Food and Nutrition Sciences
Vol.3 No.2(2012), Article ID:17510,7 pages DOI:10.4236/fns.2012.32024
SRT1720, a SIRT1 Activator, Aggravates Bleomycin-Induced Lung Injury in Mice
![]()
1First Department of Internal Medicine, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Japan; 2Department of Pathology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Japan; 3Department of Radiological Sciences, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Japan.
Email: htaro@med.u-toyama.ac.jp
Received October 12th, 2011; revised November 29th, 2011; accepted December 6th, 2011
Keywords: Interstitial Lung Diseases (ILDs); Lung Injury; SIRT1; Bleomycin; X-Ray Irradiation; Oxidative Stress
ABSTRACT
Diagnosis and management of interstitial lung diseases (ILDs), caused by lung epithelial injury followed by apoptosis, are often challenging. It has been controversial whether the SIRT1 protein, a principal modulator of longevity due to caloric restriction, ameliorates or aggravates ILD in animal models. Here we examined the effect of SRT1720, a synthetic activator of SIRT1, on bleomycin-induced lung injury in a mouse model and apoptosis in cultured epithelial cells. Oral intubation of SRT1720 over a period of 15 days caused body weight loss and a high mortality rate among bleomycin-treated mice. Histological examinations showed that the SRT1720 load increased fibrosis in the bleomycin-treated lung. An analysis of bronchoalveolar lavage fluid revealed remarkably increased numbers of inflammatory cells in the SRT1720-treated group. Moreover, the apoptosis of A549 lung cancer cells, caused by X-ray irradiation and an anti-Fas activating antibody, was promoted by SRT1720. These results indicate that SRT1720 not only aggravates bleomycin-induced ILD, but stimulates the apoptosis of physically and biologically stimulated A549 cells. While SIRT1 activators are considered promising for the treatment of conditions such as diabetes mellitus, fatty liver, and chronic obstructive pulmonary diseases, an excess of food containing SIRT1 activators may be harmful depending on the disease state, especially in the case of acute inflammation.
1. Introduction
Treating interstitial lung diseases (ILDs) is one of the most challenging fields of medicine [1]. ILDs include acute lung injury, idiopathic interstitial pneumonia, radiation pneumonitis, infections, drug-induced toxicity, and a variety of other lung diseases, so a diagnosis is often difficult [2-4]. Although the pathogenesis of pulmonary fibrosis is complex, numerous studies have implicated lung epithelial injury and apoptosis in this process [5]. Furthermore, oxidative stress plays a critical role [6].
Bleomycin is an anti-cancer agent, which kills cancerous cells by inducing oxidative stress and thereby breaking DNA strands [7]. However, oxidative stress can cause ILD. Mice with bleomycin-induced lung injury have been used to investigate the pathogenesis of lung disease [8]. Radiation pneumonitis often occurs in irradiated patients with malignant diseases such as lung cancer, esophageal cancer, breast cancer, and Hodgkin’s lymphoma [9]. Pneumonitis is due to oxidative stress [10] and lung epithelial apoptosis [11]. Therefore, prevention and treatment of this disease are urgently needed [12].
The Sirtuin (silent information regulator) family, class III histone deacetylases (HDACs), is composed of seven homologs (SIRT1-7) in mammals [13]. SIRT1 is a highly conserved protein thought to be a principal modulator of longevity due to caloric restriction [14]. SIRT1 was also reported to regulate a variety of cellular processes including chromatin stability, metabolism, inflammation, and reactions to oxidative stress [15,16]. SIRT1 modifies gene expression by deacetylating histones and transcripttion factors, including p53, FOXOs, PGC-1α, hypoxiainducible factor-1α(HIF-1α), and nuclear factor-кB (NF-кB) [17-19]. Several reports have shown that SIRT1 has not only a protective effect against oxidative stress [15,16, 20], but also anti-inflammatory properties [21]. Conversely, Kim et al. [22] reported that in mice, inhibittion of SIRT1 improved inflammation in the lungs by modifying the expression of HIF-1α and vascular endothelial growth factor (VEGF). Nevertheless, the role of SIRT1 in ILD is not fully understood. Recently, SRT1720 was synthesized as a potent and specific activator of SIRT1 [23]. This compound has more than 10-fold the effect of resveratrol found in red wine. We recently reported that SRT1720 decreased the expression of genes involved in oxidative stress and inflammatory cytokines in the livers of MSG (monosodium glutamate)-treated mice that exhibit obesity and insulin resistance [24].
As mentioned, the role of SIRT1 in ILD has been in dispute. In this study, we investigated the effects of SRT 1720 on bleomycin-treated mice and A549 lung cancer cells undergoing radiation-induced apoptosis. We concluded that SRT1720 aggravates some forms of experimentally produced ILD.
