough (S = 20 cm2) was used to prepare the monolayers and the surface pressure was precisely measured with a sensor using a Wilhelmy plate. Null ellipsometry was used to determine the excess concentration of adsorbed molecules by measuring the changes in the polarisation of light upon reflection. A house-made ellipsometer with a He-Ne laser (λ = 632.8 nm) and a polariser were used to record the ellipsometric measurements. The angle of incidence of light on the surface of solution was 1˚ away from the Brewster angle. After reflection on the water surface, the laser light was passed through a λ/4 retardation plate, analyser and a photomultiplier. In this “null ellipsometer” configuration [20], the analyser angle is multiplied by 2 to generate the ellipsometric angle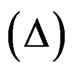 , which is defined as the phase difference between the parallel and the perpendicular polarisation of reflected light. The laser beam probed a surface of 1 mm2 and a depth of 1 µm. In the case of amphiphilic molecules, there is an excess concentration of molecules adsorbed at the interface compared to that in the bulk and therefore, the ellipsometric angle
, which is defined as the phase difference between the parallel and the perpendicular polarisation of reflected light. The laser beam probed a surface of 1 mm2 and a depth of 1 µm. In the case of amphiphilic molecules, there is an excess concentration of molecules adsorbed at the interface compared to that in the bulk and therefore, the ellipsometric angle 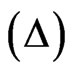 is mainly sensitive to the interfacial layer. This angle is proportional to the quantity of proteins adsorbed at the interface and hence, variation of the ellipsometric angle is a relevant probe for observing changes occurring at the interface. Using the measured ellipsometric angle
is mainly sensitive to the interfacial layer. This angle is proportional to the quantity of proteins adsorbed at the interface and hence, variation of the ellipsometric angle is a relevant probe for observing changes occurring at the interface. Using the measured ellipsometric angle 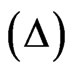 and the estimated refractive index increment of the protein to 0.2 ml/g, the surface concentration of adsorbed protein was calculated [21]. Initial values of the ellipsometric angle
and the estimated refractive index increment of the protein to 0.2 ml/g, the surface concentration of adsorbed protein was calculated [21]. Initial values of the ellipsometric angle  and the surface pressure of pure buffer solutions were recorded on the subphase/buffer for at least 30 minutes. These values were subtracted from all the data. Values of Δ and surface pressure
and the surface pressure of pure buffer solutions were recorded on the subphase/buffer for at least 30 minutes. These values were subtracted from all the data. Values of Δ and surface pressure 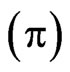 were stable and recorded every 4 s with a precision of ±0.5˚ and ±0.5 mN/m, respectively, at 20˚C.
were stable and recorded every 4 s with a precision of ±0.5˚ and ±0.5 mN/m, respectively, at 20˚C.
The lipids are spread at the surface of the trough and the quantity of spread molecules adjusted to record the starting surface pressure. In the second step, the protein is injected in the subphase (with a needle); the adsorption kinetic is followed by recording the ellipsometric angle and the surface pressure over time. The increase of ellipsometric angle 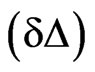 from the initial value of lipid alone
from the initial value of lipid alone 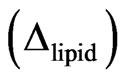 to the final value corresponding to the end of the adsorption kinetics of the protein
to the final value corresponding to the end of the adsorption kinetics of the protein 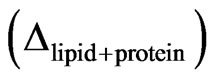 indicates the presence of protein below or inside the lipidic layer. The shear elastic constant was determined by surface rheology. The rigidity of the interfacial layers at the air/water or lipid/water interface was measured using a 10 mm diameter paraffin-coated aluminium disk float, placed at the centre of a 40 mm diameter Teflon trough [22-24]. A small magnet in the float was kept centred by permanent magnetic field. A mirror fixed on the magnet reflected the laser beam onto a photodiode that helped in sensitive angular detection of the float. When a sinusoiddal torque of 0.01 - 100 Hz was applied to the float by an oscillating field perpendicular to the permanent solenoid field, the device acted as a simple harmonic oscillator. The latter field acted as a restoring torque equivalent to an interfacial layer with a rigidity of 0.16 mN/m (set as the sensitive limit of the rheometer) and the resistance of the interfacial layers was directly measured. One important advantage of this set-up is the achievement of a high sensitivity due to very small deformations. To assess and detect rigidity of the monolayer, the amplitude and phase of the mechanical response of pure subphase was first analysed in the frequency range of 0.01 - 100 Hz. This measurement took approximately 1 hour. Then, the protein solution was directly poured in the trough and the mechanical response of the layer formed at the interface recorded at a fixed frequency of 5 Hz. At the end of the kinetics, when the shear elastic constant
indicates the presence of protein below or inside the lipidic layer. The shear elastic constant was determined by surface rheology. The rigidity of the interfacial layers at the air/water or lipid/water interface was measured using a 10 mm diameter paraffin-coated aluminium disk float, placed at the centre of a 40 mm diameter Teflon trough [22-24]. A small magnet in the float was kept centred by permanent magnetic field. A mirror fixed on the magnet reflected the laser beam onto a photodiode that helped in sensitive angular detection of the float. When a sinusoiddal torque of 0.01 - 100 Hz was applied to the float by an oscillating field perpendicular to the permanent solenoid field, the device acted as a simple harmonic oscillator. The latter field acted as a restoring torque equivalent to an interfacial layer with a rigidity of 0.16 mN/m (set as the sensitive limit of the rheometer) and the resistance of the interfacial layers was directly measured. One important advantage of this set-up is the achievement of a high sensitivity due to very small deformations. To assess and detect rigidity of the monolayer, the amplitude and phase of the mechanical response of pure subphase was first analysed in the frequency range of 0.01 - 100 Hz. This measurement took approximately 1 hour. Then, the protein solution was directly poured in the trough and the mechanical response of the layer formed at the interface recorded at a fixed frequency of 5 Hz. At the end of the kinetics, when the shear elastic constant 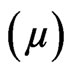 (expressed in mN/m) reached a constant value, a new measurement between 0.01 and 100 Hz was recorded to determine whether the system behaves as an elastic layer. Rigidity measurements were carried out in parallel to ellipsometry. All of the experiments were performed at 18˚C. Protein solutions were prepared in the range 1 - 80 µg/ml (i.e., ≈36 nM – 3 µM) in 50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 1 mM DTT for surface pressure, null ellipsometry and surface rheology measurements.
(expressed in mN/m) reached a constant value, a new measurement between 0.01 and 100 Hz was recorded to determine whether the system behaves as an elastic layer. Rigidity measurements were carried out in parallel to ellipsometry. All of the experiments were performed at 18˚C. Protein solutions were prepared in the range 1 - 80 µg/ml (i.e., ≈36 nM – 3 µM) in 50 mM Tris/HCl, pH 8.0, 300 mM NaCl and 1 mM DTT for surface pressure, null ellipsometry and surface rheology measurements.
3. RESULTS AND DISCUSSION
3.1. Crystal Structure of NHR1
The Neuralized NHR1 domain of Drosophila melanogaster was crystallised with a tandem of six histidine residues (His-tag) at the N-terminus. The crystal structure of NHR1 was determined to a resolution of 2.1 Å by molecular replacement and refined Rcryst of 16.1% and Rfree of 22.3%. The data processing statistics, the refinement statistics and an assessment of the model geometry are included in Table 1. The structure was validated with PROCHECK [16] and the quality of the final NHR1 model was assessed by Ramachandran plot. No electron density was observed for the first 12 residues including the His-tag. 110 water molecules were placed in the model. A magnesium ion from the crystallisation condition was found co-ordinated with Asn 116, Glu 30 and a water molecule at a distance of 2.21 Å, 2.18 Å and 2.41 Å respectively. The X-ray crystal structure comprises of two β sheets, one containing 7 and the other having 4 anti-parallel β strands, with 36.3% of β strand and 4.5% of 310 helix (Figure 1(a)). Comparison of the crystal structure to the solution structure of Drosophila NHR1 domain (PDB code: 2YUE) [8] showed the best match with the NMR model 8 (RMSD of 1.9 Å), while the NMR model 4 was least similar with an RMSD of 2.41 Å. One difference between the crystal structure and the NMR structure is the presence in the latter of an additional β strand at the C-terminus, which is defined by residues Ser162 to Tyr 165. The rest of the NMR structure is similar to that defined by X-ray protein crystallography methods.
Before we defined the crystal structure, homologues for the NHR1 domain had been identified using the sequence-structure homology recognition program, FUGUE [25]. The hit with highest Z-score (with 95% confidence) of 4.01 for NHR1 was a SPRY domain-containing SOCS box protein 2 (data not shown). This suggested that the NHR1 domain could structurally be similar to the predicted SPRY domain protein. On analysing the function of SOCS (Suppressor of Cytokine Signalling) box motif, it was found that this motif consisting of approximately 40 residues, contains a BC box that binds elongin C and then elongin B to form the E3 ubiquitin ligase complex [26]. This is consistent with the fact that both SPRY and NHR domains are found to co-occur in proteins with RING fingers. Thus, the two proteins have the same ultimate function to ubiquitinate the target protein.
