Advances in Alzheimer's Disease
Vol.3 No.1(2014), Article ID:43689,9 pages DOI:10.4236/aad.2014.31004
Mutations of TP53 Gene and Oxidative Stress in Alzheimer’s Disease Patients
Jolanta Dorszewska1*, Agata Różycka2, Anna Oczkowska1, Jolanta Florczak-Wyspiańska3, Michał Prendecki1, Mateusz Dezor1, Izabela Postrach1, Paweł P. Jagodzinski2, Wojciech Kozubski3
1Laboratory of Neurobiology, Department of Neurology, Poznan University of Medical Sciences, Poznan, Poland
2Department of Biochemistry and Molecular Biology, Poznan University of Medical Sciences, Poznan, Poland
3Chair and Department of Neurology, Poznan University of Medical Sciences, 10, Fredry Str., 61-701 Poznan, Poland
Email: *dorszewskaj@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 26 November 2013; revised 13 January 2014; accepted 31 January 2014
ABSTRACT
Alzheimer’s disease (AD) leads to the generation of β-amyloid (Aβ), which may damage DNA and thus lead to apoptosis induction by the p53 pathway. Dysfunction of the p53 protein may then be connected with the development of AD. Studies were conducted on 28 AD patients and 30 non-AD controls. Analysis of TP53 mutations in exon 7 was performed on DNA isolated from whole blood and biochemical parameters in the peripheral lymphocytes of these individuals. Our study showed a silent mutation TP53 C708T (21%) [p < 0.05] and a missense mutation TP53 C748A (4%) only in the AD patients. Moreover, in AD patients with the TP53 C748A mutation, the level of 8-oxo-2’- deoxyguanosine (8-oxo2dG) was more than 5 times higher than the average level in this study group. In AD patients with the wild-type TP53 gene, the level of 8-oxo2dG was correlated with the level of protein p53 (R = +0.7388, p < 0.05). The level of the oxoguanine DNA glycosylase 1 (OGG1) protein was similar in AD patients with the silent mutation and the wild-type gene TP53 (p < 0.05) and lower than in the controls. It appears that mutations in exon 7 of TP53 (C748A, C708T) may be associated with pathogenesis of AD.
Keywords:TP53; 8-oxo2dG; p53; OGG1; AD

1. Introduction
Alzheimer’s disease (AD) is one of the most important neurodegenerative diseases. A number of factors are involved in the pathogenesis of AD, e.g. amyloid precursor protein (APP). Mutations in the APP gene account for familial AD (FAD) which is caused by changes which result in an increase of the β-amyloid (Aβ) level in the processing of this gene [1] . An elevated level of Aβ may generate free radicals, which may then oxidize macromolecules, such as DNA [2] -[5] .
Among the markers of oxidative DNA modifications, 8-oxo-2’-deoxyguanosine (8-oxo2dG) is the most abundant product of hydroxyl-induced oxidation in the purine bases of nucleic acids. Variable levels of 8-oxo2dG have been shown in both the brain [6] [7] and peripheral blood lymphocytes of AD patients [8] [9] . Furthermore, it has been demonstrated that in humans, 8-oxoguanine DNA glycosylase 1 (OGG1) is the main DNA repair enzyme that excises 8-oxo2dG from DNA [9] . A study by Dorszewska et al. [9] showed that the expression of three isoforms of OGG-1a, 1b, and 1c changes to the levels of 8-oxo2dG in the peripheral blood lymphocytes of AD patients.
It was postulated that decreased expression of OGG1 may lead to higher background mutation frequency, e.g. GC → AT [10] . This type of mutation is commonly observed in the tumor suppressor TP53 gene [10] . According to De la Monte and Wands [11] , the p53 protein can be involved in neuronal death in AD patients, and its transcription is up-regulated at the early stages of the disease and down-regulated during the neurodegenerative process. TP53 gene mutants have also been found in various cancers. There is accumulating evidence pointing to the contribution of oxidative stress in both AD and cancer [12] . Postmortem studies have shown underreported signs of AD in patients diagnosed with brain tumors, thus disclosing a putative crosslink between the p53 pathway and degeneration [13] .
The purpose of this study was to analyze of the TP53 gene mutations in exon 7 and the extent of oxidative DNA damage (8-oxo2dG) as well as expression of p53 and OGG1 protein levels in the peripheral lymphocytes of AD patients and controls.
2. Materials and Methods
2.1. Patients and Control Subjects
The studies were conducted on 28 patients with AD, including 15 women and 13 men aged 52 - 85 years. Patients with AD were diagnosed according to the criteria of the National Institute of Neurological and Communicative Disorders–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [14] . The patients were not screened for the presence of known mutations in the APP or PSEN genes.
The control group included 30 non-AD individuals, 17 women and 13 men aged 40 - 83 years. None of the control subjects had verifiable symptoms of dementia or any other neurological disorders.
A local ethical committee approved the study and written consent of all the patients or their caregivers was obtained.
