Graphene
Vol. 1 No. 2 (2012) , Article ID: 23970 , 21 pages DOI:10.4236/graphene.2012.12005
Recent Advances in Fabrication and Characterization of Graphene-Polymer Nanocomposites
School of Chemistry, Physics and Mechanical Engineering, Faculty of Science and Engineering, Queensland University of Technology, Brisbane, Australia
Email: c2.yan@qut.edu.au
Received August 14, 2012; revised September 20, 2012; accepted October 12, 2012
Keywords: Graphene; Polymer Nanocomposites; Fabrications and Properties
ABSTRACT
Graphene has attracted considerable interest over recent years due to its intrinsic mechanical, thermal and electrical properties. Incorporation of small quantity of graphene fillers into polymer can create novel nanocomposites with improved structural and functional properties. This review introduced the recent progress in fabrication, properties and potential applications of graphene-polymer composites. Recent research clearly confirmed that graphene-polymer nanocomposites are promising materials with applications ranging from transportation, biomedical systems, sensors, electrodes for solar cells and electromagnetic interference. In addition to graphene-polymer nanocomposites, this article also introduced the synergistic effects of hybrid graphene-carbon nanotubes (CNTs) on the properties of composites. Finally, some technical problems associated with the development of these nanocomposites are discussed.
1. Introduction
Development of novel polymer-nanocomposites (PNCs) has been attracting growing research effort worldwide over last few decades. In contrast to conventional composites, PNCs are featured by the fillers with a size of less than 100 nanometers. The advantage of polymer-nanocomposite is to provide value-added properties to the pristine polymer without sacrificing its processability, inherent mechanical properties and light weight [1,2]. The key features in design and behaviour of PNCs include the size and property of nanofiller, and the interface between nanofiller and the matrix [3]. In recent past, carbon nanotubes (CNTs) based PNCs have been widely investigated. The intrinsic bundling of CNTs, the limited availability of high quality nanotubes and high cost limited their applications [2,4]. Graphene has attracted attention as a promising candidate to create new PNCs due to its excellent properties and readily availability of its precursor, graphite. The incorporation of graphene can dramatically enhance the electrical, physical, mechanical, and barrier properties of polymer composites at extremely low loadings.
The extent of the improvement is directly related to the degree of dispersion of the nanofillers in the polymer matrix [5]. Graphene is a planar monolayer of sp2 hybridized carbon atoms arranged into a two-dimensional (2D) honeycomb lattice with a carbon-carbon bond length of 0.142 nm. The adjacent graphene sheets in graphite are separated from each other by 0.335 nm, which is half the crystallographic spacing of hexagonal graphite. The adjacent graphene sheets are held together by weak Van der Waals forces and thus the graphene sheets can slide with respect to each other giving graphite its soft and lubricating properties. Electrons in graphene behave like massless relativistic particles, which contribute to very peculiar properties such as an anomalous quantum Hall effect and the absence of localization [6]. Graphene has demonstrated a variety of intriguing properties including high electron mobility at room temperature (250,000 cm2/Vs) exceptional thermal conductivity (5000 Wm−1·K−1) and superior mechanical properties with Young’s modulus of 1 TPa. Graphene can take part in certain classes of reactions including cyclo-additions, click reactions, and carbine insertion reactions [7]. However, reactions on the surfaces of graphene hamper its planar structure. The destruction of the sp2 structure leads to the formation of defects and loss of electrical conductivity [8].
Graphene can be prepared by various methods including micromechanical cleavage, epitaxial growth, chemical vapour deposition (CVD), exfoliation of graphite intercalation compounds (GICs) and chemical oxidation-reduction methods [9-11]. Among these methods, micromechanical cleavage is more reliable and effective method to produce high quality graphene. However, this approach is limited by its low production yield [8,12,13]. Both epitaxial growth and CVD techniques can also produce high quality graphene with excellent physical properties. But, with these approaches, it is difficult to obtain a high yield to satisfy the need as composite fillers. GICs are formed by the insertion of atomic or molecular layers of different chemical species between the layers of graphite. Exfoliation of GICs can produce large quantity of graphene with perfect graphene structure. However, graphene obtained from this method consists multilayered sheets because of restacking of graphene layers after deintercalation. At present, the most viable route to produce graphene in considerable quantities is reduction of graphite oxide. Graphite oxide is generally synthesized though oxidation of graphite using strong mineral acids and oxidizing agents, typically via treatment with KMnO4 and H2SO4 based on hummers method [14]. Compared to pristine graphene, graphene oxide (GO) is heavily oxygenated and its basal plane carbon atoms are decorated with epoxide and hydroxyl groups and its edge atoms with carbonyl and carboxyl groups. Hence, GO is highly hydrophilic and the presence of these functional groups reduces interplanar forces, which can improve the interfacial interaction between GO and some polymers and thus the dispersion state of GO in polymer matrices [15,16]. But, the oxidizing chemical treatment inevitably generates structural defects such as Stone-Wales (S-W) type defects, single and multiply vacancies, dislocation like defects, carbon adatoms, or accessory chemical groups. These atomic scale structural defects adversely affect the mechanical performance of graphene [17,18]. Further, the structural defects interrupt the electronic structure of graphene and change it to semi-conductive [8,13,19,20]. High temperature thermal annealing or low temperature chemical reduction processes can be carried out to make insulating GO to conductive graphene. Thermally reducing process is generally carried out by rapid heating (2000˚C/min) up to 1050˚C in vacuum or inert atmosphere while chemical reduction is based on chemical reactions of GO with chemical reducing agents [16,21]. Most commonly used chemical reducing agents are hydrazine and its derivatives [22,23], metal hydrides [24,25], HI acid [26], hydroquinone [27], p-phenylene diamine [28] etc. Different reducing processes result in different electrical properties of reduced graphene oxide (RGO). For example, Shin et al. [24] have found that the sheet resistance of graphite oxide film reduced using NaBH4 is much lower than that of films reduced using hydrazine. Generally, thermally reduced GO exhibits a higher conductivity compared to chemically reduced GO, as seen in Figure 1 [29]. More details of preparation methods and properties of graphene and its derivatives can be found in elsewhere [5,8,13,21,30].
2. Graphene-Polymer Nanocomposites
Graphene and its derivatives filled polymer nanocomposites have shown immense potential applications in the fields of electronics, aerospace, automobile, defence industries, green energy, etc., due to its exceptional reinforcement in composites. To take full advantage of its properties for applications, integration of individual graphene in polymer matrices is prime important. Compared with CNTs, graphene has a higher surface-to-volume ratio, makes graphene potentially more favourable for improving the properties of polymer matrices, such as
 (a)
(a)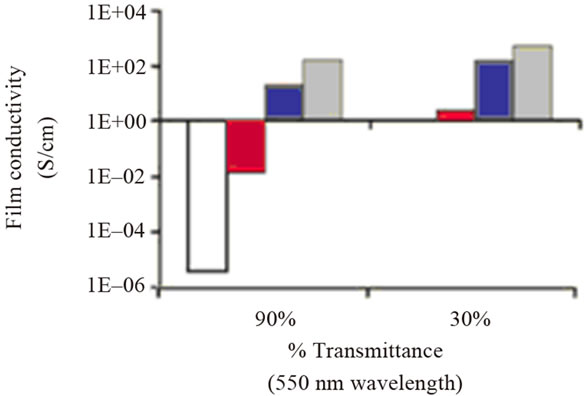 (b)
(b)
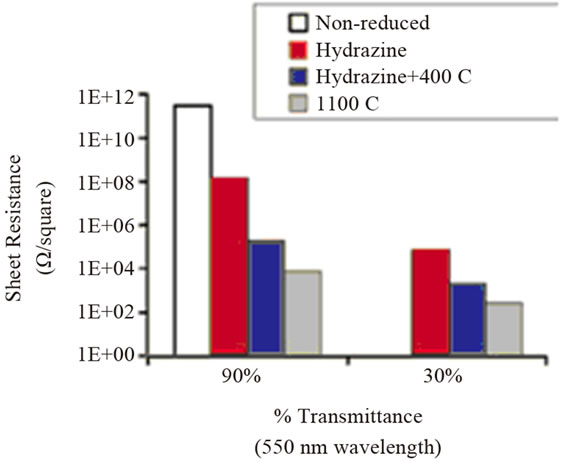
Figure 1. Comparison of the electrical properties of GO films of different optical transparency after undergoing different reduction treatment. (a) Measured sheet resistance of the films; (b) Film conductivity calculated from the sheet resistance and film thickness. Thickness of the films in the 90% transmittance group is 8.5, 5.0, 2.9 and 8.1 nm from left to right. The corresponding thickness averages are 55.3, 30.9, 66.9 nm for the films in the 30% transmittance group. Reprinted with the permission from reference [29]. Copyright 2008 American Chemical Society.
mechanical, electrical, thermal, gas permeability and microwave absorption properties. More importantly, graphene is much cheaper than CNTs, as it can be easily derived from a graphite precursor in large quantity. Many factors, including the type of graphene used and its intrinsic properties, the dispersion state of graphene in the polymer matrix and its interfacial interactions, the amount of wrinkling in the graphene, and its network structure in the matrix can affect the final properties and applications of graphene/polymer nanocomposites [20].
2.1. Synthesis of Graphene-Polymer Nanocomposites
Graphene-polymer nanocomposites have been prepared using three synthesis routes 1. Solution mixing 2. Melt blending and 3. In situ polymerization, which are most common synthesis strategies of the polymer matrix composites.
2.1.1. Solution Mixing
Solution mixing is the most straightforward method for preparation of polymer composites. The method consists three steps; dispersion of filler in a suitable solvent by, for example, ultrasonication, incorporation of the polymer and removal of the solvent by distillation or evaporation [2,30]. During the solution mixing process, polymer coats graphene sheets and when the solvent is evaporated, the graphene sheets reassemble, sandwiching the polymer to form the nanocomposite [5]. The solvent compatibility of the polymer and the filler plays a critical role in achieving good dispersion. This strategy can be employed to synthesize polymer composites with a range of polymers such as Poly (vinyl alcohol) (PVA) [31-33], Polyvinyl fluoride (PVF) [34], Polyethylene (PE) [35, 36], Poly (methylmethacrylate) (PMMA) [37], Poly (ethylmethacrylates) (PEMA) [38], Polyurethane (PU) [39]. However, solvent removal is a critical issue. Due to the oxygen functional groups, GO can be directly mixed with water soluble polymers such as PVA. Zhao et al. [30] have prepared GO-PVA composites by directly adding of PVA powder into the exfoliated aqueous dispersion of GO at 85˚C and stirring for 6h. Field Emission Scanning Electron Microscopy (FESEM) images reveal that most of the GO sheets are fully exfoliated and clearly welldispersed in the PVA matrix, while there are few restacks together. XRD observations of composites also confirmed the molecular level dispersion of GO in PVA matrix.
Chemical functionalization can improve the solubility and interaction of GO with polymers. Various types of polar polymers such as PMMA, PAA, PAN have been successfully mixed with functionalized GO (f-GO) for example, GO functionalized with isocyanate, amine [36,40] or polymer grafted GO [41] using solution mixing technique. Functionalization of graphene sheets both beneficial to disperse in water and organic solvents with reduced agglomeration and to obtain higher loading of graphene in the composites. Ultrasonication may help to obtain a homogenize dispersion of graphene sheets; however, long time exposure to high power sonication can induce defects in graphene which are detrimental to the composite properties [8].
Oxygen containing functional groups on the GO can break the conjugated structure and localize p-electrons, leading to decrease of both carrier mobility and carrier concentration. In addition, the attached groups modify the electronic structure of graphene and serve as strong scattering centers that affect the electrical transport. As a result, GO sheets are typically insulating, exhibiting a sheet resistance of about 1012 Ω/sq or higher [42]. Reduction of GO can recover the conjugated network of graphene sheets, resulting in recovery of its electrical conductivity and other properties. Conversely, reduced graphene oxide will result in irreversible restacking, which then makes dispersion of individual sheets in a polymer matrix intricate. In situ reduction can be used to both restore the conductivity and prevent restacking because of the presence of polymers in the solution mixture during the reduction [20]. Traina and co-workers [43] have prepared in situ chemically reduced GO in polyvinyl alcohol (PVOH) matrix using hydrazine hydrate in mild thermal condition. The chemically reduced GO/ PVOH composite exhibits the surface electrical resistivity of 3.1 × 105 Ω/sq at filler loading of 9.4 wt% i.e. about one order of magnitude lower than the value obtained for PVOH-GO composites at the same filler content. Dramatic enhancement of electrical conductivity for the in situ reduced GO-Nafion nanocomposites by exposure to hydrazine has been reported by Ansari et al. [44]. The graphene-Nafion nanocomposites containing 5 wt% reduced GO exhibits the electrical conductivity of 1.3 Sm−1 while the corresponding unreduced GO nanocomposite shows much lower conductivity which is below the detection limit of the experimental set up at 1 × 10−9 Sm−1. Dramatic enhancement of electrical conductivity indicated sufficient accessibility of the inorganic GO nanosheets to the reducing agent, through the nanochannels formed by the polymeric ionic domains. The chemically reducing process has been successfully used to fabricate other polymers such as vinyl acetate/vinyl chloride copolymers [45]. However, suitable reducing agents are needed to be selected depending on the type of polymer as in situ reduction may cause polymer degradation [20]. The in situ thermally reducing of GO have not been successful since the majority of polymers cannot stand high temperature that is necessary for the reduction.