Results obtained from DALI search for the NHR1 domain corroborated the FUGUE analysis. Structural superposition and alignment of the SPRY domain proteins with NHR1 using Baton/Comparer showed that the secondary structure elements, mostly β strands, were organised in a similar way. The common residues between both the domains were compared using JOY [27]. The position of Gly167 was found to be absolutely conserved (Figure 1(b)) among the NHR and SPRY domains. Arg128, Gly230 and Cys252 were also found to be conserved (Figure 1(b)). The low sequence identity (10% - 12%) between the NHR1 domain and the SPRY domains suggests that the NHR domain is probably a distant relative of the SPRY.
The SPRY domain occurs in a variety of cellular proteins that mediate protein-protein interactions. This domain is considered to be a migratory domain since it is found associated with different protein domains, and recognises a specific individual partner. SPRY is known to interact with a peptide called VASA [28]. Alignment of the amino acid sequence of VASA with Tom homologues and intracellular regions of Serrate and Jagged proteins (that are known to interact with Neuralized), suggested that both NHR1 and SPRY could recognise similar protein sequences (Figure 1(c)). Ponting and collaborators have previously showed that NHR1 domains resemble the SPRY motif in as much as they cooccur in proteins with RING finger proteins [29]. However, the low sequence conservation between NHR1 and SPRY domain proteins makes it difficult to identify functionally conserved residues. The overall fold of the NHR1 domain was a sandwich with jelly roll-like topology. The overall analysis suggests that the NHR1 domain belongs to a new super-family that is similar in structure to the Concanavalin A-like lectins/glucanases having the SPRY domain.
3.2. Binding Sites
From structural analysis of NHR1 it was evident that the overall domain is similar to SPRY domain. The NHR1 domain mediates nuclear envelope association and delta-dependent inhibition of nuclear import and is involved in interaction with Bearded and lipids [7,9,30, 31]. We used an evolutionary trace analysis method Trace Suite II [32] to identify possible binding interface of NHR1. For this, residues with the evolutionary timecut off of P07 or higher were mapped on to the X-ray
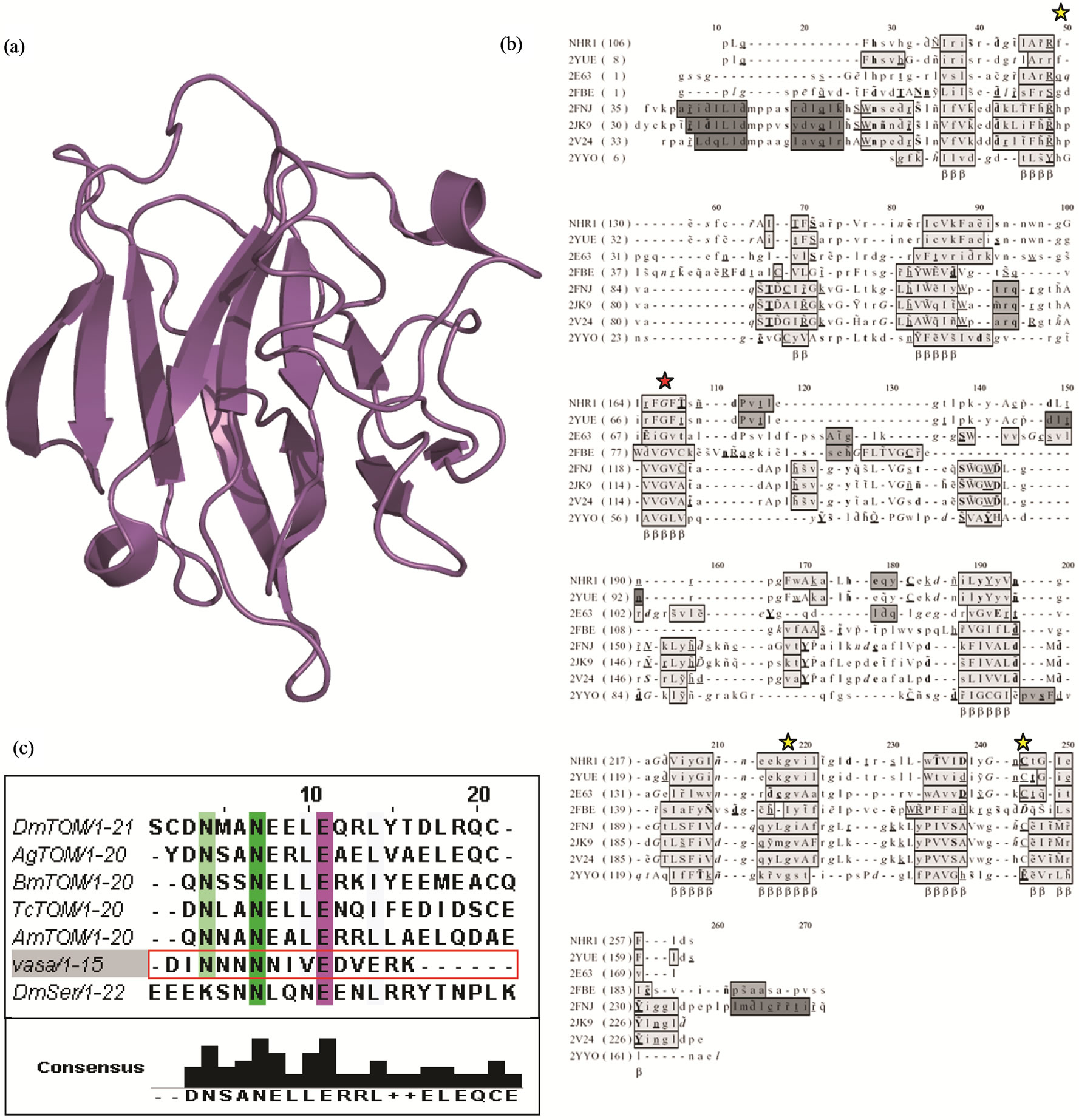
Figure 1. (a), Cartoon representation of the crystal structure of NHR1; (b), Structural alignment of NHR1 with SPRY domain using JOY [27]. NHR domain containing proteins (Drosophila Neuralized crystal (NHR1) and solution structure (2YUE) and 2E63 with the other SPRY domain structures. Position of an absolutely conserved glycine is indicated by red star and three other conserved residues are indicated by yellow stars; (c), Multiple sequence alignment of Tom homologues, intracellular domains of Notch ligands Serrate and Jagged along with the SPRY domain interacting peptide (VASApep2ih: VASA peptide, PDB code: 2IHS). The sequence include DmSer: Drosophila melanogaster Serrate; for Tom homologues, BmTom motif: Bombyx mori Tom motif 2; TcTom motif: Tribolium castaneum Tom motif 2; Dm Tom motif: Drosophila melanogaster Tom motif 2; AgTom motif: Anopheles gambiae Tom motif 2; AmTom motif: Apis mellifera Tom motif.
crystal structure of the NHR1 domain. The evolutionary trace analysis shows that conserved residues that might interact with binding partners to carry out the protein’s function clustered in the loop region of NHR1 domain.
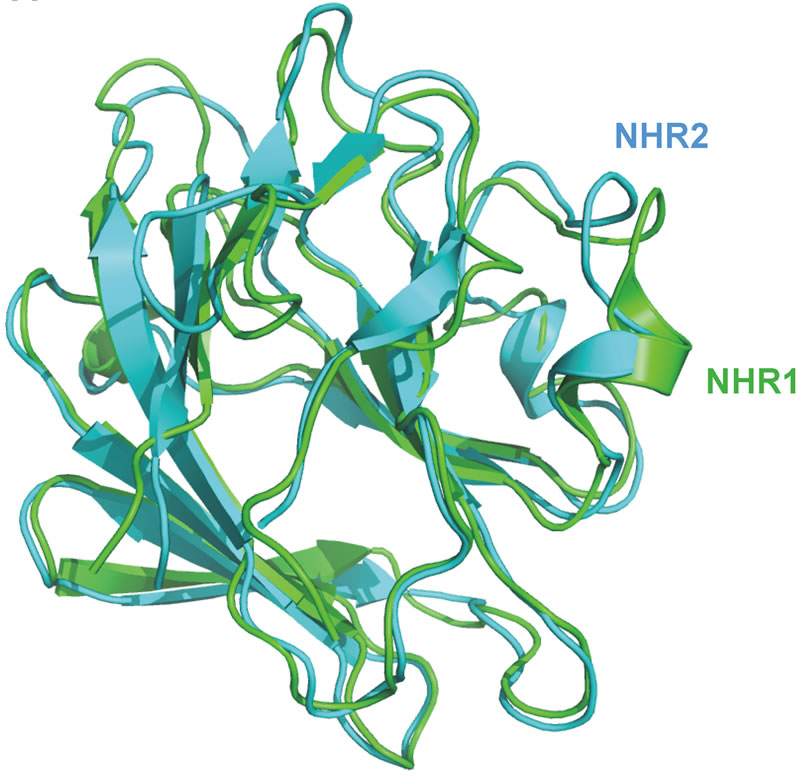
The sequence identity between NHR1 and NHR2 is 27%, allowing a comparative model of NHR2 using modeller [18] to be constructed (Figure 2(a)). Analysis of the NHR2 domain suggested that the binding region of NHR2 could be same as that of NHR1 domain, involving residues Y249, G162, S131 and F132 of NHR1, which are topologically equivalent to Y145, G58, S26 and F27 of NHR2. In addition W160 of NHR1 corresponds to Y56 of NHR2. The surface properties of NHR1 and NHR2 also showed a similar binding region with small differences between the two binding pockets. In NHR1 R165 occludes pocket B whereas NHR2 occludes pocket A with Y56.