2. Materials and Methods
2.1. Materials
The agonistic anti-Fas (CD95) monoclonal antibody (clone 7C11) was obtained from Beckman Coulter (Brea, CA). The annexin V-FITC apoptosis detection kit was purchased from Medical and Biological Laboratories (Nagoya, Japan). Bleomycin was obtained from Nippon Kayaku (Tokyo, Japan). Biochemical and other reagents were available from local suppliers.
2.2. Chemical Synthesis of SRT1720
SRT1720 was synthesized according to methods described previously [23,24]. The structure and purity of the compound were determined by nuclear magnetic resonance spectroscopy [24].
2.3. Animal Care
Eight-week-old female C57BL/6J mice were obtained from Sankyo Labo Service (Tokyo, Japan). They were given free access to laboratory chow and water, and housed in a room with a 12 h-light/12 h-dark cycle at 24˚C in the animal center of our university. The experimental protocols were approved by the Ethics Committee for Animal Use and Care of the university.
2.4. Preparation of Animal Model and Recovery of Bronchoalveolar Lavage Fluid
On day 1, mice were anesthetized with sodium pentobarbital and tracheostomized. Bleomycin was dissolved in water (to 3.3 mg/ml) and administered (150 μl/100 g of body weight) intratracheally in a germ-free manner. SRT1720 was dissolved in water, and administered 200 μl by oral gavage (20 mg/100 g of body weight) every other day from day 0 to 14. Fifteen days later, mice were anesthetized, and the trachea was cannulated with a 24- gauge IV catheter (Terumo, Tokyo, Japan). Lungs were lavaged twice with 1 ml of saline. The bronchialveolar fluid was centrifuged at 500× g for 10 min. The pellet was suspended in 100 μl of PBS (-), and a small portion was used for counting cells in a hemocytometer. The remaining cells were centrifuged onto slide glasses using a Cytospin 4 (Thermo Scientific, Kanagawa, Japan). The cells were treated with Wright-Giemsa stain to distinguish cell types. The supernatant was stored at –80˚C until used.
2.5. Lung Histology
After bronchoalveolar lavage, lungs were perfused with 10 ml of PBS (-), followed by 10 ml of phosphate-buffered 4% paraformaldehyde via the left ventricle. Lungs were removed and fixed in 10% formalin. Paraffin-embedded lung tissue was sectioned 10 μm thick, and stained with hematoxylin and eosin. The histopathological evaluation of pulmonary fibrosis was performed by the method of Ashcroft Scoring [25].
2.6. Cell Culture
A549 cells, a tumor cell line derived from a human lung carcinoma with properties of type II alveolar epithelial cells, were purchased from American Type Culture Collection (ATCC, Manassas, VA). The cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% heat-inactivated fetal calf serum (GIBCO/ Invitrogen), 50 IU/ml of penicillin, and 50 mg/ml of streptomycin. They were incubated in a 5% CO2-95% air chamber. SRT1720 was dissolved in dimethyl sulfoxide (DMSO) and diluted with each medium to make concentrations of up to 100 μM.
2.7. MTS Viability Assay
For the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) Promega, Madison, WI) viability assay, A549 and BEAS-2B cells (2 × 103 in a 96-well plate) were incubated for 24 hours with various concentrations of SRT1720. CellTiter 96 AQueous (Promega, Madison, WI) was added during the last 2 hours of the 24 h period. The color that developed was measured at 490nm.
2.8. Apoptosis Assay
On reaching 50% - 70% confluence, A549 cells were incubated with various concentrations of SRT1720 in a 60 mm plate. They were X-ray irradiated at 4 Gy/min in an MBR-15020R-3 (Hitachi Medical, Tokyo, Japan) for 30 minutes. A Fas-activating antibody (1 μg/ml) was added during the last 8 h of the 24 h culture period. After 24 h, the SRT1720-treated, X-ray-irradiated, and/or antibody-treated cells were harvested, and examined for apoptosis with an annexin V-FITC/propidium iodide (PI) kit and a FACScallibur (Becton Dickinson, Mountain View, CA), according to the manufacturer’s instructions.
2.9. Statistical Analysis
The results are expressed as the mean + SEM. The difference between groups was determined with an unpaired and a paired t-test or two-way analysis of variance (ANOVA). Survival curves for time-to-event variables were constructed using Kaplan-Meier estimates and were compared with the log-rank test. The analyses were performed using R, version 2.12.0 (The R Foundation for Statistical Computing, available from http://www.R-project.org). A value of p < 0.05 was considered significant. All p values were two-sided.