2.1.2. Melt Blending
Melt blending is a more practical and versatile technique especially for thermoplastic polymers. The technique employs a high temperature and shear force to disperse fillers in the polymer matrix. High temperature softens the polymer matrix allowing easy dispersion of reinforcement phase. This process is free from toxic solvent but less effective in dispersing graphene in the polymer matrix especially at higher filler loadings due to increased viscosity of the composites [8]. Another drawback of this technique is buckling, rolling or even shortening of graphene sheets during mixing due to strong shear forces resulting in reducing its aspect ratios which is not favourable for better dispersion [20]. Kim et al. [36] have investigated the effect of blending methods on properties of graphene/polyethylene nanocomposites. Unlikely fully isolated, single graphene sheets blended in solution, melt blended samples appear predominantly phase separated and complete exfoliation is rarely observed (Figure 2). They have also found that, melt blended composites did not display notably improved electrical conductivity nearly up to 1.2 vol% graphene loading whereas solvent blended graphene could reduced the surface resistance of polymer at even as low as 0.2 vol%. However, regardless of blending methods, tensile modulus increased with incorporation of graphene into PE matrix. Similar studies and findings have been reported for graphene/polyurethane nanocomposites by Kim and co-workers in reference [39]. However, in contrast, Bao et al. [46] have successfully prepared graphene/poly (lactic acid) (PLA) nanocomposites by melt blending with improved properties. They have adopted a master-batch strategy to disperse graphene into PLA by melt blending. The graphene was well dispersed and the obtained nanocomposites present markedly improved crystallinity, rate of crystallization, mechanical properties, electrical conductivity and fire resistance. The properties are dependent on the dispersion and loading of graphene, showing percolation threshold at 0.08 wt%. A range of composites, such as Poly (vinylidene fluoride) (PVDF) [47], Polystyrene (PS) [48], polypropylene (PP) [49,50] have been prepared using this technique.
2.1.3. In Situ Polymerization
In situ polymerization is another often used technique to fabrication graphene polymer nanocomposites such as epoxy [51-54], PMMA [55], Nylon 6 [56], PU [57], poly (butylene terephthalate) (PBT) [58], polyaniline (PANI) [59], PE [60] etc. In this method, graphene or its derivative is first swollen in the liquid monomer, and then appropriate initiator is dispersed. Polymerization is initiated either by heat or radiation. The intercalation of monomers into the layered structure of graphite, during in situ polymerization, increases interlayer spacing and exfoliates graphene platelets producing well-dispersed graphene in polymer matrix after polymerization. In situ polymerization technique makes possible the covalent bonding between the functionalized sheets and polymer matrix via various chemical reactions. Major drawback of this technique is the increase of viscosity with the progress of polymerization process that hinders manipulation and limits load fraction [2,20]. Besides, in some cases, the process is carried out in the presence of solvents, thus solvent removal is a critical issue similarly in the solvent mixing technique [20]. Zaman et al. [52] have achieved the lowest electrical conductivity percolation threshold for epoxy reported, by adopting in situ polymerization technique in preparing chemically modified graphene/epoxy composites. Their investigation showed a general approach to make highly dispersed graphene/polymer nanocomposites with good control over the structure and the properties as shown in Table 1 and Figure 3.
2.2. Properties of Graphene-Polymer Nanocomposites
2.2.1. Mechanical Properties
Experimental discovery of graphene as a nanomaterial with its intrinsic strength (~1.0 TPa) and elastic modulus (125 GPa), has opened a new and interesting area in material science in recent years. In fact, better understanding of chemistry and intrinsic properties of graphene with different approaches of making it has led scientists
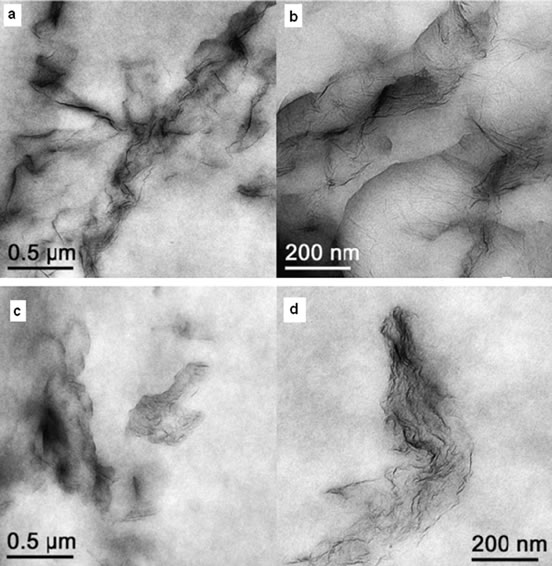
Figure 2. TEM images of 1 wt% Thermally reduced Graphene (TRG)/PE prepared by (a, b) solvent mixing (c, d) melt blending. Reprinted with the permission from reference [36]. Copyright 2011 Elsvier Ltd.
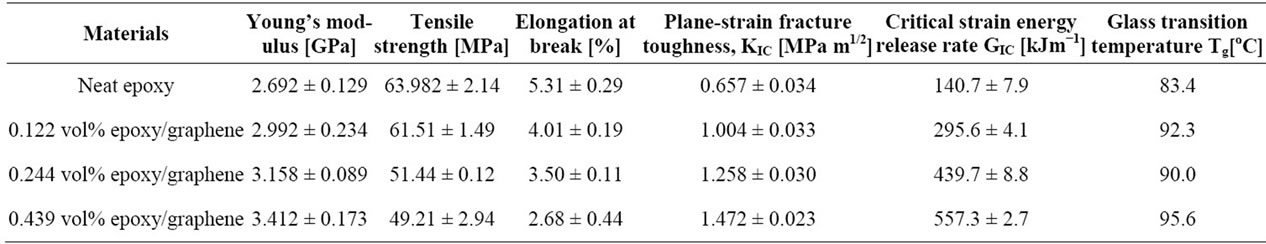
Table 1. Properties of pristine epoxy and its graphene nanocomposites. Reprinted with the permission from reference [52]. Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
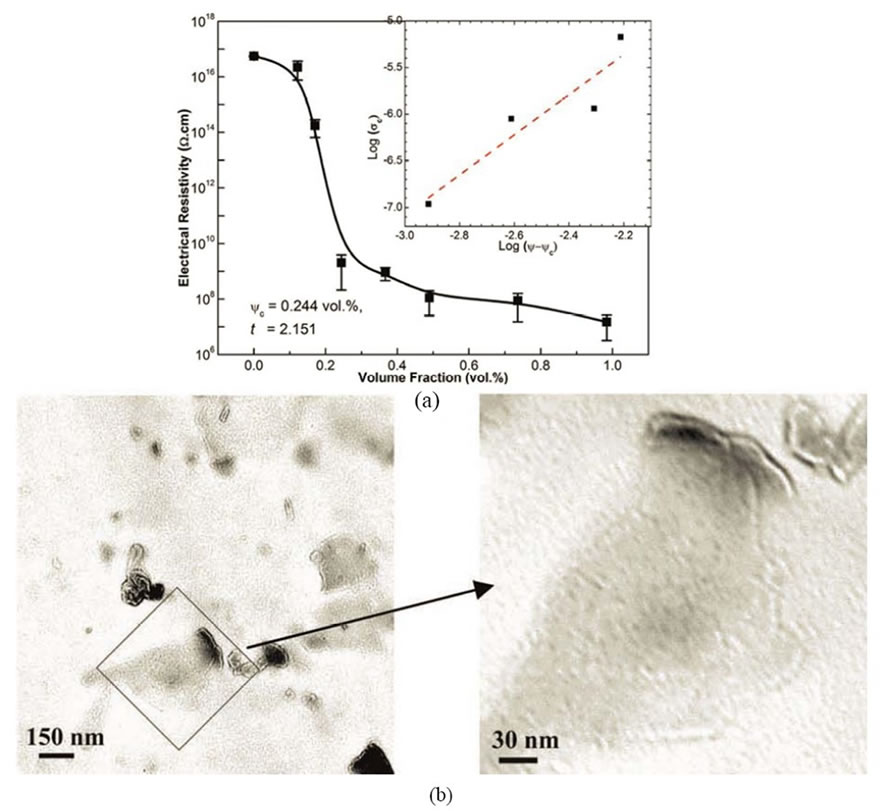
Figure 3. (a) Electrical resistivity of epoxy and its graphene nanocomposites; (b) TEM images of graphene/epoxy nanocomposites. Reprinted with the permission from reference [52]. Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
to design graphene filled polymer composites with enhanced mechanical, thermal, electrical and barrier properties. Similar to other composites, the extent of the improvement is related to many factors such as the reinforcement phase concentration and the distribution in the host matrix, interface bonding and the reinforcement phase aspect ratio. The most important aspect of these nanocomposites is that all the property enhancements are obtained at an very low filler loading in the polymer matrix [30]. Table 2 lists the percentage enhancement in the mechanical characteristics of graphene based polymer nanocomposites with respect to the base polymer matrix.
It can be observed from the table that the addition of graphene to polymer matrices can significantly influence their mechanical properties. However, the degrees of improvement are different. For an example, the tensile strength increase varies from ~0.9 for graphene/epoxy at 1.0 wt% [61], 77 for CRGO/PE at 3.0 wt% [62], and 150 for functionalized CRGO/PVA at 3.0 wt% [31]. This variation is mostly due to the structure and intrinsic properties of graphene, its surface modifications, the polymer matrix and also different polymerizing processes [12]. Although, the pristine graphene has the highest theoretical strength, it has shown poor dispersion in polymer matrices due to restacking as well as its low wettability, resulting in decreased mechanical properties of reinforced nanocomposites. GO is commonly used to improve the mechanical properties of graphene/polymer composites, for the reasons of excellent mechanical properties (e.g. Young’s modulus of monolayer of GO is 207.6 ± 23.4 GPa [63]), abundant functional groups, which facilitate strong interfacial interactions and load transfer from the host polymers to the GO and ability to significantly alter the Van der Waals interactions between the GO sheets, making them easier to disperse in polymer matrices [64]. El Achaby et al. [65] have fabricated graphene oxide nanosheets (GOn)/PVDF nanocomposite films by solution casting method with various GOn contents in dimethylformamide (DMF). Due to the strong and specific interaction between carbonyl group (C = O) in GOn surface and fluorine group (CF2) in PVDF, the GOn were homogeneously dispersed and distributed within the matrix. As shown in Figure 4, the Young’s modulus and tensile strength of PVDF were increased by 192% and 92%, respectively with the addition of 2 wt% GOn. The morphology of nanocomposites (Figure 5) where the majority of GOn has been exfoliated and uniformly dispersed throughout the polymer matrix with almost no large agglomeration is in excellent agreement with observation of improved mechanical properties. The property enhancements can be related to the strong and specific interfacial interaction that results in the adsorption of macromolecular chains of PVDF on to the GOn surface.
Strong interfacial adhesion between the graphene platelets and polymer matrix is crucial for effective reinforcement. Incompatibility between phases may lower stress transfer due to poor interfacial adhesion, resulting
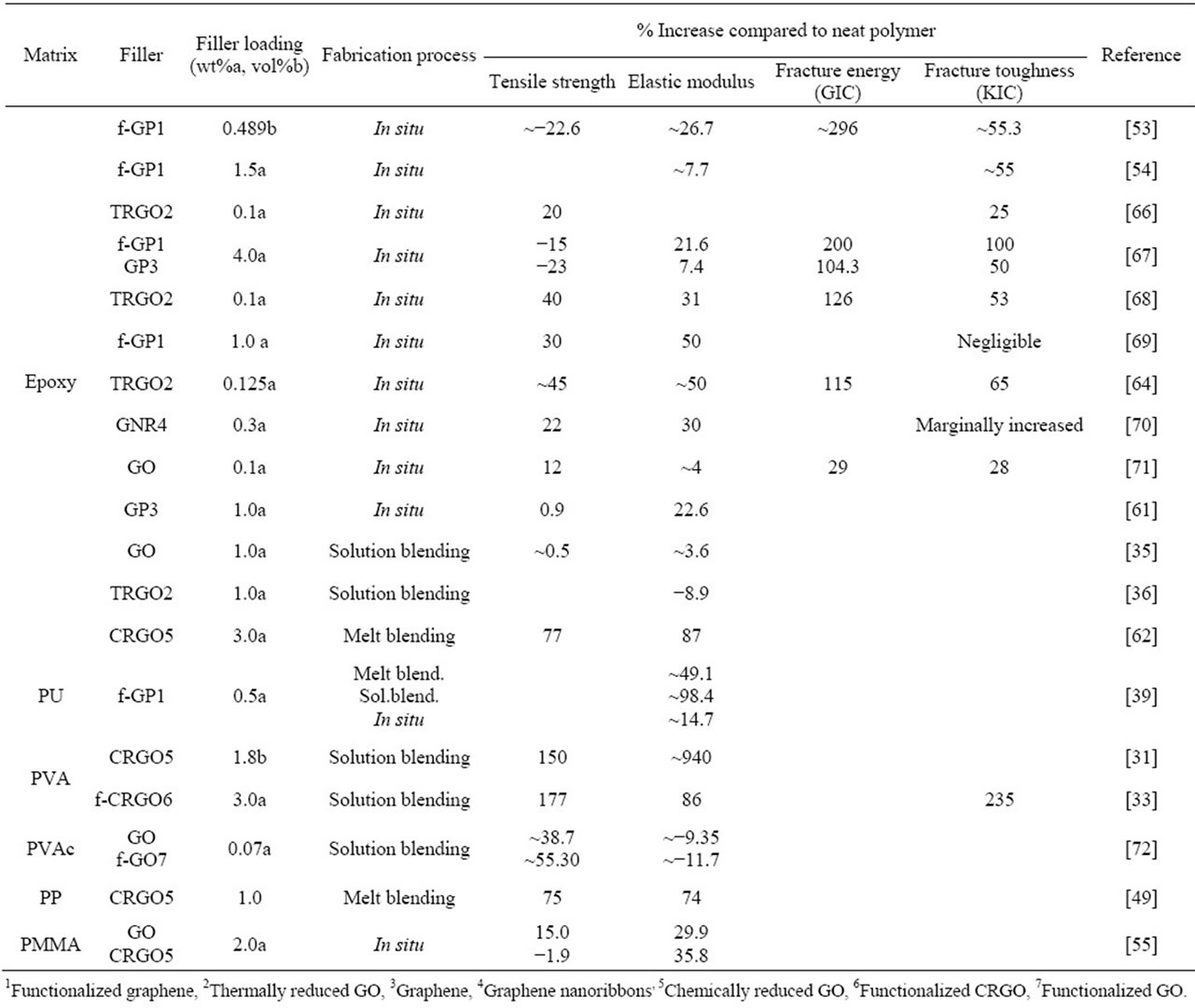
Table 2. Mechanical properties of graphene-polymer nanocomposites.