An additional approach to the study of these solvent exposed residues was adopted. This involved examining inter-molecular interactions between symmetry related NHR1 molecules in the crystals using the PICCOLO software [33]. These residues were mapped on the electrostatic surface of NHR1 and revealed hydrophobic residues that cluster around the basic/positively charged region and cover two cavities (pocket A and B) (data not shown).
Interestingly, size exclusion chromatography and mass spectrometry revealed the existence of monomeric (21.25 and 19.39 kDa, respectively) and dimeric (38.763 and 44.7 kDa, respectively) forms of NHR1 (Figure 2(b), (c)). Given the observed interactions between symmetry equivalents of NHR1 in the crystal and the predicted structural similarity between NHR1 and NHR2, we conducted functional analyses in-vivo using the yeast two-
 (a)
(a)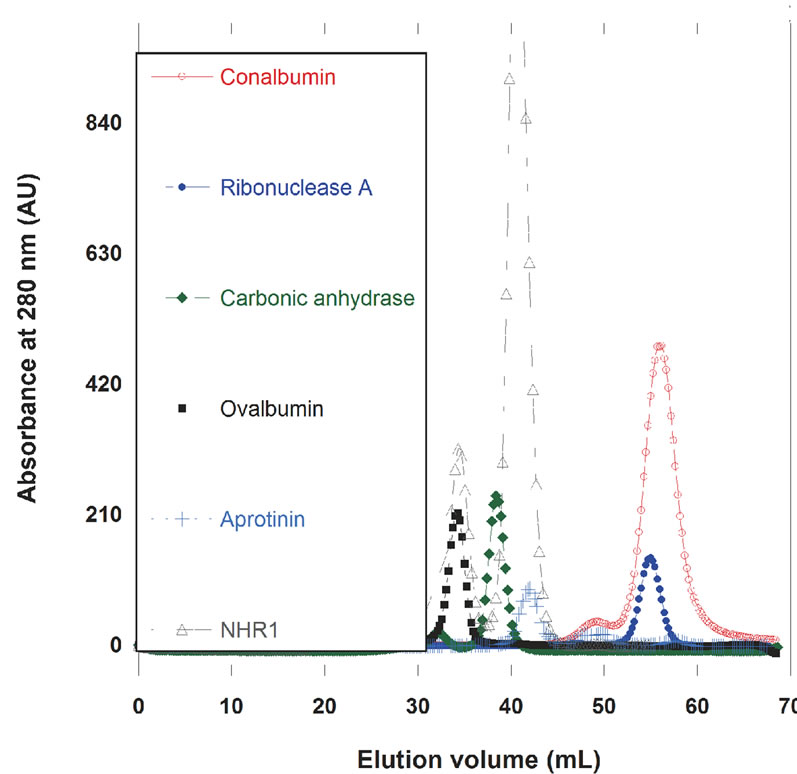 (b)
(b) (c)
(c)
Figure 2. (a), Superposition of NHR1 with structure model of NHR2 domain to compare the binding interface of NHR1:Tom peptide and SPRY: VASA peptide. The NHR1 and NHR2 chains are coloured green and cyan respectively; (b), Calibration of Superdex S75 16/60 column to determine molecular weight of NHR1. On left, the gel filteration profile all low molecular proteins are shown. Two peaks corresponding to monomer and dimer of NHR1 are marked. On right is the calibration curve; (c), Electrospray ionisation mass spectrometry (ESI-MS) analysis of purified NHR1. Peaks A correspond to dimeric and peak B to monomeric forms of NHR1.
hybrid system to establish if NHR1 can associate to forms homodimers and whether NHR1 and NHR2 physically interact. In both cases, yeast two-hybrid failed to demonstrate any interaction (data not shown).
3.3. Interaction of NHR1 with Tom
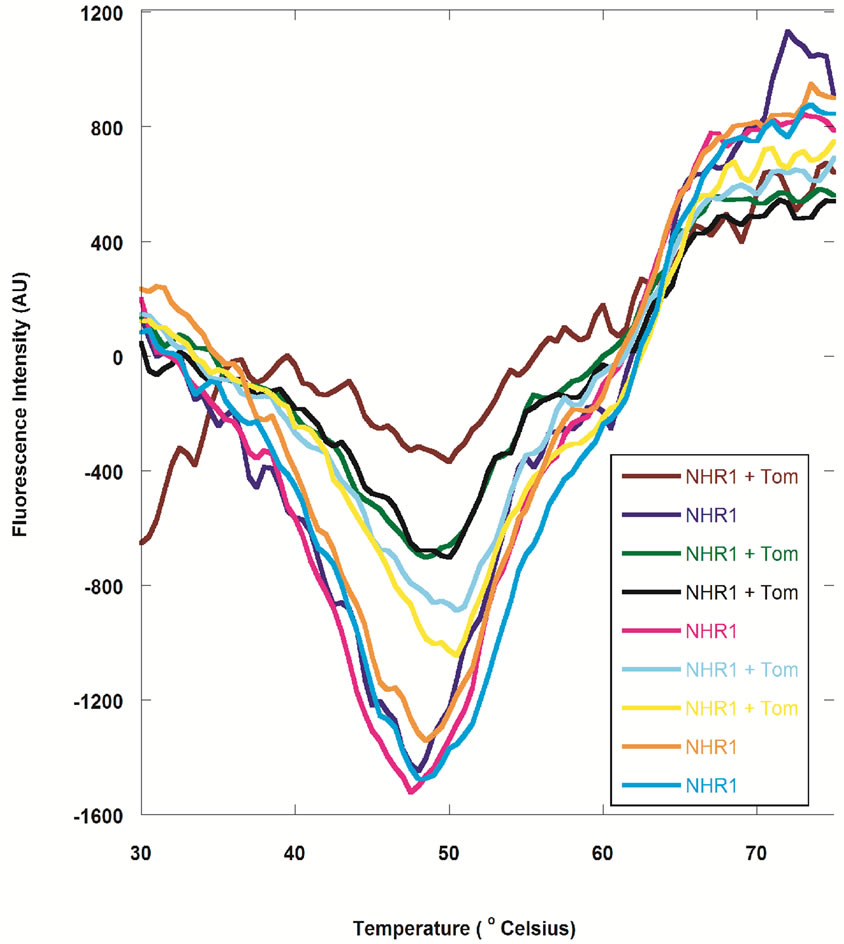
Despite numerous attempts, we were unable to obtain a soluble and folded form of Tom protein (Gupta D., PhD Thesis) [34] and therefore a 12 residue peptide of Tom corresponding to the minimal binding region was synthesized. The binding of the Tom mimic peptide to NHR1 was studied by examining the unfolding of purified NHR1 protein in the fluorescence-based thermal shift assay. The assay takes advantage of an environmentally sensitive, fluorescent Sypro orange dye that preferentially binds to the hydrophobic-core region of the protein [19]. The controlled increase of temperature by iCycler iQ causes the protein to gradually unfold resulting in the exposure of the core hydrophobic residues and hence in an increase in the fluorescence signal from the Sypro orange dye. This enables monitoring of the unfolding of protein as well as determination of its melting temperature (Tm) from the melting curve. On addition of the ligand to the protein, the midpoint of the melting curve increases or decreases relative to that for the uncomplexed state of the protein, suggesting the presence of the ligand either stabilises or destabilises the protein respectively. This technique was employed to study the change in thermal denaturation of NHR1 alone as well as in the presence of the Tom peptide. Figure 3(a) shows the melting curves of NHR1 alone and NHR1 with peptide. The average melting temperature of NHR1 alone [Tm (NHR1)] was 47.75˚C and the addition of Tom peptide causes an increase in the melting temperature to 50.13˚C. The change in melting temperature 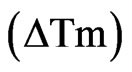 given by:
given by:
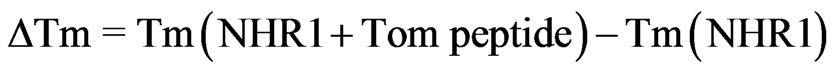
An increased melting temperature of ~2 degree of NHR1 in the presence of the peptide implies that the two molecules interact with each other and form a complex. The control peptide showed no increase in the melting temperature, suggesting that it does not bind the NHR1 protein, confirming that the interaction of NHR1 with Tom peptide is specific. Furthermore, the sigmoidal shape of the melting curve represents a cooperative, twostate transition thermal unfolding of a typical single and autonomously folding protein unit.