2.2. Mutation Analysis of the TP53 Gene in Exon 7
DNA for genotyping was isolated from whole blood by standard procedure. TP53 mutation analysis was carried out on DNA using PCR and DNA sequencing with the use of primers targeting exon 7, designed with Primer 3+ software. The following primers were used: TP53F: GCGCACTGGCCTCATCTT and TP53R: AGGGTGGCAAGTGGCTC. PCR was carried out in 20 μl of mixture containing: 11.6 μl of Mili-Q water, 2.0 μl of 10x PCR buffer without MgCl2 (Novazym, Poland), 0.8 μl of MgCl2 solution (25 mM, Novazym, Poland), 2.2 μl of primer solution, 1.1 μl dNTPs (Novazym, Poland), 0.3 μl Allegro Taq polymerase (Novazym, Poland), and 2 μl of the tested DNA. The annealing temperature was optimized to 62˚C and the PCR was performed for 35 cycles. Quality of the product was analyzed by standard gel electrophoresis.
The PCR product (see Figure 1) was purified and sequenced according to a standard protocol at the Laboratory of Molecular Biology Techniques at the Faculty of Biology, Adam Mickiewicz University, Poznan, Poland. The samples were analyzed with sequencer 3130xl Genetic Analyzer (Applied Biosystems HITACHI, USA). All samples showing the presence of mutations were re-analyzed to confirm the presence of specific changes. The sequencing results were analyzed using BioEdit software based on a reference sequence.
2.3. Determination of 8-oxo2dG
Isolation of DNA. DNA was isolated from peripheral blood lymphocytes by five-fold centrifugation in a lytic

Figure 1. Results of agarose gel electrophoresis of the PCR product of the TP53 gene in exon 7.
buffer containing 155 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, and pH 7.4 in the presence of a buffer containing 75 mM NaCl, 9 mM Na2EDTA, pH 8.0, sodium dodecyl sulfate and proteinase K (Sigma, St. Louis, MO). Subsequently, NaCl was added, the lysate was centrifuged, and DNA present in the upper layer was precipitated with 98% ethanol.
Enzymatic hydrolysis of DNA to nucleosides. DNA was hydrolyzed to nucleosides using P1 nuclease (Sigma) for 2 h at 37˚C in 10 mM NaOAc, pH 4.5. The solution was buffered with 100 mM Tris-HCl, pH 7.5. Subsequently, the DNA was hydrolyzed with alkaline phosphatase (1U/µl; Roche, Germany) for 1 h at 37˚C and the obtained nucleoside mixture was applied to a high performance liquid chromatography system with both electrochemical and ultraviolet detection (HPLC/EC/UV).
Estimation of 8-oxo2dG. To determine the 8-oxo2dG levels the nucleoside mixture was applied to the HPLC/ UV system (P580A; Dionex, Germany), coupled to an electrochemical detector (CoulArray 5600; ESA, USA). Nucleosides were separated in a Thermo Hypersil BDS C18 (250 mm × 4.6 mm × 5 µm) column (Germany). The system was controlled and data were collected and processed using Chromeleon software (Dionex, Germany). The results were expressed as a ratio of oxidized nucleosides in the form of 8-oxo2dG to unmodified 2’dG [15] .
2.4. Estimation of OGG1 and p53 Proteins Levels
Isolation of protein. Blood was gradiated onto gradisol L at a 1:1 ratio and centrifuged, followed by collection of the interphase which was then rinsed in PBS buffer (0.9% NaCl in phosphate buffer) and centrifuged. The obtained lymphocyte precipitate was rinsed with radioimmunoprotein assay (RIPA) buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1% IGEPAL CA-630, 0.05% SDS, and 1% sodium deoxycholate), supplemented with a protease inhibitor cocktail (Sigma), homogenized in a mixture of RIPA with protease inhibitor cocktail (16:1) and 0.5 µl PSMF (Sigma) in isopropanol (10 mg/100 µl), and centrifuged. The obtained supernatant then underwent further analysis [16] .
Western blot. The OGG1 protein was analyzed in 12% and the p53 protein was analyzed in 7.5% polyacrylamide gel. Equivalent amounts of protein (30 µg protein/lane) were loaded to the wells. The gel-separated proteins were electrotransferred to a nitrocellulose filter in a semidry Western Blot analysis apparatus (Apelex, France). To estimate the levels of the OGG1 protein, the filters were exposed first to an anti-OGG1/2 goat polyclonal antibody (G-20, IgG, 200 µg/1.0 ml; Santa Cruz, USA), and for the p53 protein, anti-p53 mouse monoclonal antibody (IgG-2a, 200 µg/1.0 ml; Santa Cruz, USA) diluted 1:500.