3. Results
3.1. SRT1720 Affects Viability and Body Weight Gain in Bleomycin-Treated Mice
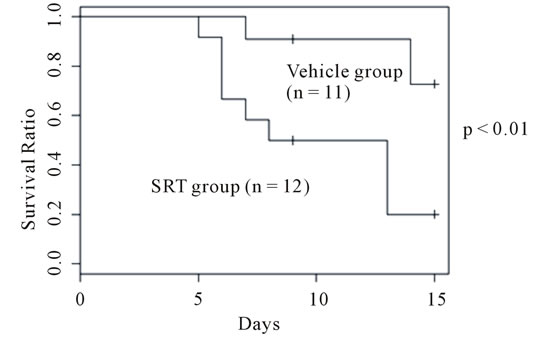
First, we examined the effect of SRT1720 on the viability of bleomycin-treated mice. Our previous study showed that oral administration of SRT1720 to mice did not affect body weight or food intake at a dose of 200 mg/kg of body weight during a period of 70 days [24]. In this study, a dose of 100 mg/kg was adopted. Bleomycinadministered mice began to die from day 6, and about 20% had died by day 15 (Figure 1(a)). SRT1720 markedly lowered the survival rate, with 80% of mice having died by day 13. The change in the body weight of mice was recorded daily (Figure 1(b)). Mice treated with bleomycin began to lose weight from day 2, and showed a maximal loss at around day 12. Thereafter, mice gained weight gradually. A similar curve, although more profound, was seen for the SRT1720-treated group.
These results indicate that SRT1720 significantly aggravated bleomycin-induced lung injury. Surviving mice in the two groups appeared to overcome bleomycin’s toxicity.
3.2. SRT1720 Enhances Lung Fibrosis in Bleomycin-Treated Mice
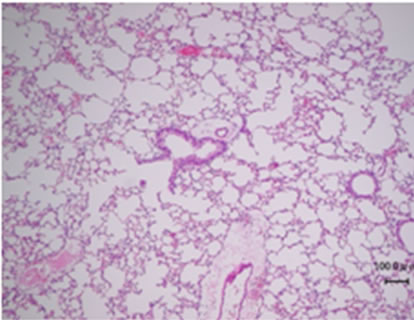
Bleomycin is known to cause pulmonary fibrosis [26]. Thus, we examined how the uptake of SRT1720 affected fibrosis in bleomycin-treated mice by histochemistry. The bleomycin treatment evoked marked fibrosis in 15 days (Figures 2(a) and (b)). This fibrosis was exaggerated by SRT1720 (Figure 2(c)). The fibrotic areas are quantified in Figure 2(d). The mean Ashcroft score was 2.41 + 0.90 for bleomycin-treated mice, and 3.80 + 0.23 for bleomycin/SRT-treated mice, indicating that SRT1720 significantly aggravated fibrosis in the bleomycin-induced model.
 (a)
(a) (b)
(b)
Figure 1. Effects of SRT1720 on survival and body weight change in bleomycin-treated mice. (a) A SRT1720 solution was given to bleomycin-treated mice and survival was recorded up to Day 15, as described in the Materials and Methods. The initial number of mice is shown in parentheses. Survival rates were calculated by the method of Kaplan-Meier. Survival was significantly lower in the SRTtreated group (p = 0.01); (b) Change in body weight of control mice (closed circles), bleomycin-treated mice (closed squares), and bleomycin plus SRT1720 mice (closed triangles). Body weight was significantly lower in the SRT1720- treated group than in the counterpart (Two-way ANOVA, p < 0.01).
3.3. SRT1720 Increases Macrophage and Lymphocyte Numbers in Bronchoalveolar Lavage Fluid
We attributed the aggravation of bleomycin-induced lung injury in mice treated with SRT 1720 to enhanced inflammation. Hence, bronchoalveolar lavage fluid was recovered from surviving mice, and macrophages, lymphocytes, neutrophils, eosinophils, and basophils were enumerated. Figure 3 clearly indicates macrophages and lymphocytes to be more abundant in the SRT1720- treated group. The number of neutrophils was also slightly larger in the SRT1720-treated mice. These results indicate that the greater mortality and weight loss in the SRT1720-treated group are due to increased numbers of
 (a)
(a) (b)
(b)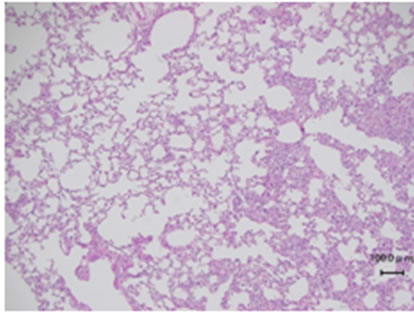 (c)
(c) (d)
(d)
Figure 2. Hematoxyline-eosin staining of the lungs of bleomycinor bleomycin plus SRT1720-treated mice. (a) A normal lung; (b) The lung of a bleomycin-treated mouse; (c) The lung of a bleomycin plus SRT1720-treated mouse. Results representative of each treatment are presented; (d) The fibrotic areas in (b) and (c) are quantified using Ashcroft scores.