3.4. Determination of Binding Constants
Isothermal titration calorimetry (ITC) was performed for determination of the NHR1-Tom binding affinity. Complete saturation of the binding sites with the Tom peptide was achieved when a high NHR1 concentration (670 μM) was used to titrate against 4 μM of the peptide. The binding curve is shown in Figure 3(b). The enthalpy 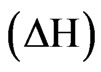 and entropy
and entropy 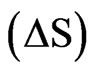 calculated from the binding curve were −4.5 kcal mol−1 and −134 entropy units, respectively. The binding affinity of the peptide with NHR1 was 6 ± 0.8 μM in 1:1 stoichiometry. To gain further insights into the nature of the NHR1-Tom peptide interaction, we performed NMR spectroscopy. The [1H, 15N]-HSQC spectra of the NHR1-Tom peptide complex reveals the extent of molecular reorganization in NHR1 upon peptide binding (Figure 3(c)).
calculated from the binding curve were −4.5 kcal mol−1 and −134 entropy units, respectively. The binding affinity of the peptide with NHR1 was 6 ± 0.8 μM in 1:1 stoichiometry. To gain further insights into the nature of the NHR1-Tom peptide interaction, we performed NMR spectroscopy. The [1H, 15N]-HSQC spectra of the NHR1-Tom peptide complex reveals the extent of molecular reorganization in NHR1 upon peptide binding (Figure 3(c)).
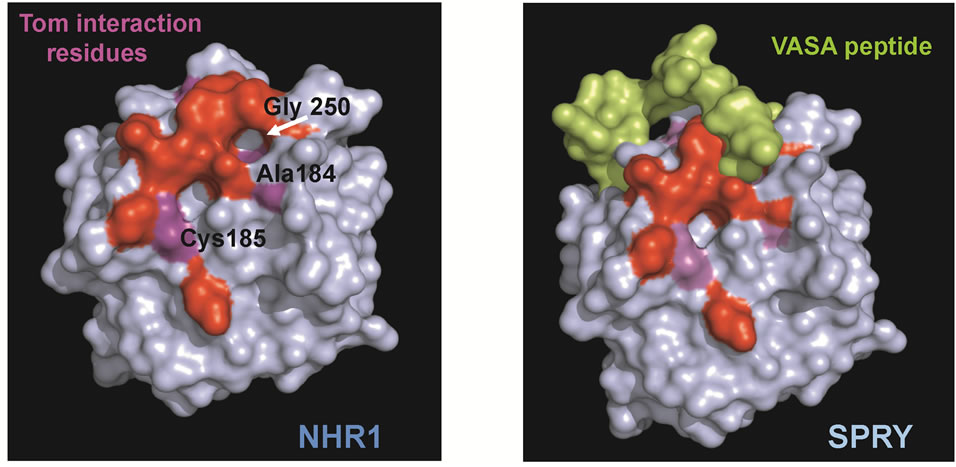
The binding interface of NHR1 along with the superimposed NHR2 model and Tom peptide were compared with the binding interface of VASA peptide with SPRY domain protein (PDB code: 2IHS) shown in Figure 4(a). Since VASA peptide has similarity to Tom peptide and the intracellular region of Serrate known to bind Neuralized/Mindbomb [35] (Figure 2(a)), the interacting interfaces of both NHR1 and SPRY proteins with Tom and VASA peptides, respectively were examined. This showed that the interacting residues on both domains lie in loop regions. In Figure 4(a) the binding region of VASA peptide is compared with the NHR1: Tom interface, where the residues that interact with the Tom peptide are coloured purple. One of the cavities that interact with Tom (see purple coloured patch of residue Gly250 marked by the arrow) is fully covered when the VASA peptide is docked on the NHR1 structure. The analysis shows that the binding interfaces of NHR1 and SPRY domain proteins are similar and therefore, they could recognise similar peptides with the motif NXXNXXXE.
3.5. Interaction of NHR1 with Lipids
The interaction of phosphoinositides (PIP) with Drosophila melanogaster Neuralized protein [9] contributes to the membrane localisation of Neuralized in the absence of Delta and plays an important regulatory role of Neuralized-mediated Delta endocytosis. Furthermore, the same study showed that an N-terminus, lysine-rich region (TKDKLSSKKKMHLLKKIKKRF; residues 69-89) of Neuralized protein binds PIP.
However, since the crystal structure of NHR1 reveals the domain contains a large hydrophobic region, we investigated whether NHR1 is also involved in binding lipids. For this, we performed lipid-binding experiments with purified NHR1 blotted onto nitrocellulose membranes previously spotted with immobilised lipids (membrane lipid strip, Echelon Biosciences). We found that purified NHR1 binds PI4P to a greater extent than phosphatidic acid (Figure 5). This finding prompted us to characterise the kinetics and surface rheology of the NHR1-PI4P interaction of protein monolayers. The results are presented in the next section.
3.6. NHR1 Interfacial Analysis
3.6.1. The Air/Liquid Interface
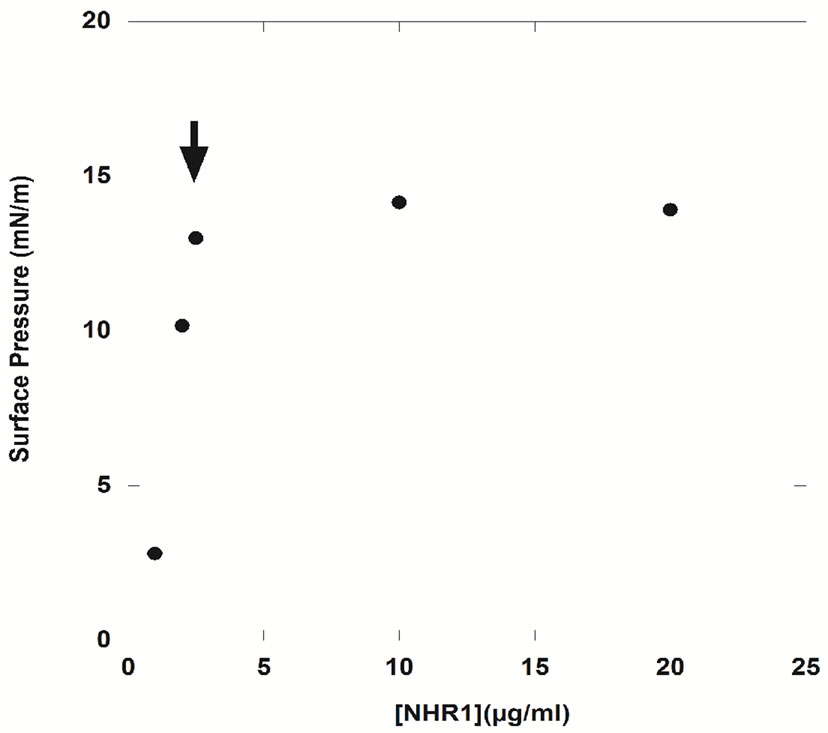
To determine whether NHR1 was surface active, and ascertain its intrinsic amphiphilic nature, experiments were first performed at the air/liquid interface, in which the air is the hydrophobic component of the system. Purified NHR1 was directly dispensed in the trough at a final concentration of 30 μg/ml (1.5 μM). Figure 6(a)
shows that NHR1 was saturated at the interface when the subphase concentration reached ≥3 µg/ml, strongly suggesting that NHR1 is surface active. Therefore, a bulk concentration lower that 3 µg/ml (i.e. 2.5 µg/ml, 0.13 μM) was chosen as the working concentration for subsequent experiments to determine the interaction of NHR1 with lipid, a value reflecting that protein-lipid interactions are favoured compared to protein-protein interactions. When NHR1 at 2.5 µg/ml in buffer solution is poured in the Langmuir trough, after 8 hours of the experiment the
 (a)
(a) (b)
(b)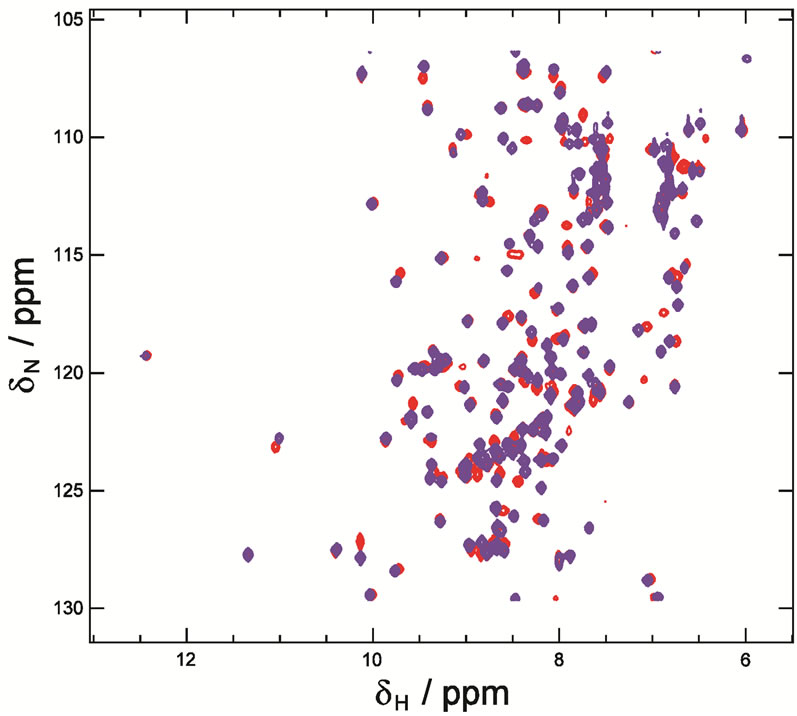 (c)
(c)
Figure 3. (a), Thermal shift assay of NHR1 alone and in presence of Tom. More than five experiments were performed as shown in the graphs with different line colours; (b), Isothermal titration calorimetry. The purified NHR1 (670 μM, in syringe) was titrated with Tom peptide (4.5 μM, in cell). 25 injections of 10 μl each were injected to saturate the peptide. Complete saturation was obtained after repeating the experiment 4 times. Both the protein and peptide were present in 50 mM Tris/HCl, 300 mM NaCl and 1 mM DTT, pH 8.0. Binding affinity of 6 μM was determined from the curve by Origin software; C, Overlay of the [1H, 15N]-HSQC spectra of NHR1 protein with Tom peptide recorded at pH 8.0. The peaks shown in blue correspond to free NHR1 protein, while those in red correspond to the complex of NHR1 with Tom. The concentration of the peptide was twice that of NHR1.
