Advances in Alzheimer's Disease
Vol.2 No.3(2013), Article ID:37285,10 pages DOI:10.4236/aad.2013.23013
S100b protects IMR-32 cells against Ab(1-42) induced neurotoxicity via modulation of apoptotic genes expression*
![]()
1CNR-ICRM Institute of Chimica del Riconoscimento Molecolare, c/o Institute of Biochemistry and Clinical Biochemistry, Catholic University School of Medicine, Rome, Italy; #Corresponding Author: e.clementi@icrm.cnr.it
2Department for Innovation in Biological, Agro-food and Forest Systems (DIBAF), Tuscia University, Viterbo, Italy
3Institute of Biochemistry and Clinical Biochemistry, Catholic University School of Medicine, Rome, Italy
Copyright © 2013 M. Elisabetta Clementi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 20 May 2013; revised 21 June 2013; accepted 2 July 2013
Keywords: S100b; Neurodegeneration; Oxidate Methionine; Apoptotic Genes Expression
ABSTRACT
Amyloid beta (1-42) peptide is considered responsible for the formation of senile plaques that accumulate in the brain of patients with Alzheimer’s disease (AD). In the past years considerable attention has been focused on identifying new protective substances that prevent or almost retard the appearance of amyloid beta (1-42)-related neurotoxic effects. In this study, human neuroblastoma cells (IMR-32) was used as system model to evaluate the protective role of S100b, a neurotrophic factor and neuronal survival protein, that is highly expressed by reactive astrocytes in close vicinity of beta-amyloid deposits, against amyloid beta (1-42)-dependent toxicity. Our results show that at nanomolar concentrations, S100b protects cells against Aβ mediated cytotoxicity, as assessed by MTS vitality test. The protective mechanism seems to be related to the effect on bcl-2 (an anti-apoptotic gene) expression, which is highly downregulated by amyloid beta (1-42) treatment, while resulted more expressed in the presence of S100b. On the contrary, Bax, a pro-apoptotic gene, resulted down-regulated by the treatment with S100 compared with the results obtained in the presence of amyloid beta (1-42) peptide. However, at micromolar doses, S100b is toxic for IMR-32 cells and its toxicity adds to that of the Aβ peptide, suggesting that additional molecular mechanisms may be involved in the neurotoxic process.
1. INTRODUCTION
S100b comes from the S100 family of EF-hand Ca2+ binding proteins, mainly expressed in astrocytes of the central nervous system, exerting intracellular and extracellular regulatory activities [1,2]. In particular the intracellular roles of S100 proteins are implicated in the regulation of protein phosphorylation, the dynamics of cytoskeleton constituents, Ca2+ homeostasis, enzyme activities, transcription factors, cell growth and differentiation, and the inflammatory response [3-5]. The extracellular implications are instead implicated in trophic maintenance, survival and outgrowth of neuronal cells, both during development of the nervous system and in neurodegenerative conditions [4].
In the brain, astrocytes release S100b constitutively but the level of protein is increased by a number of agents such as 5-HT1A receptor agonists, glutamate, adenosine, lysophosphatydic acid [6-8] and by pathological circumstances such as Alzheimer’s disease and Down’s syndrome, where S100 levels in severely affected brain regions are greater than age-matched controls [9,10].
On this subject, recent hypotheses shows that in Alzheimer’s disease, the astrocytes production of pro-inflammatory cytokines such as S100b and IL-1, regulates the early phases of neuronal degeneration [1,11-13]. In fact, although the hypothesis of amyloid and tau aggregation could explain the neuropathological alterations during the late stage of AD, strong evidences demonstrate that distress accumulated in cells, pro-inflammatory cytokines mediated, could initiate an early activation of the innate cellular immunity and the inflammatory cascade [14]. In this scenario, the S100b produced by astrocytes, seems to be one of the most important actors, since modulates both the phases.