Subsequently, individual sheets of nitrocellulose filter were incubated with the second antibody; for the OGG1 protein this was mouse anti-goat IgG-HRP (200 µg/0.5 ml; Santa Cruz, USA) and for p53 protein this was goat anti-mouse IgG-HRP (200 µg/0.5 ml; Santa Cruz, USA) at a dilution of 1:2000. Peroxidase BMB was added (BM blue POD substrate precipitation; Roche, Germany) to stain the immunoreactive bands. The surface area of the immunoreactive bands was registered using a densitometer (GS-710; Bio-Rad, Hercules, CA) in the Quantity One System [9] [17] .
2.5. Statistical Analysis of the Results
The obtained data were evaluated using Kruskal-Wallis and Fischer exact tests. Correlations between the obtained results were tested using the Spearman test.
GraphPad (Instant, USA) and Statistica for Windows (Stat Soft, USA) were used to perform statistical analyses of the results.
3. Results
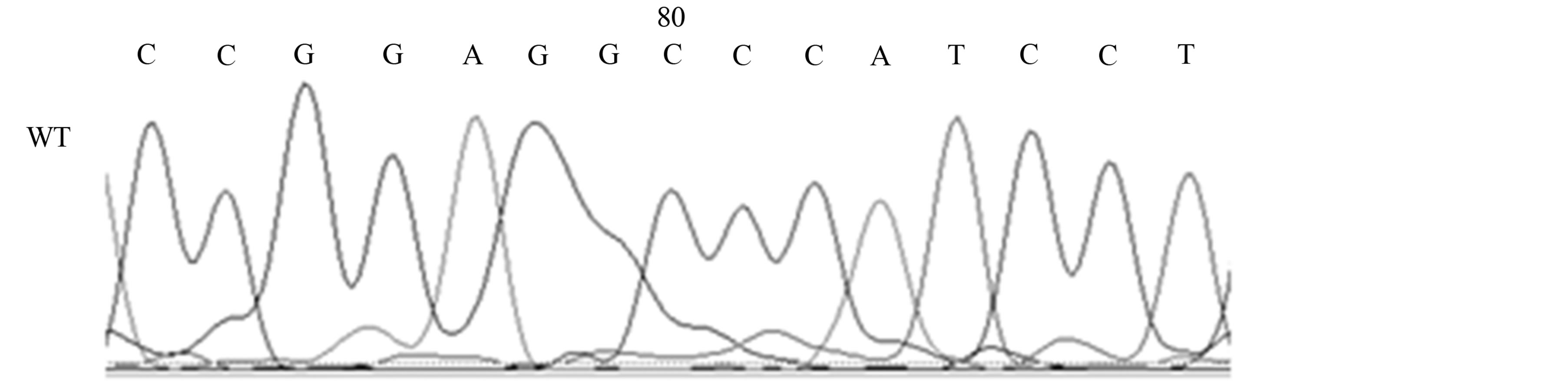
Our studies revealed the presence of two different mutations only in AD patients, a silent mutation TP53 C708T (Y236Y) in six patients (21%) [Fischer’s exact test, p < 0.05] and a missense mutation TP53 C748A (P250T) in one patient (4%) (see Table 1, Figures 2(a) and (b)).
 (a)
(a)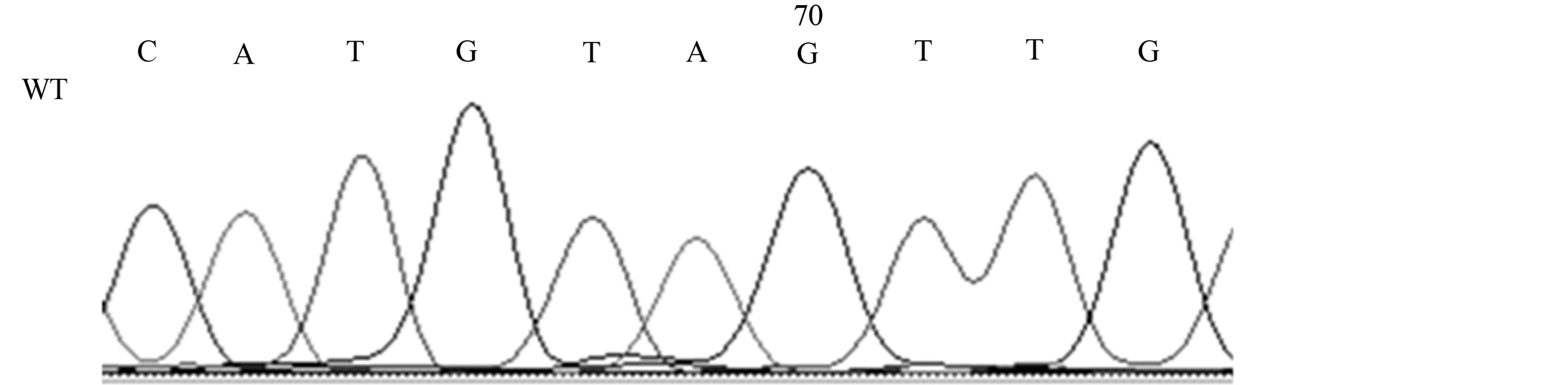 (b)
(b)
Figure 2. Mutations of the TP53 gene in exon 7 in AD patients; 2A, TP53 C748A (P250T) missense mutation (M) and wild-type (WT); 2B, TP53 C708T (Y236Y) [reverse strand] silent mutation (M) and wild-type (WT).