 (a)
(a)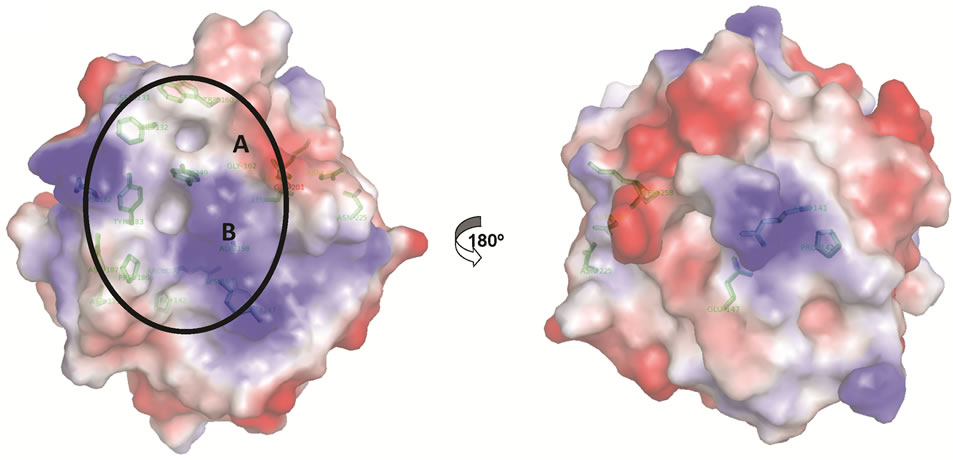 (b)
(b)
Figure 4. (a), Comparison of NHR1 binding interface with SPRY binding VASA peptide. The surface of NHR1 is coloured blue with the predicted residues of the hydrophobic patch are coloured red and the residues interacting with Tom peptide are coloured purple with residues labelled in white. The VASA peptide, in green, that binds SPRY protein is docked on the NHR1 surface. The peptide covers one of the cavities, which is the same region that interacts with Tom, suggests that the binding interface of NHR1 and SPRY is similar; (b), Potential interacting residues are mapped on the NHR1 electrostatic surface. The electrostatic surface potential of NHR1 was calculated using APBS. Functionally important residues that could be involved in interactions with NHR1 binding partners are shown as green sticks. The hydrophobic patch formed by aromatic, surface exposed and conserved residues is circled.
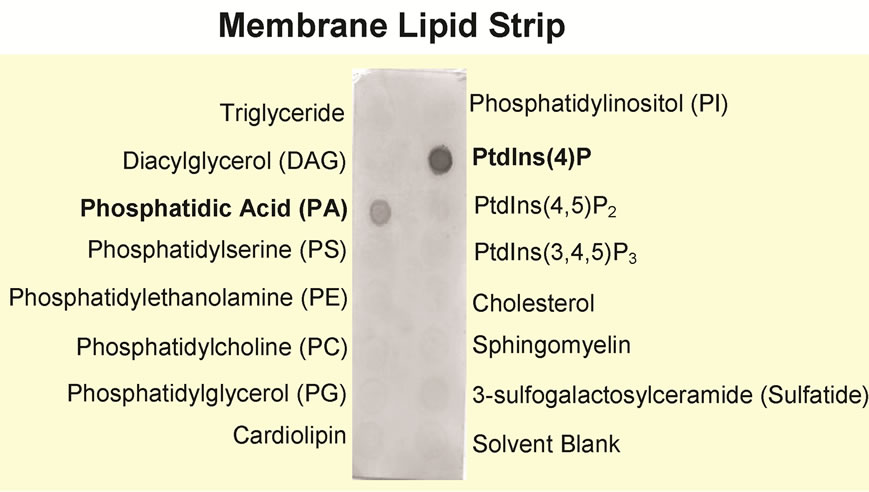
Figure 5. Lipid overlay assay. Pure NHR1 was used to assess protein binding to various lipids present on the strip. The results show that NHR1 binds strongly to Phosphatidyl inositol-4-phosphate [PtdIns (4) P] and to lesser extent to phosphatidic acid (PA).
surface pressure at the interface reached a plateau and remained constant at 13 mN/m respectively. The ellipsometric angle increases with time and reaches a plateau around 9˚, corresponding to a surface concentration of 1.8 mg/m² according to the relationship from De Feitjer: 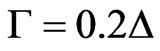 valid in case of globular proteins [21], thus confirming that the domain is surface active and forms stable monolayers (Figure 6(b)).
valid in case of globular proteins [21], thus confirming that the domain is surface active and forms stable monolayers (Figure 6(b)).
The low protein concentration allows the recording of the first adsorption events and to extract several parameters relevant to the surface activity. Two of these parameters were obtained from the plot of 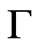 versus p (Figure 6(c)). Here,
versus p (Figure 6(c)). Here,  is the surface concentration at which the surface pressure becomes different from zero. q is extracted from the slope of the
is the surface concentration at which the surface pressure becomes different from zero. q is extracted from the slope of the 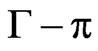 curve (i.e.,
curve (i.e.,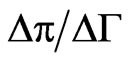 ) and corresponds to the increase of surface pressure relative to the increase of surface concentration [36]. For NHR1,
) and corresponds to the increase of surface pressure relative to the increase of surface concentration [36]. For NHR1, 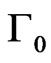 = 1.1 mg/m² and q = 25 mN/m. The value of
= 1.1 mg/m² and q = 25 mN/m. The value of 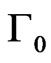 is characteristic of that seen in other globular proteins of similar size [37,38]. The rather high value of q indicates that lateral interactions develop more effectively once the protein is adsorbed at the interface and that the increase of the surface concentration induces a high change in the surface pressure.
is characteristic of that seen in other globular proteins of similar size [37,38]. The rather high value of q indicates that lateral interactions develop more effectively once the protein is adsorbed at the interface and that the increase of the surface concentration induces a high change in the surface pressure.
Surface rheology experiments were carried out to define the viscoelastic properties of NHR1 monolayers. Figure 7(a) shows the evolution of the shear elastic constant µ, measured at fixed frequency, versus time for three concentrations (1 µg/ml, 2.5µg/ml and 8.75 µg/ml). At the end of each kinetic (i.e., 14 hour, when the slope becomes lower for 2.5 µg/ml), measurements at variable frequencies in the frequency range of 0.01 - 100 Hz were performed and the real part and an elastic layer model (i.e., a simple harmonic oscillator) was adjusted to the imaginary part of the response to determine if the layer behaves as an elastic layer (Figure 7(a)). The measurements confirmed that the protein does not form elastic layers at the interface except at very low concentration (1 µg/ml), where the fit was reasonable (Figure 7(b)). At protein concentration of 2.5 µg/ml complex rheological effects take place, as evidenced by the poor fit to the equation (Figure 7(c)). It can be thus concluded that above 1 µg/ml concentration of NHR1 the layer is not viscoelastic, this in turn suggesting that the extent of short range interactions lead to the formation of a poorly connected layer.