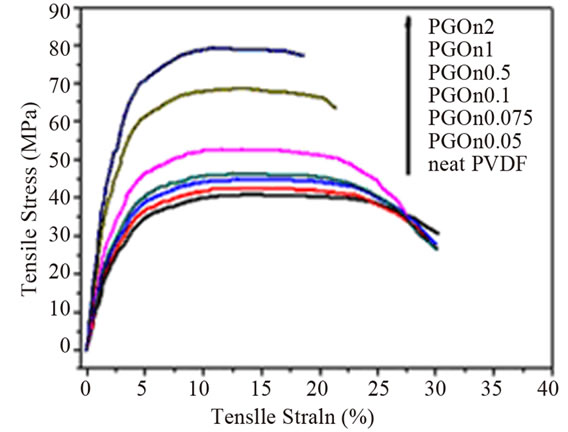
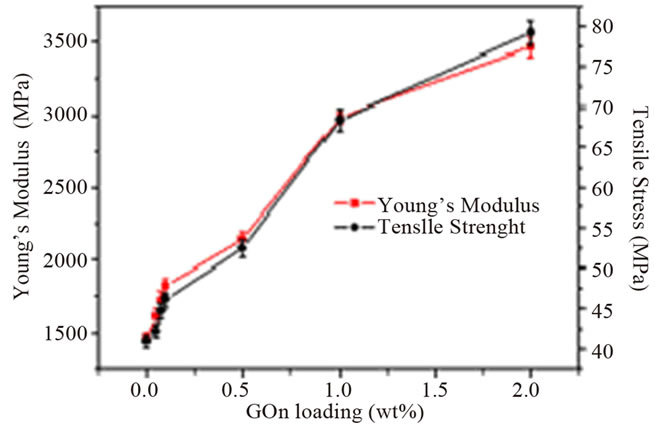
Figure 4. (a) Typical stress-strain curves of PVDF/GOn; (b) Young’s modulus and tensile strength versus GOn contents. Reprinted with the permission from reference [65]. 2012 Elsevier B.V.

Figure 5. Low (left) and high (right) magnification SEM of PVDF/GOn nanocomposite films at 2 wt% GOn. Reprinted with the permission from reference [65]. 2012 Elsevier B.V.
in a lower composite strength properties. Covalent or non-covalent functionlization of graphene based materials can be used to tailor the interface to promote stronger non-covalent interaction between the matrix and graphene platelets. Hydrogen bond interactions and Van der Waals interactions were reported as the responsible interactions for improved mechanical properties [32,73,74].
Although physical interactions can improve the properties of composites, the relative movements between the filler and matrix cannot be avoided under external stresses, which limit the attainable maximum strength. In order to alleviate this problem, chemical tailoring of the interface between filler and matrix is important which may provide the most effective means to increase the interfacial shear strength for improving stress transfer due to formation of covalent bonds between the filler and matrix [30]. For example, GO was covalently bonded to PU via the formation of urethane bonds (-NH-CO) from the reaction between the hydroxyl groups (-OH) on the surface of the GO and -NCO groups on the ends of PU chains as shown in Figure 6. This chemical bonding has led to the increase in toughness by 50% at 1 wt% loading without losing its elasticity [75]. Various chemical modifications have been reported in literature [33,48,76-79].
Other than the intrinsic properties and interfacial interaction between the graphene and host polymer, a wrinkled topology of graphene would produce an enhanced mechanical interlocking and adhesion with the polymer chains and consequently strengthens the interaction andload transfer between graphene and the polymer matrix [2,12,74,80]. Comparison of micro-mechanical predictions, utilizing Halpin-Tsai model, with experimental data shows that the theoretically predicted value for Young’s modulus of the graphene/epoxy nanocomposites is ~13% lower than the experimental results. However, the predictions for CNTs/epoxy composites are over predicted the test data by up to 12% [68]. It has been suggested that the wrinkled structure of graphene, which is different from the rectangular shape assumed by the model, may play a significant role in reinforcement. Recently, molecular dynamics and molecular mechanics simulation studies [81] showed that besides the interfacial bonding energy, the mechanical interlocking plays important roles in the interfacial bonding characteristics between the
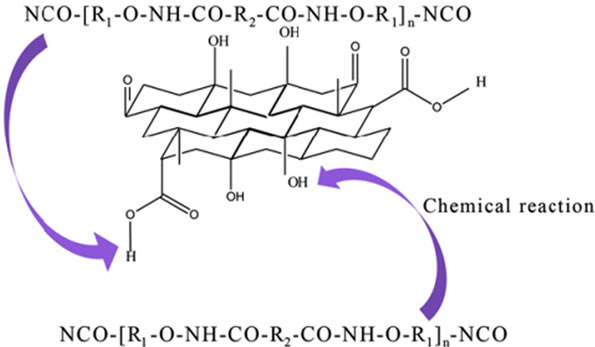
Figure 6. The schematic illustration for the formation of the covalent bonds between the GO and PU matrix. Reprinted with the permission from reference [75]. 2012 Elsevier Ltd.
graphene and polymer matrix. The study suggested nanoscale surface roughness of graphene, arise due to absorption of chemical functional groups, can more strongly interlock with the polymer molecules to arrest the polymer chains slippage and facilitate better load transfer. Rafiee et al. [64] have reported significant reinforcement from TRGO, attributed to strong interfacial bonding augmented by mechanical interlocking with matrix due to the nanoscale roughness of the platelets.
Beyond the mechanical reinforcement, other improvements in fatigue [64,82,83], creep [84], crazing [82], fracture toughness [64,68,71], impact strength [85], of the graphene-polymer nanocomposites have been reported.
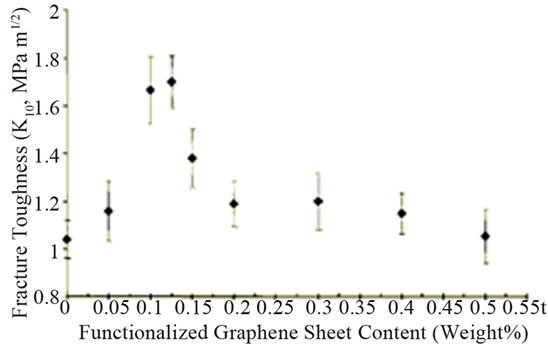
The smaller creep strain was shown in epoxy nanocomposites with 0.1 wt% graphene at the higher stress loading of 40 MPa than that of pristine epoxy [84]. This reflects the less deformation of nanocomposites compared to pristine epoxy. Further, it was found that the strain at the end of the hold period (after 36 h) was 15% smaller in the composite compared to pristine epoxy. Conversely, the creep behaviour is essentially identical for the filled and pristine epoxy at the smaller stress load of 20 MPa. Addition of 0.125 wt% TRGO into epoxy improved the fracture toughness of nanocomposite by ~65% compared to pristine epoxy [64]. It is worthy to note that to achieve comparable increase (~62%) in KIC, the required weight fraction (~14.8%) of SiO2 nanoparticles is ~120 fold larger than TRGO. Similarly, to obtain a 65% increase in KIC, the volume fraction of Al2O3 (~5%) and TiO2 (~10%) nannoparticles in epoxy is ~30 to ~60 fold larger than TRGO. For CNTs-epoxy composites, the best reported enhancement in KIC is ~43% which occurs at 4-fold higher nanofiller weight fraction [68]. However, for higher filler loading of TRGO, the enhancement in KIC diminishes and finally begins to approach the pristine epoxy value as shown in Figure 7(a). This indicates that dispersion of higher fraction of two dimensional graphene in polymer matrix is more challenging. It has also shown significant reduction in crack growth rate for the nanocomposite compared to the pristine epoxy as illustrated in Figure 7(b).
2.2.2. Conductive Properties
In Table 3, we summarize the electrical and thermal conductive properties of graphene-polymer nanocomposites from the literature with respect to base polymer matrix.
2.2.2.1. Electrical Conductivity The most fascinating property of graphene is its very high electrical conductivity. When used as fillers with insulating polymer matrix, conductive graphene may greatly enhance the electrical conductivity of the composites. The filled composite materials exhibit a nonlinear increase of the electrical conductivity as a function of the filler concentration. At certain loading fraction, known as percolation threshold, the fillers are able to form a network leading to a sudden rise of the electrical
 (a)
(a)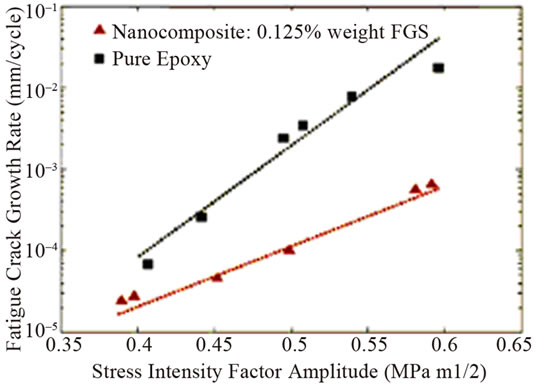 (b)
(b)
Figure 7. (a) Mode I fracture toughness (KIC) plotted as a function of the weight fraction of graphene in the epoxy matrix; (b) Crack growth rate (da/dN) plotted as a function of the stress intensity factor amplitude (ΔK) for the pristine epoxy and nanocomposite with 0.125 wt% of TRGO Reprinted with the permission from reference [64]. 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
conductivity of the composite [2]. Various factors influence the electrical conductivity and the percolation threshold of the composites such as concentration of filler, aggregation of filler, processing methods, the presence of functional groups and aspect ratio of graphene sheets, inter-sheet junction, distribution in the matrix, wrinkles and folds etc [2,8]. The filler need not be in direct contact for current flow, rather conduction can take place via tunnelling between thin polymer layers surrounding the filler particles, and this tunnelling resistance said to be the limiting factor in the composite conductivity [86]. The pristine graphene has the highest conductivity; however difficulty in producing a large amount by mechanical exfoliation limits its use. Reduction of electrically insulating graphene oxide eliminates the oxygen functional groups and partially restores the electrical conductivity, making reduced graphene oxide suitable conductive filler for composite. It is reported that thermally reduced GO has higher electrical conductivity than chemically reduced GO due to the absence of oxygenated functional groups [8]. Kim et al. [39] have studied the effect of thermal and chemical reduction of GO on electrical properties of graphene/ PU composites. The lower percolation threshold of <0.5 vol% was reported for TRGO while >2.7 vol% for graphite. However, CRGO and GO did not show decrease in surface resistance due to loss of electrical conductivity after graphite oxidation. On the contrary, recent work by Shen et al. [87] has revealed that electrical conductivity of rGO-g (2.5 × 103 S/m) (chemical reduction using glucose) is higher by four orders of magnitude compared to conductivity of TRGO (2.8 × 10−1 S/m), much higher than that of GO (2.7 × 10−7 S/m). It has been suggested that lower conductivity of TRGO is possibly due to the presence of oxygenated species and the smaller sp2 domains created by thermal reduction of GO which makes it difficult to restore the conductivity network in reduced graphene. They have also observed that significantly high electrical conductivity value for Polylactic acid (PLA)/rGO-g compared to PLA/GO nanocomposites. For example, at1.25 vol%, PLA/GO has a conductivity value of 6.47 × 10−13 S/m, while the value of PLA/rGO-g is 2.2 S/m.