Table 1. Identified TP53 mutation in exon 7 in AD patients.
n-Number of patients with mutations. Fischer’s exact test, *p < 0.05.
In the AD patient with the TP53 C748A missense mutation was shown that the level of 8-oxo2dG was more than 5 times higher (298.0 × 10−5) than the average level in the AD group (see Table 2). However, in this AD patient the level of the p53 (87.1) and OGG1 (12.2) proteins was similar to the average level in patients with AD (see Table 2).
In the AD patients with the TP53 C708T silent mutation, slightly elevated levels were shown of the 8-oxo2dG and p53 proteins as compared to patients with the wild-type TP53 gene and controls (see Table 2, Figure 3). However, the OGG1 protein level was similar in AD patients with and without the TP53 C708T mutation and at the same time in these patients was about two times lower than in the controls. This study also demonstrates that in AD patients with the wild-type TP53 gene the level of 8-oxo2dG correlated with the level of the p53 protein (Spearman test, R +0.7388, p < 0.05, see Table 2, Figure 3).
4. Discussion
In AD various factors, e.g. elevated level of Aβ [2] -[5] , may lead to the generation of oxidative stress and oxidative DNA damage [18] . It was shown that the neurotoxic 42 - 43 amino-acid-long Aβ peptide is a breakdown product of a much larger protein, i.e. the Aβ/A4 protein precursor—APP [19] . Moreover, the 4 - 4.5 kDa Aβ/A4 polypeptide is probably the major protein component of senile plaques (SPs) [20] . It was also shown that Aβ/A4-related peptides may occur in both AD and normal subjects, while their production is increased in FAD [20] [21] . Moreover, it has been indicated that intraneuronal Aβ may be the cause of mitochondrial [22] , lysosomal [23] [24] and synaptic [25] dysfunctions which possibly lead to apoptosis [26] and oxidative degeneration.
8-Oxo-2dG or its nucleoside is one of the markers of oxidative DNA modification. Moreover, 8-oxo2dG is considered to be a marker of oxidative DNA damage in degenerative diseases, cancer and aging process. As has been shown in the literature, oxidative DNA damage in AD (8-oxo2dG) may occur both in the central nervous system [6] [7] as well as in the peripheral lymphocytes [8] [9] [27] [28] . Studies by Dorszewska et al. [8] [9] [27] , Dezor et al. [17] as well as this study on the peripheral lymphocytes of AD patients have demonstrated elevated 8-oxo2dG levels.
Studies by Dezor et al. [17] , Dorszewska et al. [9] [29] and this study have also shown that nucleotide oxidation, reflected by an elevated 8-oxo2dG level in AD patients, may be associated with a decrease in the level of the OGG1 protein and/or by a decrease in the mitochondrial OGG1-1b isoform’s expression. Further studies by Iida et al. [30] indicated that the reduction in OGG1 expression in the brains of patients with AD was accompanied by the formation of neurofibrillary tangles (NFTs), axonal dystrophy and reactive astrocytes. However, Mao et al. [31] demonstrated that the decreased excision activity of OGG1 in patients with AD may be affected by mutations in the gene encoding OGG1. In Mao et al.’s study, the presence of mutations was shown in 4 out of 14 patients with AD, where two patients possessed C796 deletions that completely eliminated the activity of the OGG1 enzyme and two patients carried single point mutations that led to distinctly decreased activity of the OGG1. Moreover, literature data indicated that oxidative DNA damage may induce different types of transversions in genes coding DNA repair (OGG1) [10] [30] .
It has also been shown that oxidized guanine in DNA induces GC-to-AT transversion-type point mutations. The same type of mutation and the loss of heterozygosity have also been observed in the tumor suppressor TP53 gene and are commonly associated with a wide variety of tumors in humans and in experimental animals [10] [32] -[34] . Mutations abrogating of p53 function and allelic loss of its locus were among the first genetic lesions identified in glioblastoma multiforme [35] . TP53 mutations are also present in all grades of human astrocytoma [36] and in the murine model of astrocytoma [37] .

Figure 3. Western blot analysis of OGG1 and p53 protein levels, 1 - 8 controls and 9 - 16 AD patients.
Table 2. Level of 8-oxo2dG (8-oxo2dG/dG × 10−5), p53 and OGG1 proteins (% area immunoreactive bands) in AD patients with and without the detected TP53 C708T mutation and controls.
Mean ± SD. No significant differences in the Kruskal-Wallis test. AD patients with the wild-type TP53 gene, Spearman coefficient +0.7388 between 8-oxo2dG and p53 protein levels (p < 0.05).