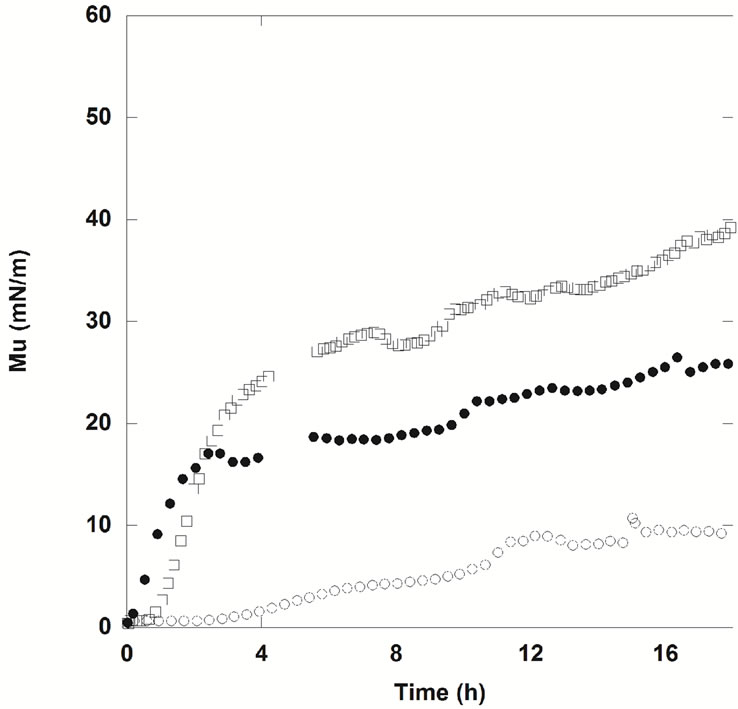
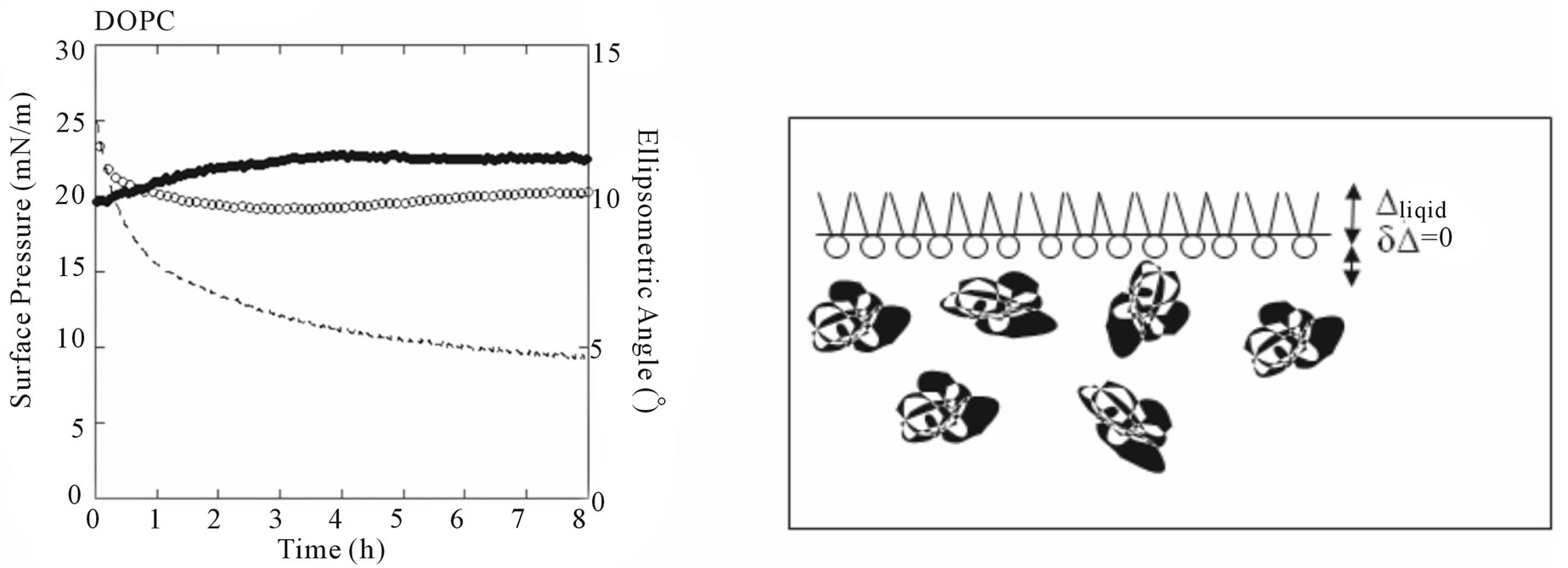
To determine whether NHR1 interacts with the lipid PI4P, two monolayers were prepared by spreading solutions of PI4P and DOPC (1,2-dioleoyl-sn-glycero-3- phosphocholine). An initial surface pressure for lipids of p = 25 mN/m was chosen because lipidic layers are rather dense at these conditions and also reduce adsorption of protein in the free space of the surface (this could happen if the initial surface pressure was lower than this value). The results of these experiments are shown in Figures 8(a) and (b).
For DOPC monolayers, the initial surface pressure
 (a)
(a) (b)
(b) (c)
(c)
Figure 6. Ellipsometric and surface pressure measurements of NHR1. (a), Determination of surface activity of NHR1 at air/water interface. Surface pressure reaches 13 mN/m with only 2.5 μg/ml of protein, indicating the protein is surface active at air/water interface; (b), Measurement of surface pressure, π (dark circles) and surface concentration, Г (open circles) at 2.5 μg/ml. The inset represents the change of ellipsometric angle of NHR1 alone, i.e. protein = 9˚; (c), The surface concentration was drawned versus ellipsometric measurements at 2 μg/ml. θ corresponds to the slope of dπ/dГ and Г0 is the surface concentration at which the surface pressure becomes different from zero.
corresponded to an ellipsometric angle of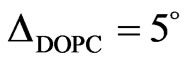 , for PI4P the corresponding value was
, for PI4P the corresponding value was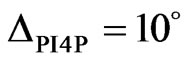 . These values remained stable over a few hours (Figure 8(b)). The ellipsometric angle for the protein alone was
. These values remained stable over a few hours (Figure 8(b)). The ellipsometric angle for the protein alone was 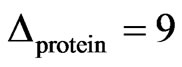 (Figure 8(a)). DOPC was chosen as negative control because lipid-binding experiments in membrane lipid strips showed that NHR1 does not interact with this lipid, as illustrated by the pattern of Figure 8(a). This observation was confirmed in studies at interfaces because there was no increase of ellipsometric angle (Figure 8(a)) and the surface pressure slightly increased at the beginning of the kinetic to then decrease regularly. This pattern resembled the evolution of the surface pressure of pure DOPC at the liquid/air interface, as shown by the thin line in Figure 8(a). The difference in surface pressure at the interface between DOPC alone and DOPC in presence of NHR1 after 3 hours can be explained by the rather low stability of DOPC when exposed to air, leading to high variability in the surface pressure behaviour during many hours.
(Figure 8(a)). DOPC was chosen as negative control because lipid-binding experiments in membrane lipid strips showed that NHR1 does not interact with this lipid, as illustrated by the pattern of Figure 8(a). This observation was confirmed in studies at interfaces because there was no increase of ellipsometric angle (Figure 8(a)) and the surface pressure slightly increased at the beginning of the kinetic to then decrease regularly. This pattern resembled the evolution of the surface pressure of pure DOPC at the liquid/air interface, as shown by the thin line in Figure 8(a). The difference in surface pressure at the interface between DOPC alone and DOPC in presence of NHR1 after 3 hours can be explained by the rather low stability of DOPC when exposed to air, leading to high variability in the surface pressure behaviour during many hours.