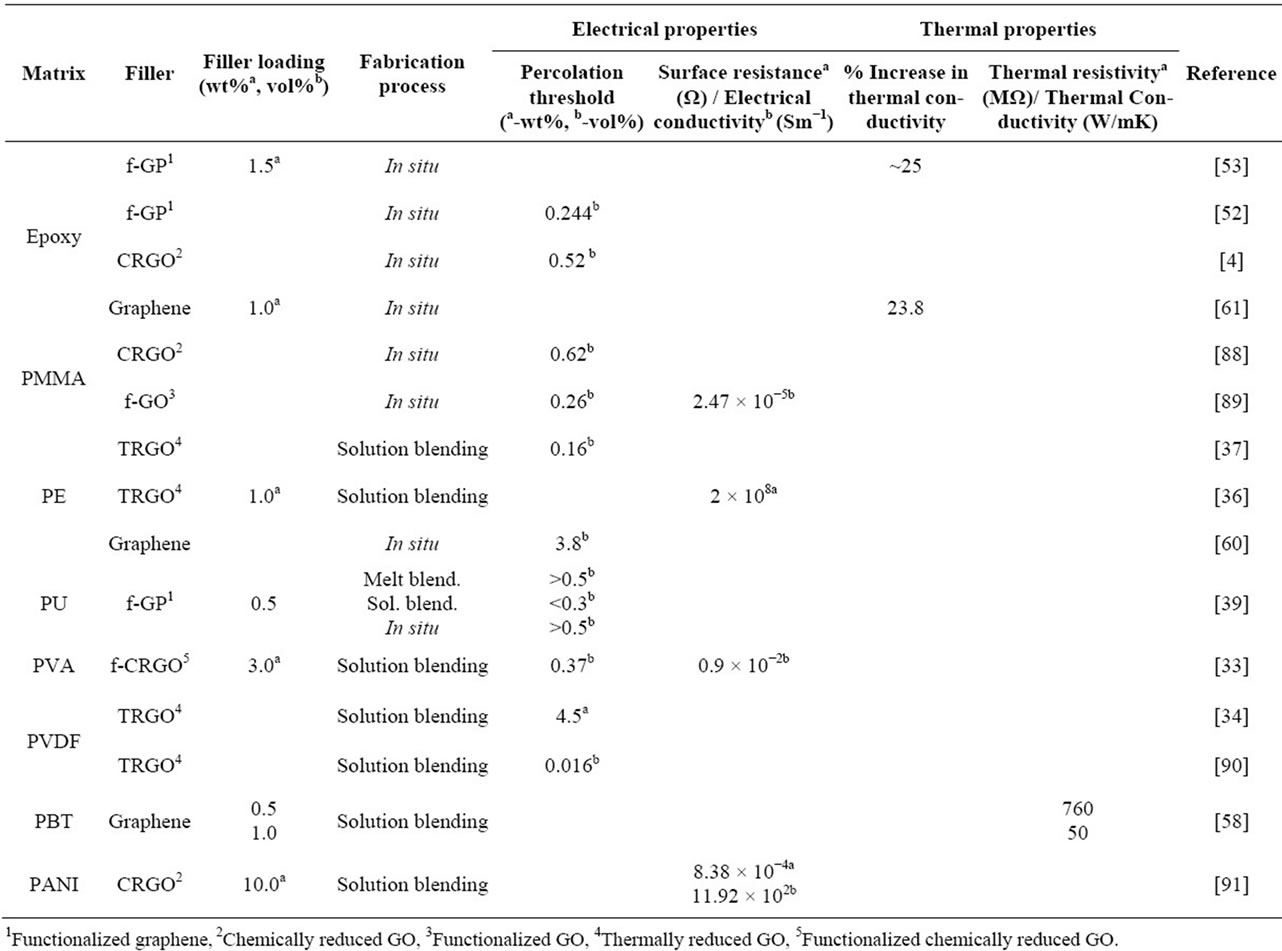
Table 3. Electrical and thermal properties of graphene/polymer nanocomposites.
Interestingly, recent work on Zhang and co-workers [37] studied the effect of surface chemistry of graphene (oxygen content of graphene sheets) on electrical property of graphene-PMMA nanocomposites. Electrical percolation threshold increases with increasing the oxygen content of graphene sheets. PMMA composites with the lowest oxygen content in graphene (graphene-13.2) show a dramatic increase in electrical conductivity of over 12 orders of magnitude, from 3.33 × 10−14 S/m with 0.4 vol% of graphene to 2.38 × 10−2 S/m with 0.8% of graphene. The conductivity reaches up to 10 S/m at 2.67 vol% (Figure 8). This rapid transition indicates the formation of an interconnected graphene network. In addition, composites with the lowest oxygen content (graphene-13.2) in graphene exhibit much higher conductiveity, in the percolation transition range than composites with higher content of oxygen (graphene-9.6 & graphene-5.0). The presence of oxygen-containing groups on graphene has been proved to disrupt its graphitic sp2 network and decrease its intrinsic conductivity. Generally, the higher the oxygen content, the lower the intrinsic conductivity.
Wang et al. [92] have reported the ability to tailor the electrical properties of the composites by altering the GO oxidation state. Each energy barrier, from either the GO surface groups or the contact between GO platelets, possesses a characteristic voltage above which the electrons can tunnel through. The total switching voltage of the composites should be the sum of those characteristic voltages. Thus adjusting the oxidation state of GO can affect the energy barriers from surface groups [93] and eventually change the total switching voltage. They have observed that by increasing the reduction temperature the
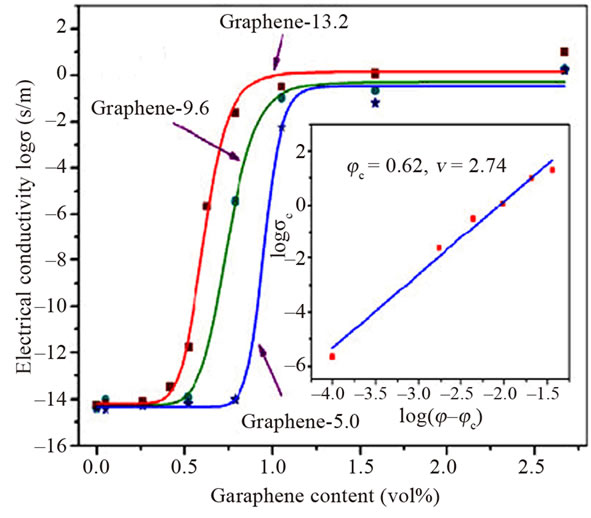
Figure 8. Electrical conductivity of graphene/PMMA composites as a function of graphene content. Reprinted with the permission from reference [37]. 2012 Elsevier Ltd.
switching field was shifted to lower electric field. It is suggested that this was due to a reduced number of oxidized surface groups, as well as the number of energy barriers. The saturated conductivity can also be changed by tuning the oxidation state of GO. This is likely due to the rearrangement of functional groups on the GO surface during the heat treatment process. Another study reported the low percolation threshold of 0.16 vol% and the highest electrical conductivity of ~64.1 S/m at 2.7 vol% for PMMA-RGO composites, prepared by a simple latex technology approach where self-assembly of positively charged PMMA latex particles and negatively charged graphene oxide sheets through electrostatic interactions, followed by hydrazine reduction [88]. The effect of temperature on electrical conductivity of graphene/PVDF composite was investigated [94]. The Graphene/PVDF composites showed a gradual increase in resistivity with temperature followed by a sharp increase when the melting point of PVDF is reached. As the temperature approaches the melting point of the polymer, the distance between particles increases (due to volume expansion of the matrix), leading to a sharp increase in resistance. In contrast, the TRGO/PVDF nanocomposites show its resistivity decreases gradually with temperature with a dramatic decrease in resistivity above the melting point. This negative temperature coefficient behaviour of TRGO/PVDF composite was attributed to the higher aspect ratio of TRGO which leads to contact resistance predominating over tunnelling resistance. Usually, contact resistance can predominate as the number of contacts increases either because of an increase in the number of particles or an increase in the aspect ratio.
2.2.2.2. Thermal Conductivity Thermal conductivity (К) of the material is governed by the lattice vibrations (phonon). High thermal conductive graphene (~3000 Wm−1·K−1, at room temperature) has been used as filler to improve the thermal conductivity and thermal stability of polymer. CNTs show similar intrinsic thermal conductivity, but sheet-like 2D structure of graphene may provide lower interfacial thermal resistance and hence produce better conductivity enhancement in polymer composites [8,86]. Other factors such as aspect ratio, orientation and dispersion of graphene sheets will also affect thermal properties of composites. Thermal conductivity of graphene based composites with different polymer matrices such as epoxy [20,53,54,61, 95,96], PMMA [37,97], PP [51], PC [98] etc. has been extensively investigated (Table 3). Shahil et al. [99, 100], have fabricated thermal interface materials (TIMs) based on epoxy and a mixture of graphene and multilayer graphene (MLG). TIMs showed cross plane thermal conductivity (K) up to ~5.1 W/mK at 10 vol% loading, which corresponds to thermal conductivity enhancement of ~2400% compared to pristine epoxy as shown in the Figure 9. This unusual enhancement has been explained by means of high intrinsic thermal conductivity and geometrical shape of graphene/MLG flakes, low thermal resistance at the graphene/matrix interface, high flexibility of MLG flakes and optimum mix of graphene and MGL with different thickness and lateral size. Chatterjee and co-workers [53], prepared amine functionalized graphene by mixing dodecylamine with expanded graphene nanoplatelets (EGNPs) under N2 atmosphere at 80˚C. These functionalized EGNPs were dispersed in epoxy using three-roll mill calendaring and resulting nanocomposites showed steady increase of thermal conductivity with EGNPs loading. At 2 wt% of EGNP loading an increment by 36% is observed as compared to pristine epoxy. The increasing trend promises higher thermal conductivity at larger EGNP concentrations. Since efficient heat propagation in EGNPs is mainly due to acoustic phonons, a uniform dispersion and network of EGNPs in the polymer matrix may contribute to the steady increase in thermal conductivity in the composites.
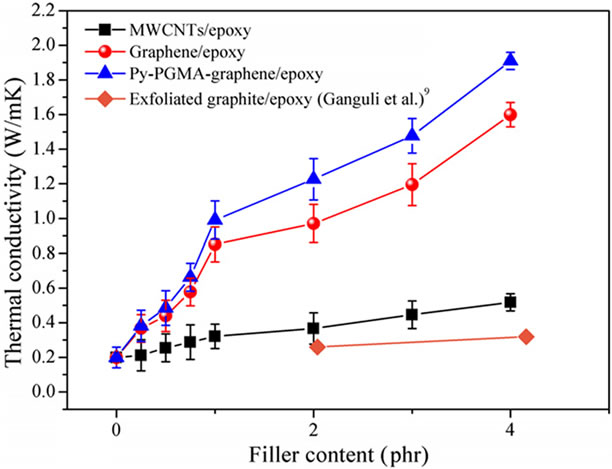
Teng and co-workers [54] have reported significant increase in thermal conductivity of epoxy composites with the increasing graphene content, which is superior to the MWCNT/epoxy composites, as illustrated in Figure 10(a). Further, chemically modified graphene (CMG)/ epoxy composite exhibited the highest improvement in thermal conductivity. For example, at 1 phr loading of CMG, thermal conductivity of composite improved by 208.7%. This significant enhancement can be because of better graphitic structure of graphene (non covalent functionalization can preserve the structure of graphene com-
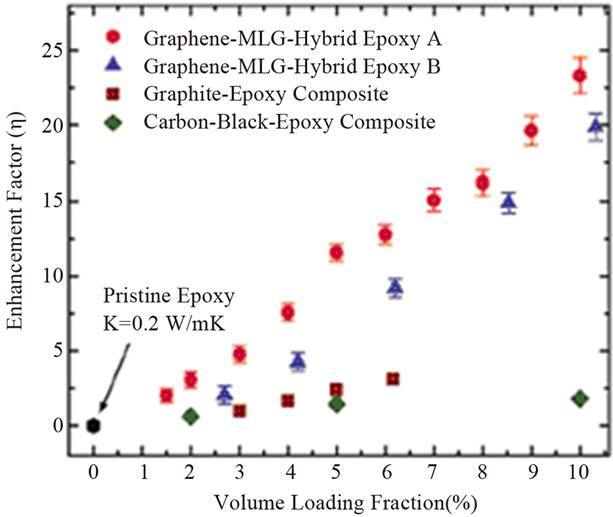
Figure 9. Thermal conductivity enhancement factor as a function of the filler volume loading fraction. Reprinted with the permission from reference [99]. 2012 American Chemical Society.
pared to thermal reduction), reduced interfacial thermal resistance due to strong interactions between CMG and epoxy matrix, and increased contact area between graphene and the matrix caused by homogeneous dispersion of CMG in the matrix. A hybrid of graphene (MGP) and multi wall carbon nanotubes (MWCNTs) was fabricated to generate the synergetic effect on thermal conductivity of epoxy nanocomposites by Yang et al. [61]. As seen in the Figure 10(b), MGP/epoxy composite showed the least improvement in thermal conductivity of all composites. By contrast, the hybrid carbon fillers/ epoxy composite exhibited a significant improvement in thermal conductivity (~147%). They proposed that this synergetic effect originated from the contact geometry changes by bridging planar graphene sheets by the MWCNTs which increases the contact surface area within hybrid nanofillers and decreased interfacial resistance within hybrid nanaofillers resulting reduced phonon scattering. A synergistic effect of hybrid of graphite nanoplatelets (GNP) and SWCNTs was reported by Yu et al. [101]. The experimental data showed a pronounced maximum of thermal conductivity of 1.75 Wm−1·K−1 at a GNP: SWCNT filler ratio of 3:1 (7.5 wt% GNPs and 2.5 wt% SWCNTs in epoxy).
2.2.3. Other properties
Thermal stability is another important property that can be improved by embedding graphene in polymer matrices. Because of high thermal stability and layered structure of graphene, incorporation of it in polymer matrices can significantly improve their thermal stability and other thermal properties like flame retardancy, thermal expansion etc. A significant number of works has reported improved thermal stability of polymers using graphene and its derivatives [37,71,102-107]. As it can be seen in Figure 11, inclusions of carbon nanofillers i.e. graphene nanosheets (GNS) and CNTs, into rigid polyurethane foam (RPUF) increase the Tg whereas decrease the Tan δ of PU [102]. Both Tg and Tan δ interpret the mobility and movement capacity of polymer molecule chain segments. The presence of GNS and CNTs highly impedes the polymer chain motion via strong interfacial interactions and acts as “physical crosslink” during the glass transition, which evidently improves the stiffness and heat resistance of the nanocomposites [102,103]. Further in reference [102], the observed amplitude of the variation in Tg and Tan δ is high for GNS nanocomposites compared with that of CNT nanocomposites (Figure 11), which is also ascribed to the greater interfacial interacttion between the matrix PU and wrinkled GNSs with unique two-dimensional geometrical morphology.