Furthermore, the mutations in AD patients that were shown in this study turned out to be similar to mutations that were previously described in various cancers. Simultaneously, as in this study, in exon 7 of the TP53 gene of AD patients with no history of cancer the synonymous variant TP53 C708T was reported by Zhang et al. [38] to be present in rectal cancer. Moreover, the missense mutation TP53 C748A was found by Boersma et al. [39] in breast cancer. A TP53 missense mutation in exon 7 was also found in an adrenocortical carcinoma patient. Although the phenotype was not clinically distinct, the authors suspected a hereditary background due to early onset of the disease [40] .
It seems that in AD the missense TP53 C748A mutation may be associated with an increase in oxidative stress, as in this study the tendency for a significant increase in the 8-oxo2dG level was observed only in the patient with this mutation. It also appears that in this AD patient elevated level of 8-oxo2dG could have induced the repair system of oxidative DNA damage by a tendency to increase the p53 protein level.
P53 is a key regulator of multiple cellular processes, and depending on the cell type it is activated by different stressors to induce apoptosis or autophagic cell death; but is also responsible for reversible and irreversible cell cycle arrest, or senescence [41] . The induction of cellular aging by elevated p53 levels in response to stress is designed to prevent proliferation of damaged cells. Two main groups of signals influence the p53 pathway. These include DNA damage and oncogenic stress as a result of cancer and/or aging which may be induced by p53 mutation [13] .
There have been reports in the literature indicating that the p53 protein participates in neuronal apoptosis in the brains of AD patients [11] and can typically be associated with increased expression of p53. An increase in the p53 protein level has been shown both in the cultures of human and rat neurons and astrocytes as well as in the peripheral blood lymphocytes and brains of patients with AD [17] [29] .
A study by Ohyagi et al. [26] suggested that in AD patients, p53-dependent apoptosis leads directly to neuronal loss through Aβ42 binding and activation of the p53 promoter. The accumulation of both Aβ42 and p53 is manifested in some degenerating-shape neurons in AD.
A study by Dorszewska et al. 2013, unpublished data] demonstrated the presence of Aβ in PS/APP mice along with a high p53 level as compared to younger mice, which may indicate the possible induction of apoptosis. It has been shown that p53-dependent neuronal apoptosis may also result from decreased activity of antiapoptotic PS1 caused by p53 protein-protein interactions or by pro-apoptotic presenilin-2 (PS2), which down-regulates PS1 expression [18] [42] . It seems that the elevated p53 level influences PS1-mediated abnormalities of intracellular calcium levels [43] . On the other hand, it seems that in this study the silent mutation C708T in the TP53 gene causes a slight (although varied) increase in p53 protein levels in AD patients. However, p53 protein levels were more varied in patients without the mutation than in patients with the mutation, probably due to the impact of other factors.
It is known that mutations in DNA cause a change of the sequence in the corresponding mRNA and may influence its stability, thus affecting the number of mRNA copies translated into protein (e.g. p53). At the same time, the presence of SNPs has been associated with changes of methylation pattern affecting the genes expression [44] . Moreover, has been suggested that the change in mRNA may alter the target of expression modulating factors which would result in misbalanced expression of p53.
However, a study by Uberti et al. [45] on fibroblasts from seven sporadic AD patients did not reveal the presence of mutations in exons 1 - 11 of the TP53 gene. This difference might be explained by the small number of studied AD patients.
In conclusion, it seems that both of the analyzed mutations of TP53 (C748A, C708T) gene in exon 7 may be involved in neurodegenerative processes in this study in AD patients. It is possible that the missense mutation, C748A, may be responsible for generating oxidative stress, which is represented by an elevated level of 8-oxo2dG. Moreover, the synonymous mutation, TP53 C708T, may lead to modification of p53 protein activity.
However, the suggested action mechanisms of both variants in AD require further studies, an analysis of both exons 1 - 11 of the TP53 gene and the biochemical parameters of oxidative stress on a cohort of AD patients with varying degrees of dementia. In vitro point mutation studies are also required.
Acknowledgements
This study was supported by grant No 502-01-11111-45-07-467, Poznan University of Medical Sciences.
References
- Hardy, J. and Selkoe, D.J. (2002) The amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science, 297, 353-356. http://dx.doi.org/10.1126/science.1072994
- Armstrong, R.A. (2009) The Molecular Biology of Senile Plaques and Neurofibrillary Tangles in Alzheimer’s Disease. Folia Neuropathologica, 47, 289-299.
- Armstrong, R.A. (2010) A Spatial Pattern Analysis of Beta-Amyloid (Abeta) Deposition in the Temporal Lobe in Alzheimer’s Disease. Folia Neuropathologica, 48, 67-74.