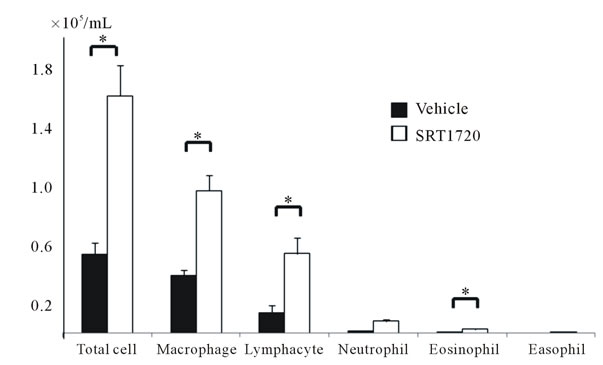
Figure 3. The cell numbers in bronchoalveolar lavage from bleomycin-treated mice (closed column), and bleomycin plus SRT1720-treated mice (open column). The total cell number was lower in the SRT1720-treated group with a value of *p < 0.10.
inflammatory cells and fibrosis.
3.4. Determination of the Toxic Concentration of SRT1720 in Cultured Cells
The results above suggest SRT1720 to be harmful in animal models of ILD such as bleomycin-induced lung injury. Because apoptosis plays an important role in ILD [5], we evaluated the influence of SRT1720 on apoptosis in cultured cells. To this end, we first determined the toxic concentration of the compound in a lung cell line, A549. SRT1720 had no effect on cell viability at up to 1 μM in either cell line (Figure 4). Milne et al. [23] found that SRT1720 was effective in deacetylating the p53 protein at 0.15 μM in a human osteosarcoma cell line. Thus, A549 cells were used for the subsequent experiment with a SRT1720 concentration of 1 μM.
3.5. SRT1720 Enhances Apoptosis in Lung Epithelial Cells
Apoptosis of A549 cells, a lung epithelial cell line, was reported to be induced by ionizing radiation and treatment with an agonistic Fas antibody [27,28].
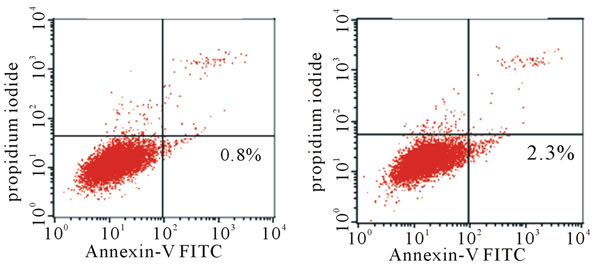
Here, we assessed the role of SRT1720 on apoptosis in A549 cells. Apoptosis was evaluated by flow cytometry in which early apoptotic cells were stained with FITClabeled annexin-V, and without propidium iodide. X-ray irradiation induced apoptosis in A549 cells (Figures 5(a) and (b)). Treatment with the Fas-activating antibody promoted apoptosis (Figure 5(c)). SRT1720 treatment markedly enhanced the apoptosis induced by irradiation and Fas activation. It should be noted that late apoptosis (and necrosis) occurs profoundly in these cells, as seen in the upper right quadrant (Figure 5(d)). SRT1720 alone gave a result similar to that of untreated cells (data not shown). These results clearly indicate that SRT1720 significantly promotes apoptosis in physically damaged and

Figure 4. Cell viability as a function of the SRT1720 concentration. A549 cell viability was examined by the MTS assay in the presence of SRT1720, as described in the Materials and Methods. Three determinations were made at each concentration.