In fact, in early triggering events of AD, astrocytes produce S100b at nM mount, exerting “in vitro” a pro-survival effect on neurons and a stimulation of neurite outgrowth. Instead, during the late phases of neurodegeneration, when levels of S100 increase (mRNA were elevated 10 - 20-fold in extracts of temporal lobe from autopsy samples of Alzheimer’s disease patients) it prevails neuronal death via apoptosis [15-17]. Recent results [18- 20], evidence that beta amyloide peptide, the major protein component of senile brain plaques in Alzheimer’s disease, determines both an oxidative stress and a cascade mechanism apoptotic triggering that depend also on the oxidative state of Met-35. We investigated in the present study, the relationships between the S100b and Aβ(1-42) peptide. In particular we have studied, in human neuroblastoma (IMR-32) cells, the effect of low (nanomolar) and higher (micromolar) S100b concentration previous treated with Aβ peptides carrying Methionine 35 both in reduced and oxidative state, on cellular vitality and on the expression of major apoptotic gene expression. The obtained results indicate that an important defensive mechanism exists to preserve the Aβ(1-42) induced neuronal degeneration carried out by S100 protein at nanomolar concentration and this effect is more evidenced with methionine in reduced state. On the contrary, at micromolar doses S100 adds its toxicity to that induced by the Aβ peptide, suggesting that additional molecular mechanisms may be involved in the neuropathology of Alzheimer Disease.
2. MATERIALS AND METHODS
2.1. Cell Culture and Treatments
Human neuroblastoma IMR-32 cells were grown in minimum essential medium (Biochrom KG, Berlin, Germany) supplemented with 10% heat inactivated fetal bovine serum (HyClone, Logan, UT), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Grand Island, NY, USA), and cultured at 37˚C in an atmosphere of 5% CO2 in air. Cell differentiation was induced by 1 mM dibutyryl cAMP and 2.5 μM 5-bromodeoxyuridine (Sigma, St. Louis, MO, USA), which were added to the culture medium three times per week, starting from the day after plating. After a week, the differentiated cells were plated at an appropriate density according to each experimental procedure.
Aβ peptides were obtained by Peptide Speciality Laboratories GmbH (Heidelberg, Germany). Analysis of the peptide by reverse phase high performance chromatography (HPLC) and mass spectrometry revealed a purity >98%. Stock solution of Aβ peptide, 2.5 mM in DMSO were prepared according to the manufacturer’s instructions and stored at −80˚C (in previous studies [21,22] these conditions have been shown to lead to the predominance of the soluble monomeric form of this peptide). Human recombinant S100b was purchased by Sino Biological Inc; stock solutions of S100b was solved in minimum essential medium, filtered and stored at –20˚C. S100b was added to the cultures 12 hr before exposure to Aβ peptide and to conform control experiments, DMSO concentration (<0.5%) was the same in all solutions used.
2.2. Cell Availability Measurement
For vitality determination, IMR-32 cells were plated in 96-well plates at a density of 10,000 cells/well and incubated for 24 and 48 h with and without (as control) 10 μM Aβ and with different concentration of S100b protein. Cell survival was evaluated by the 3-[(4,5-dimethylthiazol-2-yl)-5,3-carboxymethoxyphenyl]-2-(4-sulfophenyl)-2H tetrazolium, inner salt (MTS) reduction assay (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega, Madison, WI, USA). The MTS assay is a sensitive measurement of normal cellular metabolic status, particularly that in mitochondria, which reflects early cellular redox changes [23]. After exposure to the different treatments, cells were incubated with MTS solution (2 mg/ml) for 4 h at 37˚C in a 5% CO2 incubator. The intracellular soluble formazan produced by cellular reduction of the MTS was determined by recording the absorbance of each 96-well plate using the automatic microplate photometer (SpectraCount, Packard Bioscience Company, Groningen, The Netherlands) at a wavelength of 490 nm. The morphological features of apoptotic degeneration observable afterwards 48 h of incubation with the different treatments (10 μM amyloid and plus S100b) were analyzed and photographed by phasecontrast microscopy (Nikon Eclipse TS100) 40×.