The use of polymer in high temperature applications limits by their degradation at low temperature as compared to ceramics or metals. The degradation behaviour
 (a)
(a)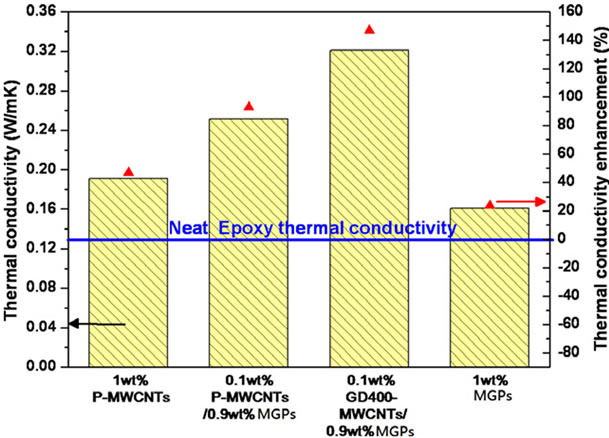 (b)
(b)
Figure 10. (a) Thermal conductivity with various filler contents of MWCNTs/epoxy, graphene/epoxy, and Py-PGMA– graphene. Reprinted with the permission from reference [54]. 2011 Elsevier Ltd. (b) Thermal conductivity of epoxy composites with 1 wt% p-MWCNTs, 0.1 wt% p-MWCNTs/ 0.9 wt% MGPs, 0.1 wt% GD400-MWCNTs/ 0.9 wt%MGPs and 1 wt% MGPs. Reprinted with the permission from reference [61]. 2010 Elsevier Ltd.
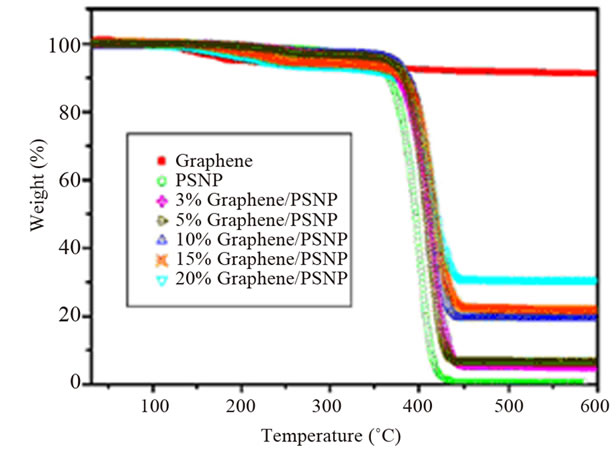
of polymers is commonly evaluated in terms of three parameters: 1) the onset temperature, considered as the temperature at which the system starts to degrade, 2) the degradation temperature, considered as the temperature at which the maximum degradation rate occurs, and 3) the degradation rate, seen in the derivative weight loss as a function of temperature curve [2]. Graphene and functionalized graphene oxide improved the thermal degradation stability of several polymer matrices, such as epoxy [105,108], HDPE [109], poly (arylene ether nitrile) (PEN) [106], polycarbonate (PC) [110]. In one study, the degradation temperature of PS composite increased with graphene content. A maximum increase of 16˚C was observed for the 20 wt% composite (Figure 12) [111]. Al though, non-reduced GO did not significantly influence
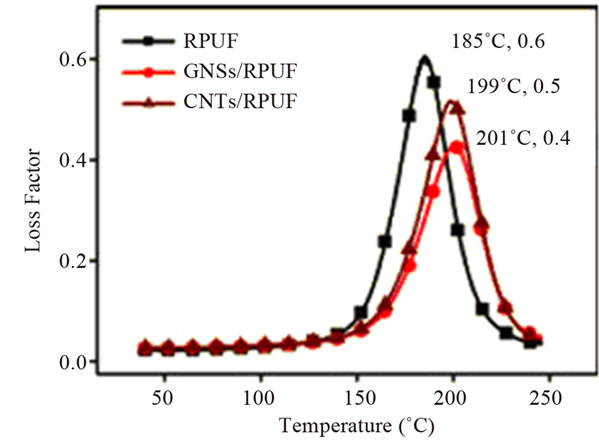
Figure 11. Temperature dependence of loss factor (tan δ) for pristine RPUF and GNSand CNT-filled RPUF nanocomposites with 0.3 wt% content. Reprinted with the permission from reference [102]. 2012 Society of Chemical Industry.
 (a)
(a)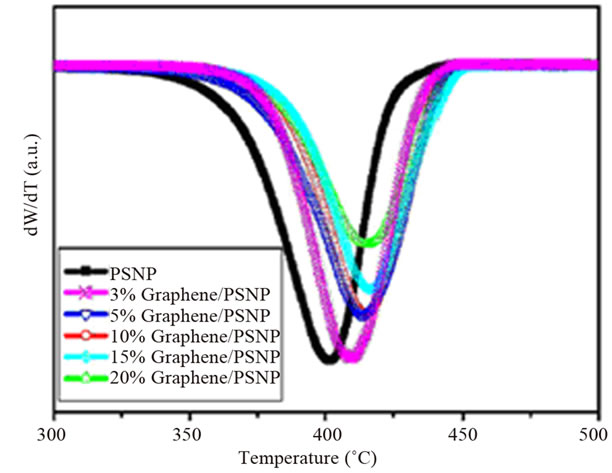 (b)
(b)
Figure 12. Thermal properties of the graphene/PS nanocomposites. (a) TGA and (b) DTG curves. Reprinted with the permission from reference [111]. 2010 Elsevier Inc.
the thermal stability of different polymers like polycarbonate (PC), acrylonitrile-butadiene-styrene (ABS), and high-impact polystyrene (HIPS), GO showed some promise toward the fabrication of polymer nanocomposites in which decreased flammability is desired [112]. Similar to SWCNTs, the negative coefficient of thermal expansion (CTE) of graphene can significantly lower the CTE of polymer matrix. Wang et al. [113] showed that the SWCNT and graphene have similar affect in decreasing CTE in epoxy matrix. More significant reduction of CTEs below Tg was observed for incorporation of 5% GO into epoxy. The reduction is as high as 31.7%. However, at above glass transition temperature (Tg), CTEs of the composites showed slight variation in comparison to the pristine resin.
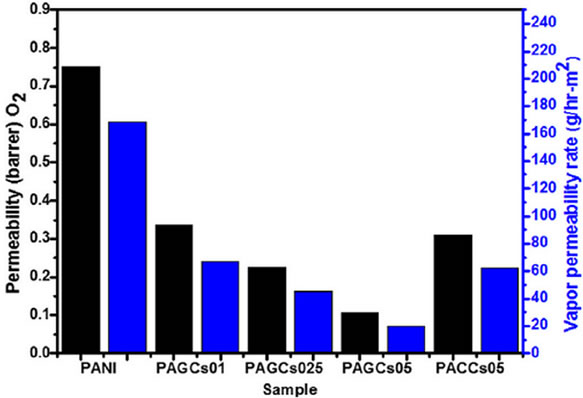
In addition to improved mechanical, electrical and thermal properties, incorporation of graphene can significantly reduce gas permeability of polymer composite relative to the pristine polymer. Various studies [8,114- 117] showed that the reduction of gas permeability is probably associated with the high aspect ratio and surface area of graphene which provide a tortuous path for the diffusing gas molecules, enhancing the gas barrier properties compared to pristine polymer. Pinto et al. [118] investigated the resistance of PLA/graphene (GNP) and PLA/GO composites to oxygen and nitrogen. The gas permeability decreased by threefold towards oxygen and a fourfold towards nitrogen at 0.4 wt% loading of GO or GNP. Though, it could be expected that more planar configuration of GNP would be more efficient in creating a tortuous path for permeation than GO particles, this was not observed, and both fillers showed similar effects. They explained this as the absence of orientation of the GNP platelets along the film plane, which does not contribute to increasing the tortuosity in the direction perpendicular to the film plane. Kim et al. [39], reported comparison study of gas permeability of various forms of graphene reinforced PU by different processing techniques. They have found that in situ polymerized TRGO was not as effective as solvent blended TRGO in reducing gas permeability. Further, the incorporation of isocyanate treated GO showed a 90% reduction in nitrogen permeability at 1.6 vol% loading. Detailed investigation by Chang group [116], on permeability of oxygen and water through graphene reinforced PANI nanocomposites have revealed the significant improvement in barrier properties compared to that of the nanoclay reinforced PANI as illustrated in Figure 13.
3. Graphene-CNTs Hybrid-Polymer Nanocomposites
Carbon nanotubes (CNTs) and graphene which are representatives of one and two dimensional nanostructure have attracted considerable attention over last two decades due to their excellent properties and wide applications. Graphene, a single-atomic layer of carbon hexagons, can be stacked into graphite or rolled up into cylindrical CNTs. They are mutually complementary in both structure and properties and yet share many common properties such as ultrahigh mechanical strength and electrical conductivity. However, they have their own drawbacks. CNT have superior mechanical properties but must be dispersed uniformly and form a network to achieve sufficient percolation for electrical conductivity. On the other hand, graphene has remarkably high electron mobility at room temperature but causes problem of its restacking property [119,120]. Zhang et al. [121] classified the graphene-CNT hybrids into three types, CNTs adsorbed horizontal to the graphene sheets (GNS), CNTs adsorbed perpendicular to the GNS and CNT wrapped with GNS.
Such hybrid structures show excellence flexibility and stretching ability and is expected to have electrical conductivity and thermal dissipation in all directions. Further, irreversible agglomeration of graphene via Van der waals interaction is found to be hindered in the presence of
 (a)
(a)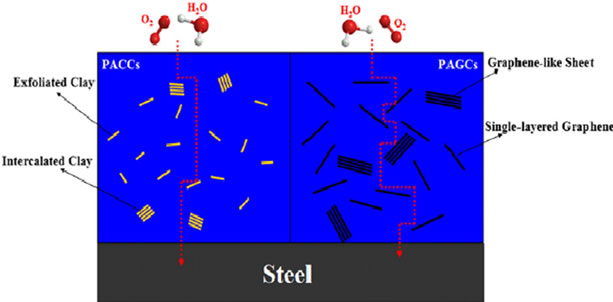 (b)
(b)
Figure 13. (a) Permeability and vapour permeability rates of PANI and nanocomposites (b) Schematic representation of O2 and H2O following a tortuous path through a polyaniline/clay composites (PACCs) and polyaniline/graphene composites (PAGCs). Reprinted with the permission from reference [116]. 2012 Elsevier Ltd.
CNTs [122]. In recent years, integrate them into a hybrid structure created a wide interest to establish synergistic effects between these two different carbon structures in composite materials.
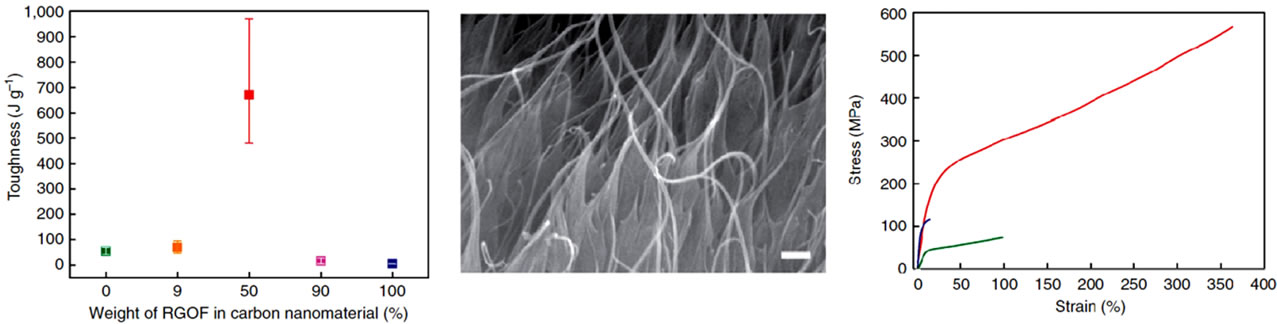
Shin et al. [123] fabricated PVA tough fibres by wet spinning of hybridized reduced GO flakes (RGOFs) and single-walled CNTs (SWNTs) into PVA solution. The fabricated fibres exhibit the toughness in the range of 480 - 970 Jg−1, far exceeding toughness of silk or Kevlar (Figure 14(a)). This synergistic toughness enhancement arises for the optimal combination of SWNTs and RGOFs (1:1), and no synergistic toughness enhancement was observed for other ratios of carbon nanoparticles. The results show that this optimal ratio of SWNT and RGOF leads to a high degree of nanoparticle self-alignment (Figure 14(b))and hinder RGOFs stacking during wet spinning which provides strong interaction with the PVA matrix, enhances crack deflection, and promotes plastic deformation (Figure 14(c)) of the stretched PVA.
Wang et al. [124] prepared SWCNT, GO and their hybrid PVA fibres and reported high strength and high conductive PVA fibre with hybrid SWCNTs and GO at 2:1 ratio. Intercalation of GO sheets into CNTs forms a well dispersed GO-CNTs network in PVA matrix which facilitates the stress transfer between the nanocarbons and PVA molecules resulting synergistic enhancement of strength properties. In addition to strength, a better dispersion state enhances the conductivity of the fibres. One study reported a marked improvement in fracture toughness and flexural modulus for different ratios of CNT and graphene with the highest improvement for CNT:graphene ratio of 9:1 [125].