 (a)
(a)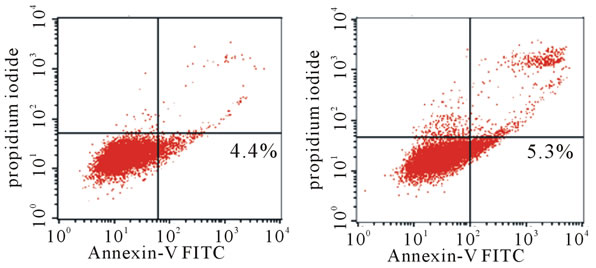 (b)
(b)
Figure 5. Apoptosis of variously treated A549 cells as analyzed by flow cytometry. Apoptosis was induced in A549 cells by X-ray irradiation, the addition of Fas-activating antibody, and/or the addition of SRT1720, as described in the Materials and Methods. Early apoptotic cells were stained by FITC-labeled annexin V but not by PI (lower right quadrants with the percentage of the total population). (a) Nonirradiated cells; (b) X-ray-irradiated cells; (c) Irradiated and Fas-activating antibody-treated cells; (d) Irradiated and Fas-activating antibody in the presence of 1 μM SRT 1720. Results representative of three determinations are shown.
biologically stimulated cells.
4. Discussion
Bleomycin-induced lung injury or pulmonary fibrosis in rodents is the best characterized animal model with which the pathogenesis of ILDs can be unravelled. Intratracheal delivery of antibiotics damages alveolar epithetlial cells through the degradation of DNA, which results from a direct attack by ferrous ions and oxygen [26,29]. Following injury to the epithelial cells, various cytokines, chemokines, and growth factors generate inflammation, eventually leading to pulmonary fibrosis. In this study, we examined the involvement of SIRT1 in an ILD-like model by using a specific activator, SRT1720. SIRT1 has been reported to participate in a myriad of physiological phenomena, such as longevity, diabetes mellitus, metabolism, cardiac function, neurodegeneration, memory and plasticity, inflammation, circadian rhythms, and cancer [30,31]. SIRT1 is believed to act as an alleviator of systemic, tissue, or cellular dysfunction. However, the present results do not support the ameliorating effects of SIRT1 activators. Kim et al. [22] reported that inhibition of SIRT1 improved lung inflammation through the modification of HIF-1α and VEGF expression in mice. Lu et al. [32] also showed that bleomycin caused the expression of HIF-1α and VEGF, which induced fibroblast proliferation and collagen production. Bleomycin treatment augments the number of inflammatory macrophages, lymphocytes, and neutrophils, and then fibrosis in the lung. Additional treatment with SRT1720 enhanced the number of inflammatory cells and fibrosis (Figure 3). Our results support these two papers suggesting the inhibition of SIRT1 to be effective at preventing or treating allergic and inflammatory airway diseases. Conversely, Sener et al. [33] reported that resveratrol, a naturally occurring SIRT1 activator, alleviated bleomycin-induced lung injury in rats. They explained that this effect was due to the antioxidative properties of the SIRT1 activator. It is necessary to apply our experimental conditions to rats to examine whether such a difference is due to the species used.
Irradiation-induced lung injury is another model of ILD that is clinically relevant. It is also considered to occur via lung epithelial injury [34] and oxidative stress [35]. While pulmonary Fas signaling is important for apoptosis of lung epithelial cells during lung injury [36], ionizing radiation also up-regulates Fas expression on the cell surface via activation of p53 [37]. We previously reported that oxidative stress induced the up-regulation of Fas expression in A549 cells [27] and ionizing radiation promoted the cell surface expression of Fas by suppressing FAP-1 mRNA levels [28]. In this context, we investigated the effects of SRT1720 on X-ray irradiation-induced and Fas-activating antibody-induced apoptosis in A549 cells. SRT1720 actually increased the number of apoptotic cells (Figure 5). Yeung et al. [38] reported that SIRT1 has a paradoxical effect of “pro-survival or pro-death” depending on the stimuli used in individual experiments. Resveratrol, considered to be capable of protecting cells from p53-induced apoptosis, increases apoptosis in response to TNFα that deacetylates to inhibit the transactivation potential of the RelA/p65 protein. It remains to be clearly shown whether the p53 pathway or the NF-kB pathway is actually driven in our in vivo and in vitro experiments.
Overall, the results of our experiments in vivo and in vitro are consistent with the notion that SRT1720 aggravates the lung damage induced by bleomycin, and X-ray irradiation/anti-Fas antibody treatment. As mentioned above, various SIRT1 activators have been considered promising compounds for treating diabetes mellitus, fatty liver, and COPD. These compounds activate SIRT1 deacetylase and/or function as antioxidants [23]. However, the present study, together with previous reports [22,32] indicates the activation of SIRT1 to have potentially harmful effects depending on the disease or organ, especially in a state of acute inflammation.
5. Acknowledgements
We would like to thank Tomomi Kubo for her assistance with the animal experiments, Takako Matsushima, Youichi Matsukura for their assistance with cytological and histological examinations, Akihiko Kawahara for his assistance with flow cytometry, Yuji Matsuya for SRT 1720 synthesis and Isao Usui for the significant discussion.