2.3. Reverse Transcription and Polymerase Chain Reaction
Total RNA was isolated using SV total RNA isolation System (Promega) that includes the elimination of any genomic DNA by DNAse treatment. The purity and quantity of the resulting RNA were determined via the measurement of the absorbance at 280 and 260 nm respectively. The A260/A280 ratio was about 1.8. The RNA was concentrated by precipitation and re-dissolved in water RNAse free. Total RNA (1 μg) from each sample was used for first strand cDNA synthesis using the MMLV Reverse and Oligo-dT, as random primer (SIGMA). The PCR was performed with 200 ng of cDNA using Gene-spin Taq-Polymerase with 2.0 mM MgCl2, 0.4 μm primer, 0.4 μm dNTPs and 2 units polymerase for each reaction (25 μl) under primer specific conditions.
The primers used for amplification were:
GAPDH sense primer;
5’-AAGAAGATGCGGCTGACTGTCGAGCCACAT-3’GAPDH anti-sense primer;
5’-TCTCATGGTTCACACCCATGACGAACATG-3’Bcl2 sense primer;
5’-CGACGACTTCTCCCGCCGCTACCGC-3’Bcl2 anti-sense primer;
5’-CCGCATGCTGGGGCCGTACAGTTCC-3’Bax sense primer;
5’-ACCAAGAAGCTGAGCGAGTGTC-3’Bax anti-sense primer;
5’-ACAAAGATGGTCACGGTCTGCC-3’Caspase3 sense primer;
5’-AGAAGATCACAGCAAAAGGAGC-3’Caspase3 anti-sense primer;
5’-TCAAGCTTGTCGGCATACTG-3’
The PCR-products had the following sizes: GAPDH 457 bp, Bcl2 319 bp, Bax 367 bp, Caspase3 378 bp. The following experimental protocol for PCR reaction (35 cycles, 45 cycles for HTert) was used: denaturation: 3 min; 95˚C; amplification: denaturation: 30 s, 95˚C; annealing: 20 s, at 60˚C (GAPDH), 67˚C (Bcl2), 56˚C (Bax) and 66˚C (Caspase-3) respectively; elongation: 30 s, 72˚C, final elongation: 72˚C, 90 s. PCR products were analyzed by electrophoresis in agarose 1.8% with ethidium bromide (1 μg/ml) in TBE 1× buffer (Tris 40 mM, EDTA 1 mM, boric acid 44 mM) for 2 h at 80 V (constant voltage) with 123 bp ladder as molecular weight markers.
Images of gels were acquired (Bio-Rad Gel Doc 2000, Hercules, CA, USA) and scanned (Bio-Rad GS800, Hercules, CA, USA) using Bio-Rad Quantity One software. The density of the PCR bands were divided by that of the housekeeping gene and expressed as percent of the control band density.
2.4. Measurement of Caspase-3 Activity
Caspase-3 activity was measured by using a specific assay kit from Sigma Chemical Co. (St. Louis, MO, USA) following manufacture’s instructions. DEVD-pNA was used as a colorimetric substrate. IMR32 cells were plated at a density of 2.9 × 106 cells/35 mm dish: after treatment with Aβ peptides and S100b, cells were harvested by centrifugation. The pellets were washed with PBS, lysed in 50 ml of chilled cell lysis buffer and left on ice for 10 min. Lysate was centrifuged at 10,000 × g for 1 min at 4˚C, and supernatant was used for the caspase-3 assay.
The protein concentration was confirmed by the BCA assay. The protease activity was determined by the spectrophotometric detection at 405 nm of the chromophore p-nitroanilide (pNA) after its cleavage by caspase-3 from the labeled caspase-3-specific substrate (DEVD-pNA). Additional control assays in the presence of specific caspase-3 inhibitor (DEVD-CHO) and in the absence of recombinant human caspase were performed for measuring the non-specific hydrolysis of the substrate (data not shown).