Figure 8(b) shows the adsorption kinetics of NHR1 below the lipidic layer for PI4P. The ellipsometric angle increased from 9.8˚ (the value of PIP4 alone at the interface) to a final value of 11.3˚, thus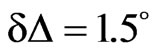 . This indicated that NHR1 was adsorbed at the liquid-PIP4 interface. During the same time, the surface pressure decreased from the starting value of pure PIP4 (25 mN/m), as does pure PIP4 when spread onto the buffer. However, the surface pressure became stable after c.a. 20 min, contrary to pure PIP4 for which surface pressure was not
. This indicated that NHR1 was adsorbed at the liquid-PIP4 interface. During the same time, the surface pressure decreased from the starting value of pure PIP4 (25 mN/m), as does pure PIP4 when spread onto the buffer. However, the surface pressure became stable after c.a. 20 min, contrary to pure PIP4 for which surface pressure was not
 (a)
(a)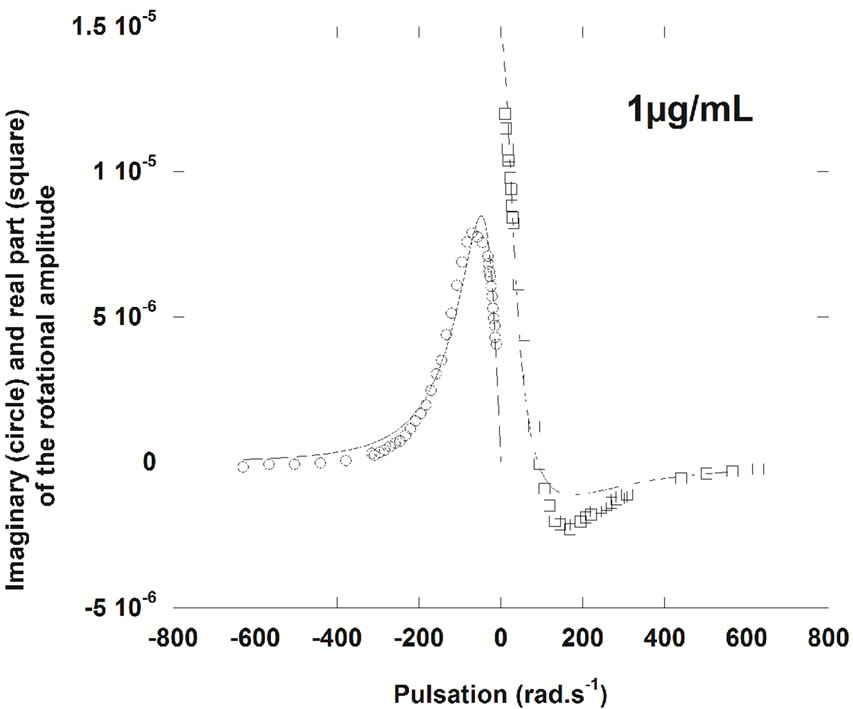 (b)
(b)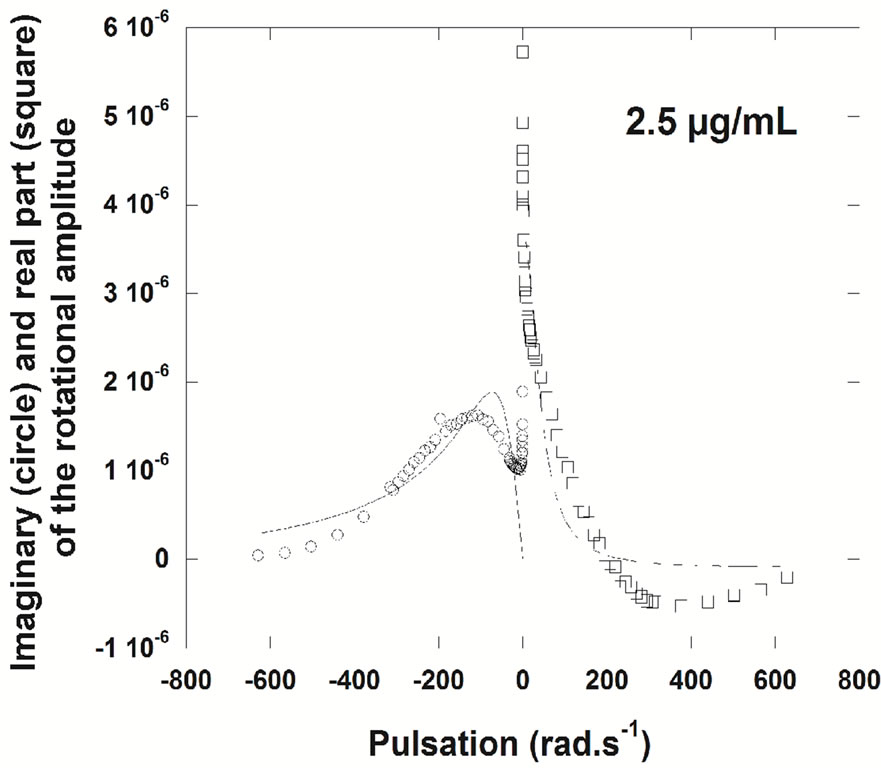 (c)
(c)
Figure 7. (a), Surface rheology measurements of NHR1 at 1 μg/ml (hollow circles), 2.5 μg/ml (squares) and 8.75 μg/ml (filled circles). The graph shows the shear elastic constant, µ, versus time measured at the fixed frequency of 5 Hz, during protein adsorption at the interface. The angular deviation versus pulsation was measured and plotted in the graphs shown below with the imaginary and real parts of the response fit a viscoelastic layer model (damped harmonic oscillator) for 1µg/ml (b) and 2.5 µg/ml (c).
stable at the interface as shown by the thin line in Figure 8(b). This behaviour proved that NHR1 was not inserted in the PIP4 monolayer (otherwise the surface pressure would increase) but was localised below the PIP4 layer thus stabilising the surface pressure. The fact that  was lower than the ellipsometric angle of a dense NHR1 monolayer (9˚) suggested that NHR1 layer was not continuous, as illustrated by the pattern of Figure 8(b). Hence, the localisation of NHR1 below the lipidic layer was attributed to the interaction of NHR1 with the head-group of PIP4.
was lower than the ellipsometric angle of a dense NHR1 monolayer (9˚) suggested that NHR1 layer was not continuous, as illustrated by the pattern of Figure 8(b). Hence, the localisation of NHR1 below the lipidic layer was attributed to the interaction of NHR1 with the head-group of PIP4.
4. FINAL REMARKS
Using a multidisciplinary approach that combines biochemical, biophysical and structural methods we have shown that the NHR1 domain physically interacts with Tom and with the phospholipid PIP4. In the former case, minor conformational changes were detected in NHR1 upon ligand binding whereas for the latter, the interaction involves the head-group of PIP4. Our studies suggest that Drosophila Neuralized can mediate its membrane localisation not only through its N-terminus lysine rich region but also through its NHR1 domain. This feature could also explain why Drosophila Neuralized isoform NeurPC that lacks the N-terminal basic region can recruit itself to the plasma membrane in presence of Delta. Taken together, our data provides new insights into NHR1 interaction that are important in Notch signaling.
5. ACKNOWLEDGEMENTS
We are grateful to Dr. Matthias Ehebauer, Prof. Alfonso Martinez Arias and Dr. Dimitri Chirgadze for their help. V. M. B.-G. thanks Ox-
 (a)
(a)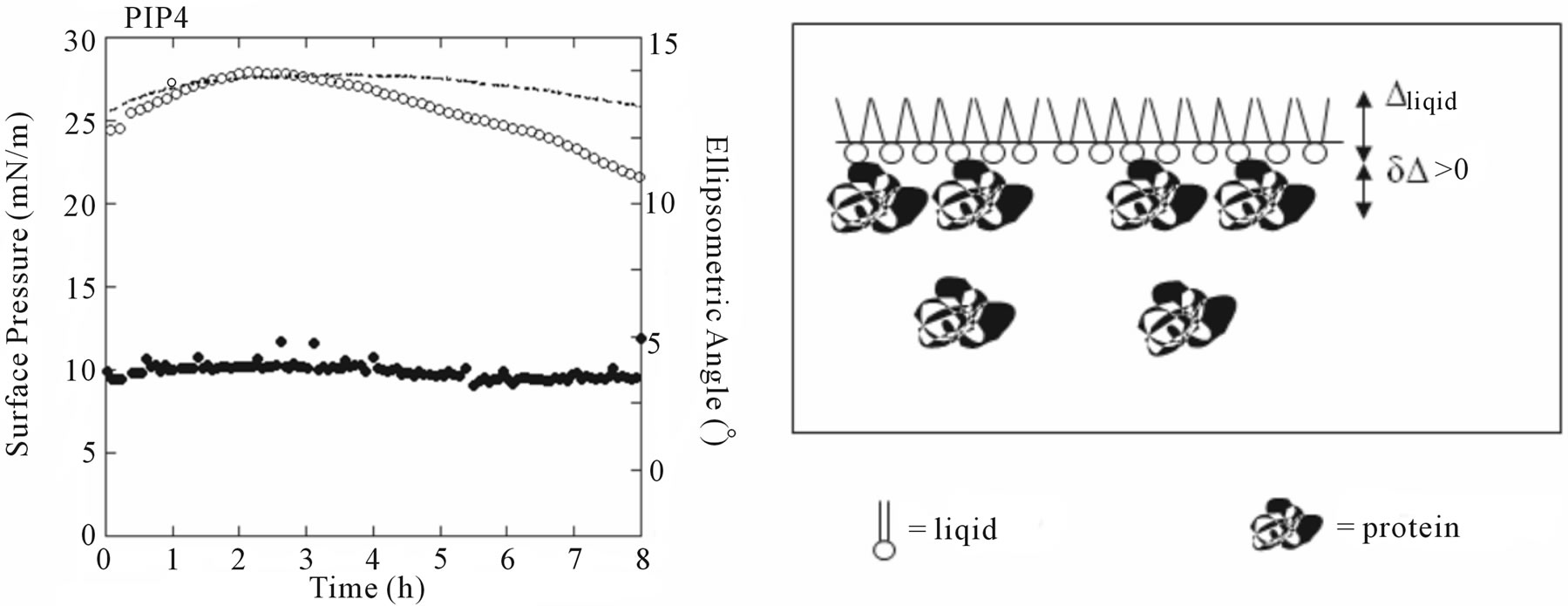 (b)
(b)
Figure 8. Ellipsometric and surface pressure measurements of NHR1 with DOPC (a) and PI4P (b). The cartoons on the right hand side illustrate the mode of interaction between NHR1 in each lipid monolayer.
ford Brookes University for financial support. D. G. thanks Cambridge Commonwealth Trusts for the generous PhD studentship.