- Parks, J.K., Smith, T.S., Trimmer, P.A., Bennett, J.P. and Parker, W.D. (2001) Neurotoxic Abeta Peptides Increase Oxidative Stress in Vivo through NMDA-Receptor and Nitric-Oxide-Synthase Mechanisms, and Inhibit Complex IV Activity and Induce a Mitochondrial Permeability Transition in Vitro. Journal of Neurochemistry, 76, 1050-1056. http://dx.doi.org/10.1046/j.1471-4159.2001.00112.x
- Pluta, R., Ułamek, M. and Jabłoński, M., (2010) Consideration of the Ischaemic Basis and Treatment of Alzheimer’s Disease. Folia Neuropathologica, 48, 11-26.
- Wang, J., Xiong, S., Xie, C., Markesbery, W.R. and Lovell, M.A. (2005) Increased Oxidative Damage in Nuclear and Mitochondrial DNA in Alzheimer’s Disease. Journal of Neurochemistry, 93, 953-962. http://dx.doi.org/10.1111/j.1471-4159.2005.03053.x
- Wang, J., Markesbery, W.R. and Lovell, M.A., (2006) Increased Oxidative Damage in Nuclear and Mitochondrial DNA in Mild Cognitive Impairment. Journal of Neurochemistry, 96, 825-832. http://dx.doi.org/10.1111/j.1471-4159.2005.03615.x
- Dorszewska, J., Florczak, J., Różycka, A., JaroszewskaKolecka, J., Trzeciak, W.H. and Kozubski, W. (2005) Polymorphisms of the CHRNA4 Gene Encoding the Alpha4 Subunit of Nicotinic Acetylcholine Receptor as Related to the Oxidative DNA Damage and the Level of Apoptotic Proteins in Lymphocytes of the Patients with Alzheimer’s Disease. DNA and Cell Biology, 24, 786-794. http://dx.doi.org/10.1089/dna.2005.24.786
- Dorszewska, J., Kempisty, B., Jaroszewska-Kolecka, J., Różycka, A., Florczak, J., Lianeri, M., Jagodzinski, P.P. and Kozubski, W. (2009) Expression and Polymorphisms of Gene 8-Oxoguanine Glycosylase 1 and the Level of Oxidative DNA Damage in Peripheral Blood Lymphocytes of Patients with Alzheimer’s Disease. DNA and Cell Biology, 28, 579-588. http://dx.doi.org/10.1089/dna.2009.0926
- Hirano, T. (2008) Repair System of 7, 8-Didydro-8-Oxoguanine as a Defense Line against Carcinogenesis. Journal of Radiation Research, 49, 329-340. http://dx.doi.org/10.1269/jrr.08049
- De la Monte, S.M. and Wands, J.R. (2006) Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. Journal of Alzheimer’s Disease, 9, 167-181.
- Frain, L., Driver, J., Gaziano, J.M., Lu, K.P., Kowall, N., Gagnon, D., Cho, K., Betensky, R. and Swanson, D. (2013) A Reduced Risk of Alzheimer Disease Is Associated with the Majority of Cancers in a National Cohort of Veterans. Alzheimer’s Association International Conference, Boston, 13-18 July 2013, 3-175.
- Lanni, C., Racchi, M., Memo, M., Govoni, S. and Uberti, D. (2012) p53 at the Crossroads between Cancer and Neurodegeneration. Free Radical Biology & Medicine, 52, 1727-1733. http://dx.doi.org/10.1016/j.freeradbiomed.2012.02.034
- McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D. and Stadlan, E.M. (1984) Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology, 34, 939-944. http://dx.doi.org/10.1212/WNL.34.7.939
- Olsen, A., Siboska, G.E., Clark, B.F. and Rattan, S.I. (1999) N6-Furfuryladenine, Kinetin, Protects against Fenton Reaction-Mediated Oxidative Damage to DNA. Biochemical and Biophysical Research Communications, 265, 499-502. http://dx.doi.org/10.1006/bbrc.1999.1669
- Ohnishi, T., Inoue, N., Matsumoto, H., Omatsu, T., Ohira, Y., Nagaoko, S. (1996) Cellular Content of p53 Protein in rat Skin after Exposure to the Space Environment. Journal of Applied Physiology, 81, 183-185.
- Dezor, M., Dorszewska, J., Florczak, J., Kempisty, B., Jaroszewska-Kolecka, J., Różycka, A., Półrolniczak, A., Bugaj, R., Jagodziński, P.P. and Kozubski, W. (2011) Expression of 8-Oxoguanine DNA Glycosylase 1 (OGG1) and the Level of p53 and TNF-Alpha Proteins in Peripheral Lymphocytes of Patients with Alzheimer’s Disease. Folia Neuropathologica, 49, 123-131.