2.5. Statistical Analysis
The data were analysed using one-way ANOVA, followed by post hoc Newman-Keul test for multiple comparisons among group means, using a Prism TM computer program (Graph-Pad, San Diego, CA, USA), and differences were considered statistically significant if P < 0.05. All results are presented as the mean ± S.E.M. of at least three different experiments performed in triplicate, unless otherwise specified
3. RESULTS
The first point investigated in this study was to clarify the experimental conditions to evidence the exerted role by S100b on Aβ (with methionine both in reduced and oxidative state) induced cellular toxicity. Thus, the toxic effects of Aβ and Aβ-Met-35OX in the presence and in the absence of different concentrations of S100b protein were evaluated on IMR-32 cells (a cellular line of human neuroblastoma).
Figure 1 shows the survival of IMR-32 cells (measured by the reduction of MTS activity), after the exposure to Aβ peptides with 5nM (panel (a)) and 5 μM (panel (b)) S100b. It appears evident that after 48 h of incubation, Aβ and Aβ-Met-35OX peptides have a toxic effect on availability of IMR32 cells inducing a significant cellular death with respect to the control. It should also be noted that the major extent of cellular death is induced by Aβ where methionine-35 is in the reduced form according to our previous data [18]. About the S100b role, it is evident that the protein alone does not interfere at nanomolar dose with the IMR32 vitality but exerts an important protective effect on the cells treated with Aβ peptides, showing availability values very similar to the untreated cells. At micromolar concentration instead S100b is neurotoxic and its effect potentiate the toxic effect of beta peptides. Hence, for studying the molecular interaction between S100b and Aβ we used a concentration of 5 nM and 10 μM respectively on all experimental procedures [24].
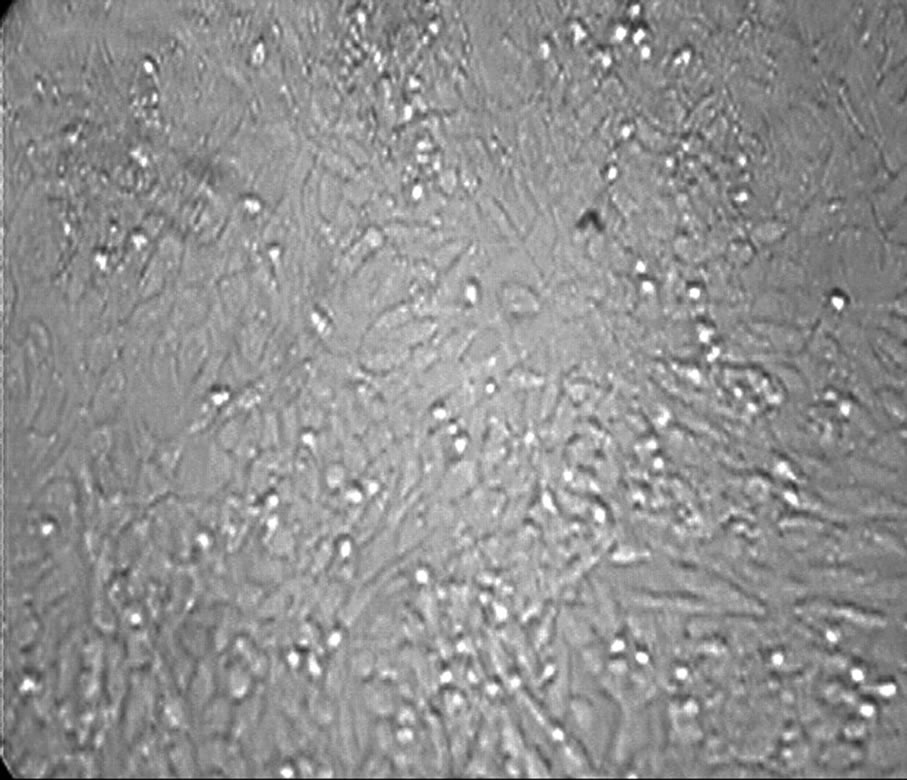
In phase-contrast observation (Figure 2), control cells (panel (a)) and cells treated with 5 nM S100b (panel (b)) appear generally healthy, with round cell bodies and a well developed network of neurites throughout the experiments. Aβ treatment was performed in the presence of 10 μM Aβ peptides, both with reduced (panel (c)) and
 (a)
(a) (b)
(b)
Figure 1. Panel (a): Effects of S100b 5 nM (Panel (a)) and 5 μM (Panel (b)) in the absence and in the presence of 10 ¼M Aβ2 (with methionine in reduced and oxidative state) on IMR- 32 cells after 48 h of incubation. The cell survival is expressed as percent of cells untreated. Cells (10,000 cells/well) were cultured with substances under analysis (experimental conditions are reported materials and methods), and the availability of cells was measured by MTS assay. All values indicate means ± S.E. of seven independent experiments. Significantly different from cells untreated: *P < 0.01. Significantly different from cells treated with beta amyloid peptides: #P < 0.01.