REFERENCES
- O. J. Dempsey, K. M. Kerr, H. Remmen and A. R. Denison, “How to Investigate a Patient with Suspected Interstitial Lung Disease,” British Medical Journal, Vol. 340, 2010, p. c2843. doi:10.1136/bmj.c2843
- O. Eickelberg and M. Selman, “Update in Diffuse ParenChymal Lung Disease 2009,” American Journal of Respiratory and Critical Care Medicine, Vol. 181, No. 9, 2010, pp. 883-888. doi:10.1164/rccm.201001-0124UP
- L. E. Vanfleteren and C. F. Linssen, “Role of MicroorgaNisms in Interstitial Lung Disease,” Current Opinion in Pulmonary Medicine, Vol. 16, No. 5, 2010, pp. 489-495. doi:10.1097/MCP.0b013e32833b1c54
- P. A. Wijnen, O. Bekers and M. Drent, “Relationship between Drug-Induced Interstitial Lung Diseases and CytoChrome p450 Polymorphisms,” Current Opinion in Pulmonary Medicine, Vol. 16, No. 5, 2010, pp. 496-502. doi:10.1097/MCP.0b013e32833c06f1
- M. Chopra, J. S. Reuben and A. C. Sharma, “Acute Lung Injury, Apoptosis and Signaling Mechanisms,” Experimental Biology and Medicine, Vol. 234, No. 4, 2009, pp. 361- 371. doi:10.3181/0811-MR-318
- A. Bast, A. R. Weseler, G. R. Haenen and G. J. den Hartog, “Oxidative Stress and Antioxidants in Interstitial Lung Disease,” Current Opinion in Pulmonary Medicine, Vol. 16, No. 5, 2010, pp. 516-520. doi:10.1097/MCP.0b013e32833c645d
- J. W. Sam, S. Takahashi, I. Lippai, J. Peisach and D. L. Rousseau, “Sequence-Specific Changes in the Metal Site of Ferric Bleomycin Induced by the Binding of DNA,” Journal of Biological Chemistry, Vol. 273, No. 26, 1998, pp. 16090-16097. doi:10.1074/jbc.273.26.16090
- J. Fukumoto, C. Harada, T. Kawaguchi, S. Suetsugu, T. Maeyama, I. Inoshima, N. Hamada, K. Kuwano and Y. Nakanishi, “Amphiregulin Attenuates Bleomycin-Induced Pneumopathy in Mice,” American Journal of Physiology —Lung Cellular and Molecular, Vol. 298, No. 2, 2010, pp. L131-138. doi:10.1152/ajplung.90576.2008
- P. Camus, A. Fanton, P. Bonniaud, C. Camus and P. Foucher, “Interstitial Lung Disease Induced by Drugs and Radiation,” Respiration, Vol. 71, No. 4, 2004, pp. 301- 326. doi:10.1159/000079633
- K. H. Lee and K. H. Rhee, “Radioprotective Effect of Cyclo(l-phenylalanyl-l-prolyl) on Irradiated Rat Lung,” Journal of Microbiology and Biotechnology, Vol. 18, No. 2, 2008, pp. 369-376.
- B. Movsas, T. A. Raffin, A. H. Epstein and C. J. Link Jr., “Pulmonary Radiation Injury,” Chest, Vol. 111, No. 4, 1997, pp. 1061-1076. doi:10.1378/chest.111.4.1061
- P. R. Graves, F. Siddiqui, M. S. Anscher and B. Movsas, “Radiation Pulmonary Toxicity. From Mechanisms to Management,” Seminars in Radiation Oncology, Vol. 20, No. 3, 2010, pp. 201-207. doi:10.1016/j.semradonc.2010.01.010
- G. Blander and L. Guarente, “The Sir2 Family of Protein Deacetylases,” Annual Review of Biochemistry, Vol. 73, 2004, pp. 417-435.