- Pastorcic, M. and Das, H.L., (2000) Regulation of Transcription of the Human Presenilin-1 Gene by Ets Transcription Factors and the p53 Protooncogene. The Journal of Biological Chemistry, 275, 34938-34945. http://dx.doi.org/10.1074/jbc.M005411200
- Kang, J., Lemaire, H.G., Unterbeck, A., Salbaum, J.M., Masters, C.L., Grzeschik, K.H., Multhaup, G., Beyreuther, K. and Müller-Hill, B. (1987) The Precursor of Alzheimer’s Disease Amyloid A4 protein Resembles a Cell-Surface Receptor. Nature, 325, 733-736. http://dx.doi.org/10.1038/325733a0
- Matsuoka, Y., Picciano, M., Malester, B., LaFrancois, J., Zehr, C., Daeschner, J.M., Olschowka, J.A., Fonseca, M.I., O’Banion, M.K., Tenner, A.J, Lemere, C.A. and Duff, K. (2001) Inflammatory Responses to Amyloidosis in a Transgenic Mouse Model of Alzheimer’s Disease. The American Journal of Pathology, 158, 1345-1354. http://dx.doi.org/10.1016/S0002-9440(10)64085-0
- Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., McConlogue, L., Hung, A.Y., Seubert, P., Vigo-Pelfrey, C., Lieberburg, I. and Selkoe, D.J. (1992) Mutation of the Beta-Amyloid Precursor Protein in Familial Alzheimer’s Disease Increases Beta-Protein Production. Nature, 360, 672-674. http://dx.doi.org/10.1038/360672a0
- Busciglio, J., Pelsman, A., Wong, C., Pigino, G., Yuan, M., Mori, H. and Yankner, B.A. (2002) Altered Metabolism of the Amyloid Beta Precursor Protein Is Associated with Mitochondrial Dysfunction in Down’s Syndrome. Neuron, 33, 677-688. http://dx.doi.org/10.1016/S0896-6273(02)00604-9
- Glabe, C. (2001) Intracellular Mechanisms of Amyloid Accumulation and Pathogenesis in Alzheimer’s Disease. Journal of Molecular Neuroscience, 17, 137-145. http://dx.doi.org/10.1385/JMN:17:2:137
- Shie, F.S., LeBoeuf, R.C. and Jin, L.W. (2003) Early Intraneuronal Abeta Deposition in the Hippocampus of APP Transgenic mice. Neuroreport, 14, 123-129. http://dx.doi.org/10.1097/00001756-200301200-00023
- Takahashi, R.H., Milner, T.A., Li, F., Nam, E.E., Edgar, M.A., Yamaguchi, H., Beal, M.F., Xu, H., Greengard, P. and Gouras, G.K. (2002) Intraneuronal Alzheimer Abeta42 Accumulates in Multivesicular Bodies and Is Associated with Synaptic Pathology. The American Journal of Pathology, 161, 1869-1879. http://dx.doi.org/10.1016/S0002-9440(10)64463-X
- Ohyagi, Y., Asahara, H., Chui, D.H., Tsuruta, Y., Sakae, N., Miyoshi, K., Yamada, T., Kikuchi, H., Taniwaki, T., Murai, H., Ikezoe, K., Furuya, H., Kawarabayashi, T., Shoji, M., Checler, F., Iwaki, T., Makifuchi, T.T., Takeda, K., Kira, J. and Tabira, T. (2005) Intracellular Abeta42 Activates p53 Promoter: A Pathway to Neurodegeneration in Alzheimer’s Disease. The FASEB Journal, 19, 255-257.
- Dorszewska, J., Florczak, J., Różycka, A., Kempisty, B., Jaroszewska-Kolecka, J., Chojnicka, K., Trzeciak, W.H. and Kozubski, W. (2007) Oxidative DNA Damage and Level of Thiols as Related to Polymorphisms of MTHFR, MTR, MTHFD1 in Alzheimer’s and Parkinson’s Diseases. Acta Neurobiolgiae Experimentalis, 67, 113-129.
- Migliore, L., Fontana, I., Trippi, F., Colognato, R., Coppede, F., Tognoni, B., Nucciarone, B. and Siciliano, G. (2005) Oxidative DNA Damage in Peripheral Leukocytes of Mild Cognitive Impairment and AD Patients. Neurobiology of Aging, 26, 567-573. http://dx.doi.org/10.1016/j.neurobiolaging.2004.07.016
- Dorszewska, J., Oczkowska. A., Florczak, J., Dezor, M. and Kozubski, W. (2013) Mutations of TP53 C708T and C748A, Oxidative DNA Damage, and p53 and OGG1 Protein Levels in Peripheral Lymphocytes of the People with Alzheimer’s Disease. Alzheimer’s & Dementia, 9, Supp., 567, Alzheimer’s Association International Conference, Boston, 13-18 July 2013, 3-044.