with oxidative 35-Methionine (panel (d)). After 48-h of treatment, a significant number reduction of living cells is evident and the cell bodies of a large amount of neurons were diminished, with condensed nuclei and retracted processes especially with the treatment with oxidative peptide. On the contrary, cells incubated with Aβ peptides, in the presence of S100b (panel (e) and (f)) were very similar to the control.
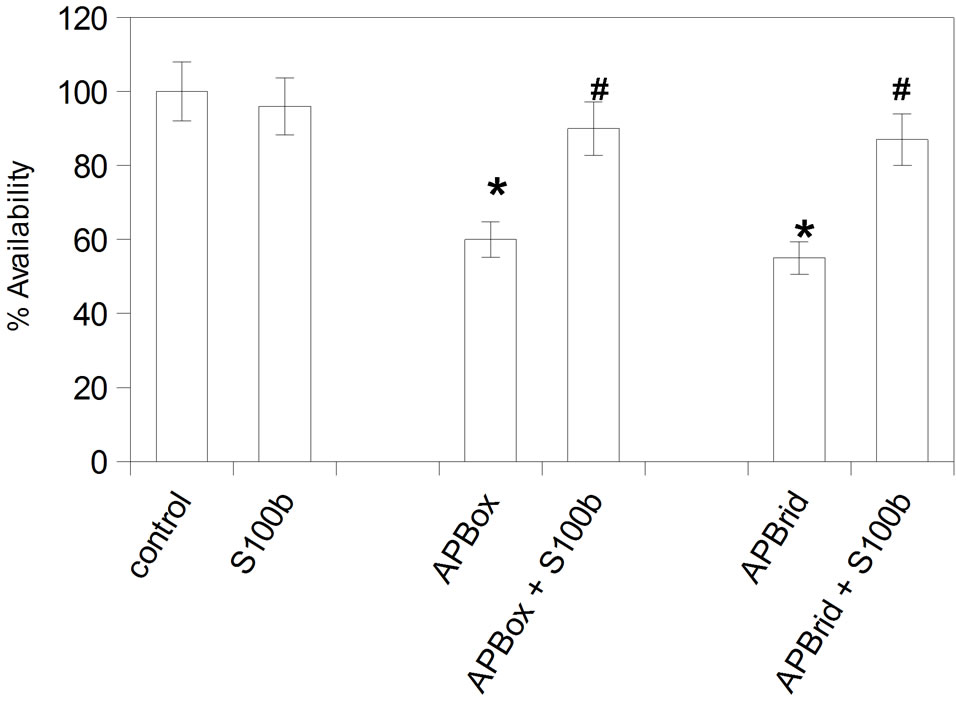
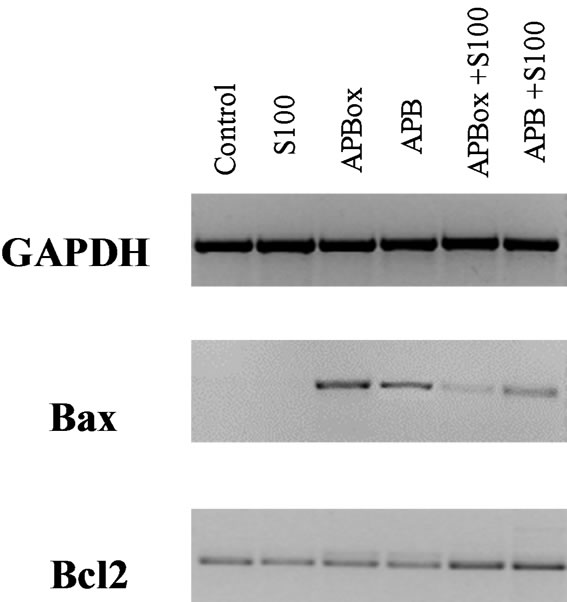
As was demonstrated in previous work S100b promotes cellular survival by increasing the expression of the antiapoptotic factor [25,26], we analyzed the gene expression of pro-apoptotic gene Bax and anti-apoptotic genes Bcl-2 in untreated neuroblastoma cells (as control) and exposed for 48 h to 10 μM with both Aβ reduced and oxidative and with 5 nM S100b (Figure 3). It is evident that Aβ treatment is able to bring about the synthesis of mRNA for pro-apoptotic gene Bax particularly when the peptide has methionine-35 in reduced state. The simultaneous treatment with beta amilod peptides and S100b determines a strong decreasing of Bax expression, according to the cellular availability but mostly an overexpression of anti apoptotic gene Bcl2. In fact, while the RNA for Bcl2 is blank for cells treated with beta amyloid peptides such as in the controls, the presence of S100b stimulates significantly the expression of this gene demonstrating an ability to modulate both Bax and Bcl2 genes.
Finally, as Bcl-2 has been shown to prevent caspase activation and subsequently cell death also the expression of caspase 3 gene and its enzimatic activity were investigated. As reported in Figure 4, the treatment with Aβ peptides increased caspase-3 expression and activity in IMR 32 cells and these outcomes are strongly prevented by the protective effect of S100b.