Kumar et al. [126] have reported a remarkable increase in thermal and electrical conductivities of Polyetherimide (PEI) containing the hybrid ternary systems of GNPs and MWCNTs in equal amounts at a fixed loading of 0.5 wt%. In the case of thermal conductivity, composites containing hybrid fillers exhibited a 45% increase whereas composites with only GNPs or MWCNTs exhibited improvement of 22% and 9%, respectively as compared to pure PEI. The surface resistivity of hybrid composite showed 8 orders of magnitude lower than that of a composite with 0.5 wt% GNPs alone and an order of magnitude lower than MWCNT/PEI composite. The formation of an interconnected hybrid network structure between MWCNTs and GNPs may facilitate the better electron transport throughout the polymer result in reduced surface resistivity. Another possible reason for reduced surface resistivity of the hybrid composite was improved dispersion and damage prevention of carbon nanotubes in the presence of graphite nanoplatelets during the fabrication process. The preserved long length of nanotubes can bridge the gap between graphite nanoplatelets thereby allowing the greater mean free path for the electron flow. Synergistic effect of hybrid graphene-CNTs in various polymer matrices has been reported in literature [61,127-129].
3. Conclusions
We have reviewed the recent advances in fabrication and properties of graphene-polymer nanocomposites. We have also discussed the recent studies and progress of synergistic property improvement in hybrid grapheneCNT polymer nanocomposites. Based on the review, it is clear that the reinforcement of graphene and its derivatives in polymer matrices has shown very promising results in improving mechanical strength and elastic modulus, enhancing electrical conductivity at a low percolation threshold, increasing thermal conductivity, stability and flame resistance, and reducing gases and water vapour permeation. All of these enhancements have a great potential for applications in many fields either as structural or functional materials. For example, high
 (a) (b) (c)
(a) (b) (c)
Figure 14. (a) Toughness values of fibres with different weight percents of RGOF in total carbon nanomaterials. (b) SEM image of the cross-sectional area of a RGOF/SWNT/PVA fibre (1:1 weight ratio of RGOF to SWNT), which clearly shows the co-assembly of RGOFs and SWNTs. Scale bar equals 1 μm (c) Stress—strain curves of hybrid (1:1 weight fraction of RGOF to SWNT, red line) SWNT/PVA (green line) and RGOF/PVA (blue line) composite fibres. Reprinted with the permission from reference [123]. 2012 Macmillan Publishers Limited.
strength and light weight structural polymer composites can be used in aerospace and automobile industries. Mechanically reinforced thin film composites find their applications in petrochemical and biomedical industries. Thermally conductive and stabilized composites can be used in the structures requiring thermal management. Electrically conductive composites have been widely used for making various sensors, conductive electrode for solar cells, antistatic coatings, electromagnetic interference shielding, etc.
However, to further commercialize graphene-polymer composites, many technical challenges need to be overcome. Most importantly, synthesis routes for mass production of graphene are urgently required. The preparation and transfer of high quality graphene is still not practicable in a cost effective manner. At present, large amount of graphene is prepared by exfoliation followed by reduction of graphite oxide. Usually, sonication and thermal shock techniques are employed to exfoliate GO but they can reduce the aspect ratio of exfoliated GO, and adversely affect the reinforcing efficiency. Moreover, various defects and impurities are often introduced into graphene during the processing and these impurities may strongly influence the electrical, mechanical and thermal properties of graphene. In addition, structure, aspect ratio, surface chemistry and number of layers of GO/RGO are all dependent on the exfoliation and reduction procedures. Therefore, reinforcement of polymer with GO or reduced GO may exhibit undesirable properties as compared to pristine graphene-polymer composites. As such, methods for synthesis of graphene at low fabrication cost are urgently required.
Generally, the properties of polymer composites depend mainly on the dispersion state of discrete filler phase in continuous polymer matrix phase. The restacking of flat graphene sheets during fabrication makes uniform dispersion difficult and limits the available surfaces to interact effectively with polymer matrix, deteriorating the reinforcing effectiveness. Strong interfacial interacttions between graphene and the host polymers and interaction within the graphene sheets are other important factors to be considered in fabricating high performance composites. Further, property enhancement of graphenepolymer composites can be achieved by morphological control of graphene. Wrinkles and surface roughness in graphene may increase the reinforcement due to mechanical interlocking but may degrade electrical and thermal properties. Therefore, the core issues such as homogeneous dispersion of graphene sheets, their connectivity and orientation, interfacial interaction with host polymer matrix still deserve further research. In addition, possible risks associated with use of graphene and its derivatives need to be evaluated.
REFERENCES
- R. A. Vaia and H. D. Wagner, “Framework for Nanocomposites,” Materials Today, Vol. 7, No. 11, 2004, pp. 32-37. doi:10.1016/S1369-7021(04)00506-1
- R. Verdejo, M. M. Bernal, L. J. Romasanta and M. A. Lopez-Manchado, “Graphene Filled Polymer Nanocomposites,” Journal of Materials Chemistry, Vol. 21, No. 10, 2011, pp. 3301-3310. doi:10.1039/c0jm02708a
- M. Terrones, et al., “Interphases in Graphene PolymerBased Nanocomposites: Achievements and Challenges,” Advanced Materials, Vol. 23, No. 44, 2011, pp. 5302- 5310. doi:10.1002/adma.201102036
- J. Liang, et al., “Electromagnetic Interference Shielding of Graphene/Epoxy Composites,” Carbon, Vol. 47, No. 3, 2009, pp. 922-925. doi:10.1016/j.carbon.2008.12.038
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, “Recent Advances in Graphene Based Polymer Composites,” Progress in Polymer Science, Vol. 35, No. 11, 2010, pp. 1350-1375. doi:10.1016/j.progpolymsci.2010.07.005
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, “Experimental Observation of the Quantum Hall Effect and Berry’s Phase in Graphene,” Nature, Vol. 438, No. 7065, 2005, pp. 201-204.doi:10.1038/nature04235
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, “The Chemistry of Graphene,” Journal of Materials Chemistry, Vol. 20, No. 12, 2010, pp. 2277-2289. doi:10.1039/b920539j
- V. Singh, et al., “Graphene Based Materials: Past, Present and Future,” Progress in Materials Science, Vol. 56, No. 8, 2011, pp. 1178-1271. doi:10.1016/j.pmatsci.2011.03.003
- K. S. Kim, et al., “Large-Scale Pattern Growth of Graphene Films for Stretchable Transparent Electrodes,” Nature, Vol. 457, No. 7230, 2009, pp. 706-710. doi:10.1038/nature07719
- S. Grandthyll, et al., “Epitaxial Growth of Graphene on Transition Metal Surfaces: Chemical Vapor Deposition Versus Liquid Phase Deposition,” Journal of Physics: Condensed Matter, Vol. 24, No. 31, 2012, p. 314204. doi:10.1088/0953-8984/24/31/314204
- M. Gao, et al., “Epitaxial Growth and Structural Property of Graphene on Pt(111),” Applied Physics Letters, Vol. 98, No. 3, 2011, p. 033101. doi:10.1063/1.3543624
- J. Du and H.-M. Cheng, “The Fabrication, Properties, and Uses of Graphene/Polymer Composites,” Macromolecular Chemistry and Physics, Vol. 213, No. 10-11, 2012, pp. 1060-1077. doi:10.1002/macp.201200029
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, et al., “Synthesis of Graphene and Its Applications: A Review,” Critical Reviews in Solid State and Materials Sciences, Vol. 35, No. 1, 2010, pp. 52-71. doi:10.1080/10408430903505036
- W. S. Hummers and R. E. Offema, “Preparation of Graphite Oxide,” Journal of the American Chemical Society, Vol. 80, No. 6, 1958, p.1339.
- D. C. Marcano, et al., “Improved Synthesis of Graphene Oxide,” ACS Nano, Vol. 4, No. 8, 2010, pp. 4806-4814. doi:10.1021/nn1006368
- J. Du and H.-M. Cheng, “The Fabrication, Properties, and Uses of Graphene/Polymer Composites,” Macromolecular Chemistry and Physics, Vol. 213, No. 10-11, 2012, pp. 1060-1077. doi:10.1002/macp.201200029
- M. C. Wang, C. Yan, L. Ma and N. Hu, “Effect of Defects on Fracture Strength of Graphene Sheets,” Computational Materials Science, Vol. 54, 2012, pp. 236-239. doi:10.1016/j.commatsci.2011.10.032
- M. C. Wang, C. Yan and L. Ma, “Graphene Nanocomposites,” In: M. C. Wang, Ed., Composites and Their Properties, Ning Hu, In Tech, Shanghai, 2012, pp. 17-36.
- W. Lu, et al., “High-Yield, Large-Scale Production of Few-Layer Graphene Flakes Within Seconds: Using Chlorosulfonic Acid and H2O2 as Exfoliating Agents,” Journal of Materials Chemistry, Vol. 22, No. 18, 2012, pp. 8775-8777. doi:10.1039/c2jm16741g
- X. An, et al., “Stable Aqueous Dispersions of Noncovalently Functionalized Graphene from Graphite and Their Multifunctional High-Performance Applications,” Nano Letters, Vol. 10, No. 11, 2010, pp. 4295-4301. doi:10.1021/nl903557p
- S. Park and R. S. Ruoff, “Chemical Methods for the Production of Graphenes,” Nat Nano, Vol. 4, No. 4, 2009, pp. 217-224. doi:10.1038/nnano.2009.58
- S. Park, et al., “The Effect of Concentration of Graphene Nanoplatelets on Mechanical and Electrical Properties of Reduced Graphene Oxide Papers,” Carbon, Vol. 50, No. 12, 2012, pp. 4573-4578. doi:10.1016/j.carbon.2012.05.042
- T. N. Huan, T. V. Khai, Y. Kang, K. B. Shim and H. Chung, “Enhancement of Quaternary Nitrogen Doping of Graphene Oxide via Chemical Reduction Prior to Thermal Annealing and an Investigation of Its Electrochemical Properties,” Journal of Materials Chemistry, Vol. 22, No. 29, 2012, pp. 14756-14762. doi:10.1039/c2jm31158e
- H.-J. Shin, et al., “Efficient Reduction of Graphite Oxide by Sodium Borohydride and Its Effect on Electrical Conductance,” Advanced Functional Materials, Vol. 19, No. 12, 2009, pp. 1987-1992. doi:10.1002/adfm.200900167
- C. Caifeng, T. Chen, H. Wang, G. Sun and X. Yang, “A Rapid, One-Step, Variable-Valence Metal Ion Assisted Reduction Method for Graphene Oxide,” Nanotechnology, Vol. 22, No. 40, 2011, pp. 405602. doi:10.1088/0957-4484/22/40/405602
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, “Direct Reduction of Graphene Oxide Films into Highly Conductive and Flexible Graphene Films by Hydrohalic Acids,” Carbon, Vol. 48, No. 15, 2010, pp. 4466-4474. doi:10.1016/j.carbon.2010.08.006
- G. Wang, et al., “Facile Synthesis and Characterization of Graphene Nanosheets,” The Journal of Physical Chemistry C, Vol. 112, No. 22, 2008, pp. 8192-8195. doi:10.1021/jp710931h
- N. Hu, et al., “Gas Sensor Based on p-Phenylenediamine Reduced Graphene Oxide,” Sensors and Actuators B: Chemical, Vol. 163, No. 1, 2012, pp. 107-114. doi:10.1016/j.snb.2012.01.016
- H. A. Becerril, et al., “Evaluation of Solution-Processed Reduced Graphene Oxide Films as Transparent Conductors,” ACS Nano, Vol. 2, No. 3, 2008, pp. 463-470. doi:10.1021/nn700375n
- X. Huang, X. Qi, F. Boey and H. Zhang, “GrapheneBased Composites,” Chemical Society Reviews, Vol. 41, No. 2, 2012, pp. 666-686. doi:10.1039/c1cs15078b
- X. Zhao, Q. Zhang and D. Chen, “Enhanced Mechanical Properties of Graphene-Based Poly (Vinyl Alcohol) Composites,” Macromolecules, Vol. 43, No. 5, 2010, pp. 2357-2363. doi:10.1021/ma902862u
- L. Jiang, X. P. Shen, J. L. Wu and K. C. Shen, “Preparation and Characterization of Graphene/Poly (Vinyl Alcohol) Nanocomposites,” Journal of Applied Polymer Science, Vol. 118, No. 1, 2010, pp. 275-279. doi:10.1002/app.32278
- R. K. Layek, S. Samanta and A. K. Nandi, “The Physical Properties of Sulfonated Graphene/Poly (Vinyl Alcohol) Composites,” Carbon, Vol. 50, No. 3, 2012, pp. 815-827. doi:10.1016/j.carbon.2011.09.039
- Y. Jinhong, X. Huang, C. Wu and P. Jiang, “Permittivity, Thermal Conductivity and Thermal Stability of Poly (Vinylidene Fluoride)/Graphene Nanocomposites,” IEEE Transactions on Dielectrics and Electrical Insulation, Vol. 18, No. 2, 2011, pp. 478-484.