- Iida, T., Furuta, A., Nishioka, K., Nakabeppu, Y. and Iwaki, T. (2002) Expression of 8-Oxoguanine DNA Glycosylase Is Reduced and Associated with Neurofibrillary Tangles in Alzheimer’s Disease Brain. Acta Neuropathologica, 103, 20-25. http://dx.doi.org/10.1007/s004010100418
- Mao, G., Pan, X., Zhu, B.B., Zhang, Y., Yuan, F., Huang, J., Lovell, M.A., Lee, M.P., Markesbery, W.R., Li, G.M. and Gu, L. (2007) Identification and Characterization of OGG1 Mutations in Patients with Alzheimer’s Disease. Nucleic Acids Research, 35, 2759-2766. http://dx.doi.org/10.1093/nar/gkm189
- Bougeard, G., Brugieres, L., Chompret, A., Gesta, P., Charbonnier, F., Valent, A., Martin, C., Raux, G., Feunteun, J., Bressac-de Paillerets, B. and Frébourg, T. (2003) Screening for TP53 Rearrangements in Families with the Li-Fraumeni Syndrome Reveals a Complete Deletion of the TP53 Gene. Oncogene, 22, 840-846. http://dx.doi.org/10.1038/sj.onc.1206155
- Gavino, C. and Richard, S. (2011) Loss of p53 in Quaking Viable Mice Leads to Purkinje Cell Defects and Reduced Survival. Scientific Reports, 1, 84.
- Hollstein, M., Sidransky, D., Vogelstein, B. and Harris, C.C. (1991) p53 Mutations in Human Cancers. Science, 253, 49-53. http://dx.doi.org/10.1126/science.1905840
- Nigro, J.M., Baker, S.J., Preisinger, A.C., Jessup, J.M., Hostetter, R., Cleary, K., Bigner, S.H., Davidson, N., Baylin, S. and Devilee, P. (1989) Mutations in the p53 Gene Occur in Diverse Human Tumour Types. Nature, 342, 705-708. http://dx.doi.org/10.1038/342705a0
- Watanabe, K., Sato, K., Biernat, W., Tachibana, O., von Ammon, K., Ogata, N., Yonekawa, Y., Kleihues, P. and Ohgaki, H. (1997) Incidence and Timing of p53 Mutations during Astrocytoma Progression in Patients with Multiple Biopsies. Clinical Cancer Research, 3, 523-530.
- Reilly, K.M., Loisel, D.A., Bronson, R.T., McLaughlin, M.E. and Jacks, T. (2000) Nf1;Trp53 Mutant Mice Develop Glioblastoma with Evidence of Strain-Specific Effects. Nature Genetics, 26, 109-113. http://dx.doi.org/10.1038/79075
- Zhang, R., Takahashi, S., Orita, S., Yoshida, A., Maruyama, H., Shirai, T. and Ohta, N. (1998) p53 Gene Mutations in Rectal Cancer Associated with Schistosomiasis Japonica in Chinese Patients. Cancer Letters, 131, 215-221. http://dx.doi.org/10.1016/S0304-3835(98)00154-2
- Boersma, B.J., Howe, T.M., Goodman, J.E., Yfantis, H.G., Lee, D.H., Chanock, S.J. and Ambs, S. (2006) Association of Breast Cancer Outcome with Status of p53 and MDM2 SNP309. Journal of the National Cancer Institute, 98, 911- 919. http://dx.doi.org/10.1093/jnci/djj245
- Waldmann, J., Patsalis, N., Fendrich, V., Langer, P., Saeger, W., Chaloupka, B., Ramaswamy, A., Fassnacht, M., Bartsch, D.K. and Slater, E.P. (2012) Clinical Impact of TP53 Alterations in Adrenocortical Carcinomas. Langenbeck’s Archives of Surgery, 397, 209-216. http://dx.doi.org/10.1007/s00423-011-0868-6
- Sakamoto, Y., Kato, S., Takahashi, M., Okada, Y., Yasuda, K., Watanabe, G., Imai, H., Sato, A. and Ishioka, C. (2011) Contribution of Autophagic Cell Death to p53-Dependent Cell Death in Human Glioblastoma Cell Line SF126. Cancer Science, 102, 799-807. http://dx.doi.org/10.1111/j.1349-7006.2011.01857.x
- Alves da Costa, C., Paitel, E., Mattson, M.P., Amson, R., Telerman, A., Ancolio, K. and Checler, F. (2002) Wild-Type and Mutated Presenilins 2 Trigger p53-Dependent Apoptosis and Down-Regulate Presenilin 1 Expression in HEK293 Human Cells and in Murine Neurons. Proceedings of the National Academy of Sciences of the United States of America, 99, 4043-4048. http://dx.doi.org/10.1073/pnas.062059899
- Das, H.K., Tchedre, K. and Mueller, B. (2012) Repression of Transcription of Presenilin-1 Inhibits γ-Secretase Independent ER Ca(2+) Leak that Is Impaired by FAD Mutations. Journal of Neurochemistry, 122, 487-500. http://dx.doi.org/10.1111/j.1471-4159.2012.07794.x
- Wang, X., Wang, W., Li, L., Perry, G., Lee, H.G. and Zhu, X.W. (2013) Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochimica et Biophysica Acta, in press. http://dx.doi.org/10.1016/j.bbadis.2013.10.015
NOTES

*Corresponding author.