4. DISCUSSION
The molecular mechanisms of Alzheimer’s disease (AD), one of the most common causes of dementia, is as yet not completely clear. AD has been identified as a protein misfolding disease due to the accumulation of abnormally folded amyloid beta protein, a peptide of 1-42 aminoacid, in the brain of Alzheimer’s patients [27,28]. Amyloid beta is an abnormal proteolytic byproduct of the transmembrane protein amyloid precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. Amyloid beta peptide is a monomeric soluble molecule which contains short regions of beta sheet and polyproline II helix secondary structures in solution [29], though they are largely alpha helical in membranes [30]; however, at sufficiently high concentration, they undergo a dramatic conformational change to form a beta sheet-rich tertiary structure which aggregates to form amyloid fibrils [31]. These fibrils deposit external neurons in dense formations known as senile plaques or neuritic plaques, in less dense aggregates as diffuse plaques, and sometimes in the walls of small blood vessels in the brain.
Another important pathogenetic event in AD is a relevant oxidative stress: the “Aβ cascade hypothesis” of Butterfield [32] in fact suggests that Aβ as small oligomers can insert into the lipid bilayer and initiate lipid peroxidation and oxidative damage to proteins and other molecules. A consequence of this event is the influx of Ca2+ into the neuron, resulting in the loss of ion homoeostasis, mitochondrial dysfunction, synapse loss and finally cell death [32-35].