- Y. Chen, et al., “Preparation, Mechanical Properties and Biocompatibility of Graphene Oxide/Ultrahigh Molecular Weight Polyethylene Composites,” European Polymer Journal, Vol. 48, No. 6, 2012, pp. 1026-1033. doi:10.1016/j.eurpolymj.2012.03.011
- H. Kim, et al., “Graphene/Polyethylene Nanocomposites: Effect of Polyethylene Functionalization and Blending Methods,” Polymer, Vol. 52, No. 8, 2011, pp. 1837-1846. doi:10.1016/j.polymer.2011.02.017
- H.-B. Zhang, W.-G. Zhang, Q. Yan, Z.-G. Jiang and Z.-Z. Yu, “The Effect of Surface Chemistry of Graphene on Rheological and Electrical Properties of Polymethylmethacrylate Composites,” Carbon, Vol. 50, No. 14, 2012, pp. 5117-5125. doi:10.1016/j.carbon.2012.06.052
- X. Li and G. B. McKenna, “Considering Viscoelastic Micromechanics for the Reinforcement of Graphene Polymer Nanocomposites,” ACS Macro Letters, Vol. 1, No. 3, 2012, pp. 388-391. doi:10.1021/mz200253x
- H. Kim, Y. Miura and C. W. Macosko, “Graphene/Polyurethane Nanocomposites for Improved Gas Barrier and Electrical Conductivity,” Chemistry of Materials, Vol. 22, No. 11, 2010, pp. 3441-3450. doi:10.1021/cm100477v
- P.-G. Ren, D.-X. Yan, T. Chen, B.-Q. Zeng and Z.-M. Li, “Improved Properties of Highly Oriented Graphene/ Polymer Nanocomposites,” Journal of Applied Polymer Science, Vol. 121, No. 6, 2011, pp. 3167-3174. doi:10.1002/app.33856
- G. Goncalves, et al., “Graphene Oxide Modified with PMMA via ATRP as a Reinforcement Filler,” Journal of Materials Chemistry, Vol. 20, No. 44, 2010, pp. 9927- 9934. doi:10.1039/c0jm01674h
- S. Pei and H.-M. Cheng, “The Reduction of Graphene Oxide,” Carbon, Vol. 50, No. 9, 2012, pp. 3210-3228. doi:10.1016/j.carbon.2011.11.010
- M. Traina and A. Pegoretti, “In Situ Reduction of Graphene Oxide Dispersed in a Polymer Matrix,” Journal of Nanoparticle Research, Vol. 14, No. 4, 2012, pp. 1-6. doi:10.1007/s11051-012-0801-0
- S. Ansari, A. Kelarakis, L. Estevez and E. P. Giannelis, “Oriented Arrays of Graphene in a Polymer Matrix by in situ Reduction of Graphite Oxide Nanosheets,” Small, Vol. 6, No. 2, 2010, pp. 205-209. doi:10.1002/smll.200900765
- T. Wei, et al., “Preparation of Graphene Nanosheet/ Polymer Composites Using in Situ Reduction-Extractive Dispersion,” Carbon, Vol. 47, No. 9, 2009, pp. 2296- 2299. doi:10.1016/j.carbon.2009.04.030
- C. Bao, et al., “Preparation of Graphene by Pressurized Oxidation and Multiplex Reduction and Its Polymer Nanocomposites by Masterbatch-Based Melt Blending,” Journal of Materials Chemistry, Vol. 22, No. 13, 2012, pp. 6088-6096. doi:10.1039/c2jm16203b
- M. El Achaby, et al., “Preparation and Characterization of Melt-Blended Graphene Nanosheets-Poly (Vinylidene Fluoride) Nanocomposites with Enhanced Properties,” Journal of Applied Polymer Science, 2012 (Online Version) doi: 10.1002/app.38081
- F. Beckert, C. Friedrich, R. Thomann and R. Mülhaupt, “Sulfur-Functionalized Graphenes as Macro-Chain-Transfer and RAFT Agents for Producing Graphene Polymer Brushes and Polystyrene Nanocomposites,” Macromolecules, Vol. 45, No. 17, 2012, pp. 7783-7090. doi:10.1021/ma301379z
- P. Song, et al., “Fabrication of Exfoliated GrapheneBased Polypropylene Nanocomposites with Enhanced Mechanical and Thermal Properties,” Polymer, Vol. 52, No. 18, 2011, pp. 4001-4010. doi:10.1016/j.polymer.2011.06.045
- M. El Achaby, et al., “Mechanical, Thermal, and Rheological Properties of Graphene-Based Polypropylene Nanocomposites Prepared by Melt Mixing,” Polymer Composites, Vol. 33, No. 5, 2012, pp. 733-744. doi:10.1002/pc.22198
- Z.-L. Mo, T.-T. Xie, J.-X. Zhang, Y.-X. Zhao and R.-B. Guo, “Synthesis and Characterization of NanoGs-PPy/ Epoxy Nanocomposites by In Situ Polymerization,” Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry, Vol. 42, No. 8, 2012, pp. 1172- 1176. doi:10.1080/15533174.2012.684259
- I. Zaman, et al., “A Facile Approach to Chemically Modified Graphene and Its Polymer Nanocomposites,” Advanced Functional Materials, Vol. 22, No. 13, 2012, pp. 2735-2743. doi:10.1002/adfm.201103041
- S. Chatterjee, et al., “Mechanical Reinforcement and Thermal Conductivity in Expanded Graphene Nanoplatelets Reinforced Epoxy Composites,” Chemical Physics Letters, Vol. 531, 2012, pp. 6-10. doi:10.1016/j.cplett.2012.02.006
- C.-C. Teng, et al., “Thermal Conductivity and Structure of Non-Covalent Functionalized Graphene/Epoxy Composites,” Carbon, Vol. 49, No. 15, 2011, pp. 5107-5116. doi:10.1016/j.carbon.2011.06.095
- J. R. Potts, et al., “Thermomechanical Properties of Chemically Modified Graphene/Poly (Methyl Methacrylate) Composites Made by in Situ Polymerization,” Carbon, Vol. 49, No. 8, 2011, pp. 2615-2623. doi:10.1016/j.carbon.2011.02.023
- F. Zhang, X. Peng, W. Yan, Z. Peng and Y. Shen, “Nonisothermal Crystallization Kinetics of in Situ Nylon 6/Graphene Composites by Differential Scanning Calorimetry,” Journal of Polymer Science Part B: Polymer Physics, Vol. 49, No. 19, 2011, pp. 1381-1388. doi:10.1002/polb.22321
- X. Wang, et al., “In Situ Polymerization of Graphene Nanosheets and Polyurethane with Enhanced Mechanical and Thermal Properties,” Journal of Materials Chemistry, Vol. 21, No. 12, 2011, pp. 4222-4227. doi:10.1039/c0jm03710a
- P. Fabbri, E. Bassoli, S. B. Bon and L. Valentini, “Preparation and Characterization of Poly (Butylene Terephthalate)/Graphene Composites by in Situ Polymerization of Cyclic Butylene Terephthalate,” Polymer, Vol. 53, No. 4, 2012, pp. 897-902. doi:10.1016/j.polymer.2012.01.015
- Y. F. Huang and C. W. Lin, “Facile Synthesis and Morphology Control of Graphene Oxide/Polyaniline Nanocomposites via in Situ Polymerization Process,” Polymer, Vol. 53, No. 13, 2012, pp. 2574-2582. doi:10.1016/j.polymer.2012.04.022
- [61] F. D. C. Fim, N. R. S. Basso, A. P. Graebin, D. S. Azambuja and G. B. Galland, “Thermal, Electrical, and Mechanical Properties of Polyethylene-Graphene Nanocomposites Obtained by in Situ Polymerization,” Journal of Applied Polymer Science, 2012 (Online Version). doi: 10.1002/app.38317
- [62] S.-Y. Yang, et al., “Synergetic Effects of Graphene Platelets and Carbon Nanotubes on the Mechanical and Thermal Properties of Epoxy Composites,” Carbon, Vol. 49, No. 3, 2011, pp. 793-803. doi:10.1016/j.carbon.2010.10.014
- [63] M. El Achaby and A. Qaiss, “Processing and Properties of Polyethylene Reinforced by Graphene Nanosheets and Carbon Nanotubes,” Materials & Design, Vol. 44, 2013, pp. 81-89. doi:10.1016/j.matdes.2012.07.065
- [64] J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, “Mechanical Properties of Monolayer Graphene Oxide,” ACS Nano, Vol. 4, No. 11, 2010, pp. 6557-6564. doi:10.1021/nn101781v
- [65] M. A. Rafiee, et al., “Fracture and Fatigue in Graphene Nanocomposites,” Small, Vol. 6, No. 2, 2010, pp. 179- 183. doi:10.1002/smll.200901480
- [66] M. El Achaby, F. Z. Arrakhiz, S. Vaudreuil, E. M. Essassil and A. Quiss, “Piezoelectric β-Polymorph Formation and Properties Enhancement in Graphene Oxide—PVDF Nanocomposite Films,” Applied Surface Science, Vol. 258, No. 19, 2012, pp. 7668-7677. doi:10.1016/j.apsusc.2012.04.118
- [67] A. Zandiatashbar, R. C. Picu and N. Koratkar, “Mechanical Behavior of Epoxy-Graphene Platelets Nanocomposites,” Journal of Engineering Materials and Technology, Vol. 134, No. 3, 2012, pp. 031011-031016. doi:10.1115/1.4006499
- [68] I. Zaman, et al., “Epoxy/Graphene Platelets Nanocomposites with Two Levels of Interface Strength,” Polymer, Vol. 52, No. 7, 2011, pp. 1603-1611. doi:10.1016/j.polymer.2011.02.003
- [69] M. A. Rafiee, et al., “Enhanced Mechanical Properties of Nanocomposites at Low Graphene Content,” ACS Nano, Vol. 3, No. 12, 2009, pp. 3884-3890. doi:10.1021/nn9010472
- [70] S. G. Miller, et al., “Characterization of Epoxy Functionalized Graphite Nanoparticles and the Physical Properties of Epoxy Matrix Nanocomposites,” Composites Science and Technology, Vol. 70, No. 7, 2010, pp. 1120-1125. doi:10.1016/j.compscitech.2010.02.023
- [71] M. A. Rafiee, et al., “Graphene Nanoribbon Composites,” ACS Nano, Vol. 4, No. 12, 2010, pp. 7415-7420. doi:10.1021/nn102529n
- [72] D. R. Bortz, E. G. Heras and I. Martin-Gullon, “Impressive Fatigue Life and Fracture Toughness Improvements in Graphene Oxide/Epoxy Composites,” Macromolecules, Vol. 45, No. 1, 2011, pp. 238-245. doi:10.1021/ma201563k
- [73] Q. Bao, et al., “Graphene-Polymer Nanofiber Membrane for Ultrafast Photonics,” Advanced Functional Materials, Vol. 20, No. 5, 2010, pp. 782-791. doi:10.1002/adfm.200901658
- [74] X. Yang, Y. Tu, L. Li, S. Shang and X.-M. Tao, “Well-Dispersed Chitosan/Graphene Oxide Nanocomposites,” ACS Applied Materials & Interfaces, Vol. 2, No. 6, 2010, pp. 1707-1713. doi:10.1021/am100222m
- [75] T. Ramanathan, et al., “Functionalized Graphene Sheets for Polymer Nanocomposites,” Nature Nanotechnology, Vol. 3, No. 6, 2008, pp. 327-331. doi:10.1038/nnano.2008.96
- [76] D. Cai, J. Jin, K. Yusoh, R. Rafiq and M. Song, “High Performance Polyurethane/Functionalized Graphene Nanocomposites with Improved Mechanical and Thermal Properties,” Composites Science and Technology, Vol. 72, No. 6, 2012, pp. 702-707. doi:10.1016/j.compscitech.2012.01.020
- [77] K. Nawaz, et al., “Observation of Mechanical Percolation in Functionalized Graphene Oxide/Elastomer Composites,” Carbon, Vol. 50, No. 12, 2012, pp. 4489-4494. doi:10.1016/j.carbon.2012.05.029
- [78] T. Kuila, et al., “Preparation of Functionalized Graphene/Linear Low Density Polyethylene Composites by a Solution Mixing Method,” Carbon, Vol. 49, No. 3, 2011, pp. 1033-1037. doi:10.1016/j.carbon.2010.10.031
- [79] J. Wang, et al., “Direct Synthesis of Hydrophobic Graphene-Based Nanosheets via Chemical Modification of Exfoliated Graphene Oxide,” Journal of Nanoscience and Nanotechnology, Vol. 12, No. 8, 2012, pp. 6460-6466. doi:10.1166/jnn.2012.5433
- [80] W. Li, et al., “Simultaneous Surface Functionalization and Reduction of Graphene Oxide with Octadecylamine for Electrically Conductive Polystyrene Composites,” Carbon, Vol. 49, No. 14, 2011, pp. 4724-4730. doi:10.1016/j.carbon.2011.06.077
- [81] X. Huang, et al., “Graphene-Based Materials: Synthesis, Characterization, Properties, and Applications,” Small, Vol. 7, No. 14, 2011, pp. 1876-1902. doi:10.1002/smll.201002009
- [82] C. Lv, Q. Xue, D. Xia and M. Ma, “Effect of Chemisorption Structure on the Interfacial Bonding Characteristics of Graphene-Polymer Composites,” Applied Surface Science, Vol. 258, No. 6, 2012, pp. 2077-2082. doi:10.1016/j.apsusc.2011.04.056
- [83] W. Zhang, I. Srivastava, Y.-F. Zhu, C. R. Picu and N. A. Koratkar, “Heterogeneity in Epoxy Nanocomposites Initiates Crazing: Significant Improvements in Fatigue Resistance and Toughening,” Small, Vol. 5, No. 12, 2009, pp. 1403-1407. doi:10.1002/smll.200801910
- [84] K. H. Kim, Y. Oh and M. F. Islam, “Graphene Coating Makes Carbon Nanotube Aerogels Superelastic and Resistant to Fatigue,” Nature Nanotechnology, Vol. 7, No. 9, 2012, pp. 562-566. doi:10.1038/nnano.2012.118
- [85] A. Zandiatashbar, C. R. Picu and N. Koratkar, “Control of Epoxy Creep Using Graphene,” Small, Vol. 8, No. 11, 2012, pp. 1676-1682. doi:10.1002/smll.201102686