- L. Bordone and L. Guarente, “Calorie Restriction, Sirt1 and Metabolism. Understanding Longevity,” Nature Reviews Molecular Cell Biology, Vol. 6, No. 4, 2005, pp. 298-305. doi:10.1038/nrm1616
- C. Cao, S. Lu, R. Kivlin, B. Wallin, E. Card, A. Bagdasarian, T. Tamakloe, W. J. Wang, X. Song, W. M. Chu, N. Kouttab, A. Xu and Y. Wan, “Sirt1 Confers Protection against Uvband h2o2-Induced Cell Death via Modulation of p53 and Jnk in Cultured Skin Keratinocytes,” Journal of Cellular and Molecular Medicine, Vol. 13, No. 96, 2009, pp. 3632-3643. doi:10.1111/j.1582-4934.2008.00453.x
- K. Hasegawa, S. Wakino, K. Yoshioka, S. Tatematsu, Y. Hara, H. Minakuchi, K. Sueyasu, N. Washida, H. Tokuyama, M. Tzukerman, K. Skorecki, K. Hayashi and H. Itoh, “Kidney-Specific Overexpression of Sirt1 Protects against Acute Kidney Injury by Retaining Peroxisome Function,” Journal of Biological Chemistry, Vol. 285, No. 17, 2010, pp. 13045-13056. doi:10.1074/jbc.M109.067728
- K. Ito, C. E. Charron and I. M. Adcock, “Impact of Protein Acetylation in Inflammatory Lung Diseases,” Pharmacology & Therapeutics, Vol. 116, No. 2, 2007, pp. 249-265. doi:10.1016/j.pharmthera.2007.06.009
- J. T. Rodgers, C. Lerin, Z. Gerhart-Hines and P. Puigserver, “Metabolic Adaptations through the Pgc-1 Alpha and Sirt1 Pathways,” FEBS Letters, Vol. 582, No. 1, 2008, pp. 46-53.
- J. H. Lim, Y. M. Lee, Y. S. Chun, J. Chen, J. E. Kim and J. W. Park, “Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1alpha,” Molecular Cell, Vol. 38, No. 6, 2010, pp. 864-878.
- Z. Ungvari, N. Labinskyy, P. Mukhopadhyay, J. T. Pinto, Z. Bagi, P. Ballabh, C. Zhang, P. Pacher and A. Csiszar, “Resveratrol Attenuates Mitochondrial Oxidative Stress in Coronary Arterial Endothelial Cells,” American Journal of Physiology—Heart and Circulatory Physiology, Vol. 297, No. 5, 2009, pp. H1876-1881. doi:10.1152/ajpheart.00375.2009
- S. R. Yang, J. Wright, M. Bauter, K. Seweryniak, A. Kode and I. Rahman, “Sirtuin Regulates Cigarette Smoke-Induced Proinflammatory Mediator Release via rela/ p65 nf-Kappab in Macrophages in Vitro and in Rat Lungs in Vivo, Implications for Chronic Inflammation and Aging,” American Journal of Physiology—Lung Cellular and Molecular, Vol. 292, No. 2, 2007, pp. L567-576.
- S. R. Kim, K. S. Lee, S. J. Park, K. H. Min, Y. H. Choe, H. Moon, W. H. Yoo, H. J. Chae, M. K. Han and Y. C. Lee, “Involvement of Sirtuin 1 in Airway Inflammation and Hyperresponsiveness of Allergic Airway Disease,” Journal of Allergy and Clinical Immunology, Vol. 125, No. 2, 2010, pp. 449-460. doi:10.1016/j.jaci.2009.08.009
- J. C. Milne, P. D. Lambert, S. Schenk, D. P. Carney, J. J. Smith, D. J. Gagne, L. Jin, O. Boss, R. B. Perni, C. B. Vu, J. E. Bemis, R. Xie, J. S. Disch, P. Y. Ng, J. J. Nunes, A. V. Lynch, H. Yang, H. Galonek, K. Israelian, W. Choy, A. Iffland, S. Lavu, O. Medvedik, D. A. Sinclair, J. M. Olefsky, M. R. Jirousek, P. J. Elliott and C. H. Westphal, “Small Molecule Activators of Sirt1 as Therapeutics for the Treatment of Type 2 Diabetes,” Nature, Vol. 450, No. 7170, 2007, pp. 712-716. doi:10.1038/nature06261
- Y. Yamazaki, I. Usui, Y. Kanatani, Y. Matsuya, K. Tsuneyama, S. Fujisaka, A. Bukhari, H. Suzuki, S. Senda, S. Imanishi, K. Hirata, M. Ishiki, R. Hayashi, M. Urakaze, H. Nemoto, M. Kobayashi and K. Tobe, “Treatment with Srt1720, a Sirt1 Activator, Ameliorates Fatty Liver with Reduced Expression of Lipogenic Enzymes in Msg Mice,” American Journal of Physiology—Endocrinology and Metabolism, Vol. 297, No. 5, 2009, pp. E1179-E1186. doi:10.1152/ajpendo.90997.2008
- T. Ashcroft, J. M. Simpson and V. Timbrell, “Simple Method of Estimating Severity of Pulmonary Fibrosis on a Numerical Scale,” Journal of Clinical Pathology, Vol. 41, No. 4, 1988, pp. 467-470. doi:10.1136/jcp.41.4.467
- B. B. Moore and C. M. Hogaboam, “Murine Models of Pulmonary Fibrosis,” American Journal of Physiology— Lung Cellular and Molecular, Vol. 294, No. 2, 2008, pp. L152-160. doi:10.1152/ajplung.00313.2007
- T. Fujita, M. Maruyama, J. Araya, K. Sassa, Y. Kawagishi, R. Hayashi, S. Matsui, T. Kashii, N. Yamashita, E. Sugiyama and M. Kobayashi, “Hydrogen Peroxide Induces Upregulation of Fas in Human Airway Epithelial Cells via the Activation of Parp-p53 Pathway,” American Journal of Respiratory Cell and Molecular Biology, Vol. 27, No. 5, 2002, pp. 542-552.