REFERENCES
- Pavlopoulos, E., et al. (2001) Neuralized encodes a peripheral membrane protein involved in delta signaling and endocytosis. Developmental Cell, 1, 807-816. doi:10.1016/S1534-5807(01)00093-4
- Liu, S., et al. (2012) Functional analysis of the NHR2 domain indicates that oligomerization of Neuralized regulates ubiquitination and endocytosis of Delta during Notch signaling. Molecular Cell Biology, 32, 4933-4945. doi:10.1128/MCB.00711-12
- Le Borgne, R., et al. (2005) Two distinct E3 ubiquitin ligases have complementary functions in the regulation of delta and serrate signaling in Drosophila. PLoS Biology, 3, e96. doi:10.1371/journal.pbio.0030096
- Lai, E.C., et al. (2001) Drosophila neuralized is a ubiquitin ligase that promotes the internalization and degradation of delta. Developmental Cell, 1, 783-794. doi:10.1016/S1534-5807(01)00092-2
- Yeh, E., et al. (2001) Neuralized functions as an E3 ubiquitin ligase during Drosophila development. Current Biology, 11, 1675-1679. doi:10.1016/S0960-9822(01)00527-9
- Lai, E.C., et al. (2000) Antagonism of notch signaling activity by members of a novel protein family encoded by the bearded and enhancer of split gene complexes. Development, 127,291-306.
- Bardin, A.J., and Schweisguth, F. (2006) Bearded family members inhibit Neuralized-mediated endocytosis and signaling activity of Delta in Drosophila. Developmental Cell, 10, 245-255. doi:10.1016/j.devcel.2005.12.017
- He, F., et al. (2009) Structural and Functional Characterization of the NHR1 Domain of the Drosophila Neuralized E3 Ligase in the Notch Signaling Pathway. Journal of Molecular Biology, 393, 478-495. doi:10.1016/j.jmb.2009.08.020
- Skwarek, L.C., et al. (2007) Neuralized contains a phosphoinositide-binding motif required downstream of ubiquitination for delta endocytosis and notch signaling. Developmental Cell, 13, 783-795. doi:10.1016/j.devcel.2007.10.020
- Weinmaster, G. and Fischer, J.A. (2011) Notch ligand ubiquitylation: What is it good for? Developmental Cell, 21, 134-144. doi:10.1016/j.devcel.2011.06.006
- Otwinowski, Z. and Minor, W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology, 276, 307-326. doi:10.1016/S0076-6879(97)76066-X
- CCP4. (1994) Collaborative Computational Project. Acta Crystallographica Section D: Biological Crystallography, D50, 760.
- Navaza, J. (1994) AMoRe: an automated package for molecular replacement. Acta Crystallographica Section A: Foundations of Crystallography, A50, 157-163. doi:10.1107/S0108767393007597
- Murshudova, G.N., Vagin, A.A. and Dodson, E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D: Biological Crystallography, D53, 240-255. doi:10.1107/S0907444996012255
- Emsley, P. and Cowtan, K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography, D60, 2126-2132. doi:10.1107/S0907444904019158
- Laskowski, R.A., et al. (1993) PROCHECK—A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26, 283-291. doi:10.1107/S0021889892009944
- Baker, N.A., et al. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Science USA, 98, 10037- 10041. (APBS)
- Sali, A. and Blundell, T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology, 234,779-815. doi:10.1006/jmbi.1993.1626
- Lo, M.C., et al. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Analytical Biochemistry, 332, 153-159. doi:10.1016/j.ab.2004.04.031
- Azzam, R.M.A. and N.M. Bashara. (1977) Ellipsometry and polarized light. In: Cowley, J.M. Ed., North Holland Personal Library, North Holland Personal Library, Amsterdam, 340.
- De Feijter, J.A., Benjamins, J. and Veer, F.A. (1978) Ellipsometry as a tool to study the adsorption behaviour of synthetic and biopolymers at the air-water interface. Biopolymers, 17, 1759-1772. doi:10.1002/bip.1978.360170711
- Venien-Bryan, C., et al. (1998) Characterization of the growth of 2D protein crystals on a lipid monolayer by ellipsometry and rigidity measurements coupled to electron microscopy. Biophysical Journal, 74, 2649-2657. doi:10.1016/S0006-3495(98)77970-6
- Renault, A., et al. (1999) Surface-induced polymerization of actin. Biophysical Journal, 76, 1580-1590. doi:10.1016/S0006-3495(99)77317-0
- Bolanos-Garcia, V.M., et al. (2005) The conserved N-terminal region of the mitotic checkpoint protein BUBR1: A putative TPR motif of high surface activity. Biophysical Journal, 89, 2640-2649. doi:10.1529/biophysj.105.063511
- Shi, J., Blundell, T.L. and Mizuguchi, K. (2001) FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. Journal of Molecular Biology, 310, 243- 257. doi:10.1006/jmbi.2001.4762
- Linossi, E.M. and Nicholson, S.E. (2012) The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life, 64, 316-323. doi:10.1002/iub.1011
- Mizuguchi, K., et al. (1998) JOY: Protein sequence-structure representation and analysis. Bioinformatics, 14, 617- 623. doi:10.1093/bioinformatics/14.7.617
- Woo, J.S., et al. (2006) Structural basis for protein recognition by B30.2/SPRY domains. Molecular Cell, 24, 967-976. doi:10.1016/j.molcel.2006.11.009
- Ponting, C.P., et al. (2001) Novel protein domains and repeats in Drosophila melanogaster: Insights into structure, function, and evolution. Genome Research, 11, 1996-2008. doi:10.1101/gr.198701
- Commisso, C. and Boulianne, G.L. (2007) The NHR1 domain of Neuralized binds Delta and mediates Delta trafficking and Notch signaling. Molecular Biology of the Cell, 18, 1-13. doi:10.1091/mbc.E06-08-0753
- Commisso, C. and Boulianne, G.L. (2008) The neuralized homology repeat 1 domain of Drosophila neuralized mediates nuclear envelope association and delta-dependent inhibition of nuclear import. Journal of Molecular Biology, 375, 1125-1140. doi:10.1016/j.jmb.2007.11.043
- Innis, C.A., Shi, J. and Blundell, T.L. (2000) Evolutionary trace analysis of TGF-beta and related growth factors: Implications for site-directed mutagenesis. Protein Engineering, 13, 839-847. doi:10.1093/protein/13.12.839
- Bickerton, G.R., Higueruelo, A.P. and Blundell, T.L. (2011) Comprehensive, atomic-level characterization of structurally characterized protein-protein interactions: The PICCOLO database. BMC Bioinformatics, 12, 313. doi:10.1186/1471-2105-12-313
- Gupta D., (2010) Structural and functional study on Notch signaling pathway and its regulatory proteins. PhD Thesis, University of Cambridge, England.
- Glittenberg, M., et al. (2006) Role of conserved intracellular motifs in serrate signaling, cis-inhibition and endocytosis. EMBO Journal, 25, 4697-4706. doi:10.1038/sj.emboj.7601337
- Damodaran, S.A.C.S.R. (2001) Molecular basis for protein adsorption at fluid-fluid interfaces. In: Dickinson, E., Miller, R., Damodaran S. and Rao, C.S., Eds., Food Colloids: Fundamental of Formulation, Royal Society of Chemistry, London, 165-180.
- Beaufils, S., et al. (2008) Characterization of the tetratricopeptide-containing domain of BUB1, BUBR1, and PP5 proves that domain amphiphilicity over amino acid sequence specificity governs protein adsorption and interfacial activity. Journal of Physical Chemistry B, 112, 7984-7991. doi:10.1021/jp711222s
- Lee, S., et al. (2012) Characterization of spindle checkpoint kinase Mps1 reveals domain with functional and structural similarities to tetratricopeptide repeat motifs of Bub1 and BubR1 checkpoint kinases. Journal of Biological Chemistry, 287, 5988-6001.