- [86] X. Jiang and L. T. Drzal, “Multifunctional High Density Polyethylene Nanocomposites Produced by Incorporation of Exfoliated Graphite Nanoplatelets 1: Morphology and Mechanical Properties,” Polymer Composites, Vol. 31, No. 6, 2010, pp. 1091-1098. doi: 10.1002/pc.20896
- [87] J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, “Graphene-Based Polymer Nanocomposites,” Polymer, Vol. 52, No. 1, 2011, pp. 5-25. doi:10.1016/j.polymer.2010.11.042
- [88] Y. Shen, et al., “Chemical and Thermal Reduction of Graphene Oxide and Its Electrically Conductive Polylactic Acid Nanocomposites,” Composites Science and Technology, Vol. 72, No. 12, 2012, pp. 1430-1435. doi:10.1016/j.compscitech.2012.05.018
- [89] V. H. Pham, T. T. Dang, S. H. Hur, E. J. Kim and J. S. Chung, “Highly Conductive Poly (Methyl Methacrylate) (PMMA)-Reduced Graphene Oxide Composite Prepared by Self-Assembly of PMMA Latex and Graphene Oxide through Electrostatic Interaction,” ACS Applied Materials & Interfaces, Vol. 4, No. 5, 2012, pp. 2630-2636. doi:10.1021/am300297j
- [90] Y.-K. Yang, et al., “Non-Covalently Modified Graphene Sheets by Imidazolium Ionic Liquids for Multifunctional Polymer Nanocomposites,” Journal of Materials Chemistry, Vol. 22, No. 12, 2012, pp. 5666-5675. doi:10.1039/c2jm16006d
- [91] H. Tang, G. J. Ehlert, Y. Lin and H. A. Sodano, “Highly Efficient Synthesis of Graphene Nanocomposites,” Nano Letters, Vol. 12, No. 1, 2011, pp. 84-90. doi:10.1021/nl203023k
- [92] C. Harish, et al., “Synthesis of Polyaniline/Graphene Nanocomposites and Its Optical, Electrical and Electrochemical Properties,” Advanced Science, Engineering and Medicine, Vol. 5, No. 2, 2013, pp. 140-148. doi:10.1166/asem.2013.1237
- [93] Z. Wang, J. K. Nelson, H. Hillborg, S. Zhao and L. S. Schadler, “Graphene Oxide Filled Nanocomposite with Novel Electrical and Dielectric Properties,” Advanced Materials, Vol. 24, No. 23, 2012, pp. 3134-3137. doi:10.1002/adma.201200827
- [94] I. Jung, D. A. Dikin, R. D. Piner and R. S. Ruoff, “Tunable Electrical Conductivity of Individual Graphene Oxide Sheets Reduced at ‘Low’ Temperatures,” Nano Letters, Vol. 8, No. 12, 2008, pp. 4283-4287. doi:10.1021/nl8019938
- [95] S. Ansari and E. P. Giannelis, “Functionalized Graphene Sheet—Poly (Vinylidene Fluoride) Conductive Nanocomposites,” Journal of Polymer Science: Part B: Polymer Physics, Vol. 47, No. 9, 2009, pp. 888-897. doi:10.1002/polb.21695
- [96] J. Li, M. L. Sham, J.-K. Kim and G. Marom, “Morphology and Properties of UV/Ozone Treated Graphite Nanoplatelet/Epoxy Nanocomposites,” Composites Science and Technology, Vol. 67, No. 2, 2007, pp. 296-305. doi:10.1016/j.compscitech.2006.08.009
- [97] S. Ganguli, A. K. Roy and D. P. Anderson, “Improved Thermal Conductivity for Chemically Functionalized Exfoliated Graphite/Epoxy Composites,” Carbon, Vol. 46, No. 5, 2008, pp. 806-817. doi:10.1016/j.carbon.2008.02.008
- [98] S. Heo, et al., “Improved Thermal Properties of Graphene Oxide-Incorporated Poly (Methyl Methacrylate) Microspheres,” Journal of Nanoscience and Nanotechnology, Vol. 12, No. 7, 2012, pp. 5990-5994. doi:10.1166/jnn.2012.6344
- [99] J. A. King, et al., “Characterization of Exfoliated Graphite Nanoplatelets/Polycarbonate Composites: Electrical and Thermal Conductivity, and Tensile, Flexural, and Rheological Properties,” Journal of Composite Materials, Vol. 46, No. 9, 2012, pp. 1029-1039. doi:10.1177/0021998311414073
- [100] K. M. F. Shahil and A. A. Balandin, “Graphene-Multilayer Graphene Nanocomposites as Highly Efficient Thermal Interface Materials,” Nano Letters, Vol. 12, No. 2, 2012, pp. 861-867. doi:10.1021/nl203906r
- [101] K. M. F. Shahil and A. A. Balandin, “Thermal Properties of Graphene and Multilayer Graphene: Applications in Thermal Interface Materials,” Solid State Communications, Vol. 152, No. 15, 2012, pp. 1331-1340. doi:10.1016/j.ssc.2012.04.034
- [102] A. Yu, et al., “Enhanced Thermal Conductivity in a Hybrid Graphite Nanoplatelet—Carbon Nanotube Filler for Epoxy Composites,” Advanced Materials, Vol. 20, No. 24, 2008, pp. 4740-4744. doi:10.1002/adma.200800401
- [103] D. Yan, et al., “Enhanced Mechanical and Thermal Properties of Rigid Polyurethane Foam Composites Containing Graphene Nanosheets and Carbon Nanotubes,” Polymer International, Vol. 61, No. 7, 2012, pp. 1107-1114. doi:10.1002/pi.4188
- [104] R. Verdejo, F. B. Bujans, M. A. R. Perez, J. A. D. Saja and M. A. L. Manchado, “Functionalized Graphene Sheet Filled Silicone Foam Nanocomposites,” Journal of Materials Chemistry, Vol. 18, No. 19, 2008, pp. 2221-2226. doi:10.1039/b718289a
- [105] S. Vadukumpully, J. Paul, N. Mahanta and S. Valiyaveetti, “Flexible Conductive Graphene/Poly (Vinyl Chloride) Composite Thin Films with High Mechanical Strength and Thermal Stability,” Carbon, Vol. 49, No. 1, 2011, pp. 198-205. doi:10.1016/j.carbon.2010.09.004
- [106] C. Bao, et al., “In Situ Preparation of Functionalized Graphene Oxide/Epoxy Nanocomposites with Effective Reinforcements,” Journal of Materials Chemistry, Vol. 21, No. 35, 2011, pp. 13290-13298. doi:10.1039/c1jm11434d
- [107] Y. Zhan, et al., “Cross-Linkable Nitrile Functionalized Graphene Oxide/Poly (Arylene Ether Nitrile) Nanocomposite Films with High Mechanical Strength and Thermal Stability,” Journal of Materials Chemistry, Vol. 22, No. 12, 2012, pp. 5602-5608. doi:10.1039/c2jm15780b
- [108] M. Stürzel, et al., “Novel Graphene UHMWPE Nanocomposites Prepared by Polymerization Filling Using Single-Site Catalysts Supported on Functionalized Graphene Nanosheet Dispersions,” Macromolecules, Vol. 45, No. 17, 2012, pp. 6878-6887. doi:10.1021/ma301376q
- [109] A. S. Wajid, et al., “High-Performance Pristine Graphene/ Epoxy Composites with Enhanced Mechanical and Electrical Properties,” Macromolecular Materials and Engineering, 2012 (Online Version). doi: 10.1002/mame.201200043
- [110] X. Jiang and L. T. Drzal, “Multifunctional High-Density Polyethylene Nanocomposites Produced by Incorporation of Exfoliated Graphene Nanoplatelets 2: Crystallization, Thermal and Electrical Properties,” Polymer Composites, Vol. 33, No. 4, 2012, pp. 636-642. doi: 10.1002/pc.22187
- [111] G. Gedler, M. Antunes, V. Realinho and J. I. Velasco, “Thermal Stability of Polycarbonate-Graphene Nanocomposite Foams,” Polymer Degradation and Stability, Vol. 97, No. 8, 2012, pp. 1297-1304. doi:10.1016/j.polymdegradstab.2012.05.027
- [112] A. S. Patole, et al., “A Facile Approach to the Fabrication of Graphene/Polystyrene Nanocomposite by in Situ Microemulsion Polymerization,” Journal of Colloid and Interface Science, Vol. 350, No. 2, 2010, pp. 530-537. doi:10.1016/j.jcis.2010.01.035
- [113] A. L. Higginbotham, J. R. Lomeda, A. B. Morgan and J. M. Tour, “Graphite Oxide Flame-Retardant Polymer Nanocomposites,” ACS Applied Materials & Interfaces, Vol. 1, No. 10, 2009, pp. 2256-2261. doi:10.1021/am900419m
- [114] S. Wang, M. Tambraparni, J. Qiu, J. Tipton and D. Dean, “Thermal Expansion of Graphene Composites,” Macromolecules, Vol. 42, No. 14, 2009, pp 5251-5255. doi:10.1021/ma900631c
- [115] O. C. Compton, S. Kim, C. Pierre, J. M. Torkelson and S. T. Yguyen, “Crumpled Graphene Nanosheets as Highly Effective Barrier Property Enhancers,” Advanced Materials, Vol. 22, No. 42, 2010, pp. 4759-4763. doi:10.1002/adma.201000960
- [116] H. Wu and L. T. Drzal, “Graphene Nanoplatelet Paper as a Light-Weight Composite with Excellent Electrical and Thermal Conductivity and Good Gas Barrier Properties,” Carbon, Vol. 50, No. 3, 2012, pp. 1135-1145. doi:10.1016/j.carbon.2011.10.026
- [117] C.-H. Chang, et al., “Novel Anticorrosion Coatings Prepared from Polyaniline/Graphene Composites,” Carbon, Vol. 50, No. 14, 2012, pp. 5044-5051. doi:10.1016/j.carbon.2012.06.043
- [118] P. Song, et al., “Permeability, Viscoelasticity, and Flammability Performances and Their Relationship to Polymer Nanocomposites,” Industrial & Engineering Chemistry Research, Vol. 51, No. 21, 2012, pp. 7255-7263. doi:10.1021/ie300311a
- [119] A. M. Pinto, J. Cabral, D. A. P. Tanaka, A. M. Mendes and F. D. Magalhaes, “Effect of Incorporation of Graphene Oxide and Graphene Nanoplatelets on Mechanical and Gas Permeability Properties of Poly (Lactic Acid) Films,” Polymer International, 2012 (online version). doi:10.1002/pi.4290
- [120] C. Li, et al., “Graphene Nano-‘Patches’ on a Carbon Nanotube Network for Highly Transparent/Conductive Thin Film Applications,” The Journal of Physical Chemistry C, Vol. 114, No. 33, 2010, pp. 14008-14012. doi:10.1021/jp1041487
- [121] A. S. Patole, et al., “Self Assembled Graphene/Carbon Nanotube/Polystyrene Hybrid Nanocomposite by in Situ Microemulsion Polymerization,” European Polymer Journal, Vol. 48, No. 2, 2012, pp. 252-259. doi:10.1016/j.eurpolymj.2011.11.005
- [122] C. Zhang and T. Liu, “A Review on Hybridization Modification of Graphene and Its Polymer Nanocomposites,” Chinese Science Bulletin, Vol. 57, No. 23, 2012, pp. 3010-3021. doi:10.1007/s11434-012-5321-x
- [123] S. S. J. Aravind, V. Eswaraiah and S. Ramaprabhu, “Facile Synthesis of One Dimensional Graphene Wrapped Carbon Nanotube Composites by Chemical Vapour Deposition,” Journal of Materials Chemistry, Vol. 21, No. 39, 2011, pp. 15179-15182. doi:10.1039/c1jm12731d
- [124] M. K. Shin, et al., “Synergistic Toughening of Composite Fibres by Self-Alignment of Reduced Graphene Oxide and Carbon Nanotubes,” Nature Communications, Vol. 3, 2012, p. 650. doi:10.1038/ncomms1661
- [125] R. Wang, J. Sun, L. Gao, C. Xu and J. Zhang, “Fibrous Nanocomposites of Carbon Nanotubes and GrapheneOxide with Synergetic Mechanical and Actuative Performance,” Chemical Communications, Vol. 47, No. 30, 2011, pp. 8650-8652. doi:10.1039/c1cc11488c
- [126] S. Chatterjee, et al., “Size and Synergy Effects of Nanofiller Hybrids Including Graphene Nanoplatelets and Carbon Nanotubes in Mechanical Properties of Epoxy Composites,” Carbon, Vol. 50, No. 15, 2012, pp. 5380- 5386. doi:10.1016/j.carbon.2012.07.021
- [127] S. Kumar, et al., “Dynamic Synergy of Graphitic Nanoplatelets and Multi-Walled Carbon Nanotubes in Polyetherimide Nanocomposites,” Nanotechnology, Vol. 21, 2010, pp. 105701-105709. doi:10.1088/0957-4484/21/10/105702
- [128] J. Yan, et al., “Preparation of Graphene Nanosheet/Carbon Nanotube/Polyaniline Composite as Electrode Material for Supercapacitors,” Journal of Power Sources, Vol. 195, No. 9, 2010, pp. 3041-3045. doi:10.1016/j.jpowsour.2009.11.028
- [129] Y. Li, T. Yang, T. Yu, L. Zheng and K. Liao, “Synergistic Effect of Hybrid Carbon Nantube-Graphene Oxide as a Nanofiller in Enhancing the Mechanical Properties of PVA Composites,” Journal of Materials Chemistry, Vol. 21, No.29, 2011, pp. 10844-10851. doi:10.1039/c1jm11359c
- [130] C. Zhang, S. Huang, W. W. Tjiu, W. Fan and T. Liu, “Facile Preparation of Water-Dispersible Graphene Sheets Stabilized by Acid Treated Multi-Walled Carbon Nanotubes and Their Poly (Vinyl Alcohol) Composites,” Journal of Materials Chemistry, Vol. 22, No. 6, 2012, pp. 2427-2434. doi:10.1039/C1JM13921E
NOTES
*Corresponding author.