- T. Yamada, M. Maruyama, T. Fujita, K. Miyabayashi, C. Shinoda, Y. Kawagishi, T. Fujishita, R. Hayashi, T. Miwa, N. Arai, S. Matsui, E. Sugiyama and M. Kobayashi, “Ionizing Radiation Suppresses Fap-1 Mrna Level in a549 Cells via p53 Activation,” FEBS Letters, Vol. 580, No. 18, 2006, pp. 4387-4391. doi:10.1016/j.febslet.2006.07.003
- E. A. Sausville, J. Peisach and S. B. Horwitz, “A Role for Ferrous Ion and Oxygen in the Degradation of DNA by Bleomycin,” Biochemical and Biophysical Research Communications, Vol. 73, No. 3, 1976, pp. 814-822. doi:10.1016/0006-291X(76)90882-2
- S. Chung, H. Yao, S. Caito, J. W. Hwang, G. Arunachalam and I. Rahman, “Regulation of SIRT1 in Cellular Functions: Role of Polyphenols,” Archives of Biochemistry and Biophysics, Vol. 501, No. 1, 2010, pp. 79-90. doi:10.1016/j.abb.2010.05.003
- E. Maury, K. M. Ramsey and J. Bass, “Circadian Rhythms and Metabolic Syndrome: From Experimental Genetics to Human Disease,” Circulation Research, Vol. 106, No. 3, 2010, pp. 447-462. doi:10.1161/CIRCRESAHA.109.208355
- Y. Lu, N. Azad, L. Wang, A. K. Iyer, V. Castranova, B. H. Jiang and Y. Rojanasakul, “Phosphatidylinositol-3-kinase/ akt Regulates Bleomycin-Induced Fibroblast Proliferation and Collagen Production,” American Journal of Respiratory Cell and Molecular Biology, Vol. 42, No. 4, 2010, pp. 432-441. doi:10.1165/rcmb.2009-0002OC
- G. Sener, N. Topaloglu, A. O. Sehirli, F. Ercan and N. Gedik, “Resveratrol Alleviates Bleomycin-Induced Lung Injury in Rats,” Pulmonary Pharmacology & Therapeutics, Vol. 20, No. 6, 2007, pp. 642-649. doi:10.1016/j.pupt.2006.07.003
- K. R. Trott, T. Herrmann and M. Kasper, “Target Cells in Radiation Pneumopathy,” International Journal of Radiation Oncology Biology Physics, Vol. 58, No. 2, 2004, pp. 463-469. doi:10.1016/j.ijrobp.2003.09.045
- J. E. Moulder and E. P. Cohen, “Future Strategies for Mitigation and Treatment of Chronic Radiation-Induced Normal Tissue Injury,” Seminars in Radiation Oncology, Vol. 17, No. 2, 2007, pp. 141-148. doi:10.1016/j.semradonc.2006.11.010
- N. Hagimoto, K. Kuwano, H. Miyazaki, R. Kunitake, M. Fujita, M. Kawasaki, Y. Kaneko and N. Hara, “Induction of Apoptosis and Pulmonary Fibrosis in Mice in Response to Ligation of Fas Antigen,” American Journal of Respiratory Cell and Molecular Biology, Vol. 17, No. 3, 1997, pp. 272-278.
- M. A. Sheard, B. Vojtesek, L. Janakova, J. Kovarik and J. Zaloudik, “Up-Regulation of Fas (CD95) in Human P53 wild-Type Cancer Cells Treated with Ionizing Radiation,” International Journal of Cancer, Vol. 73, No. 5, 1997, pp. 757-762. doi:10.1002/(SICI)1097-0215(19971127)73:5<757::AID-IJC24>3.0.CO;2-1
- F. Yeung, J. E. Hoberg, C. S. Ramsey, M. D. Keller, D. R. Jones, R. A. Frye, M. W. Mayo, “Modulation of NF-BDependent Transcription and Cell Survival by the SIRT1 Deacetylase,” EMBO Journal, Vol. 23, No. 12, 2004, pp. 2369-2380.