Methionine-35 (Met-35) side chain of AβP appears to play a critical role in peptide’s neurotoxicity; indeed, this
 (a)
(a)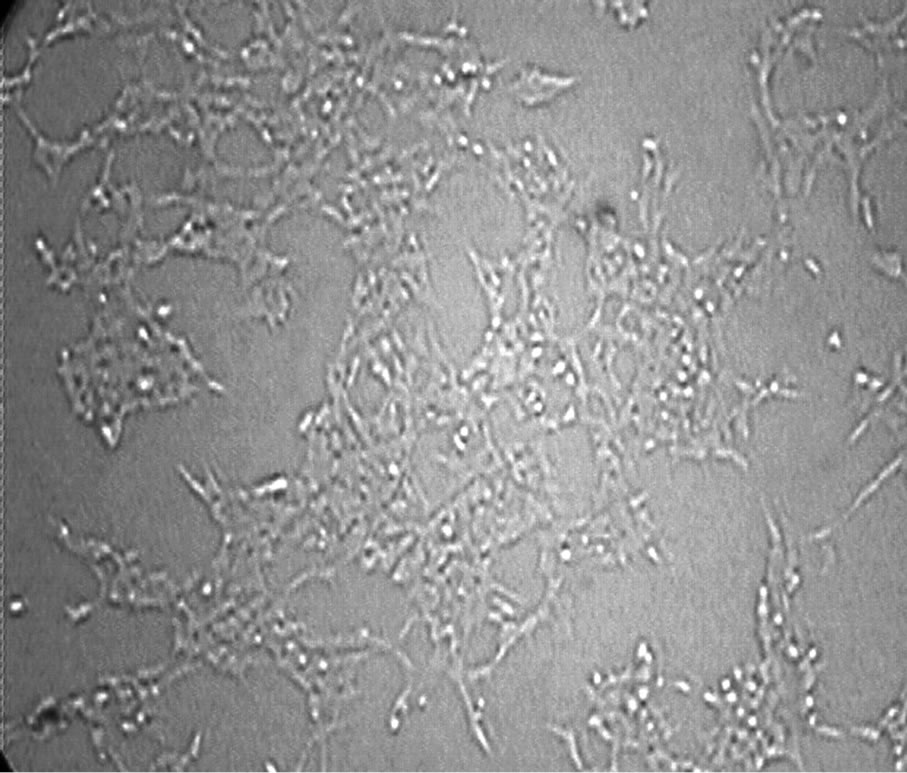 (b)
(b)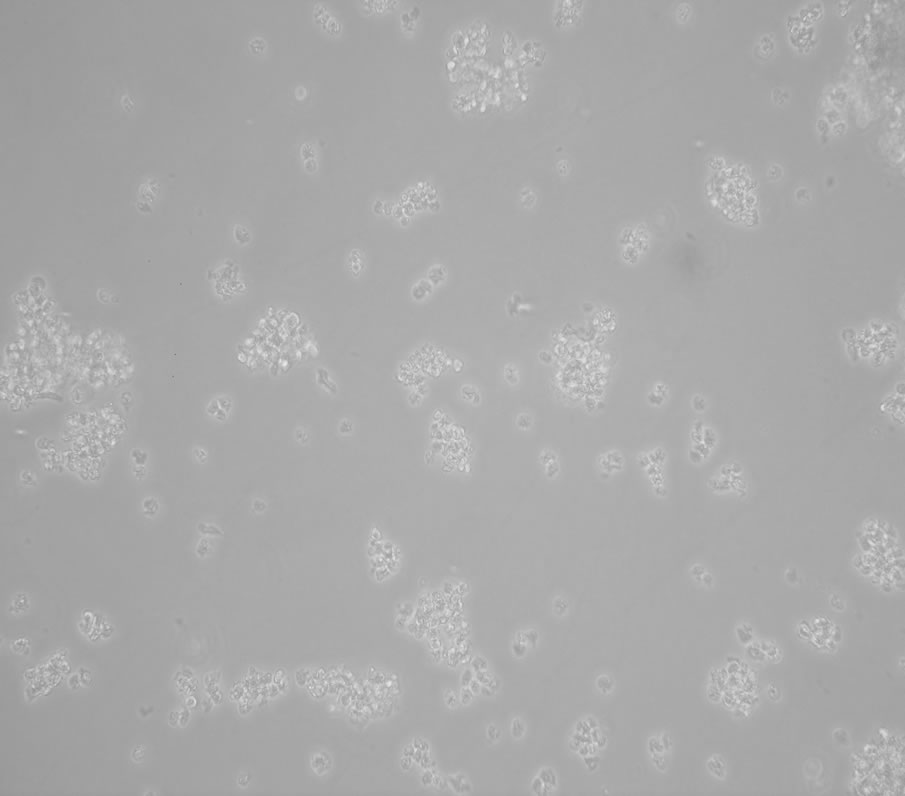 (c)
(c)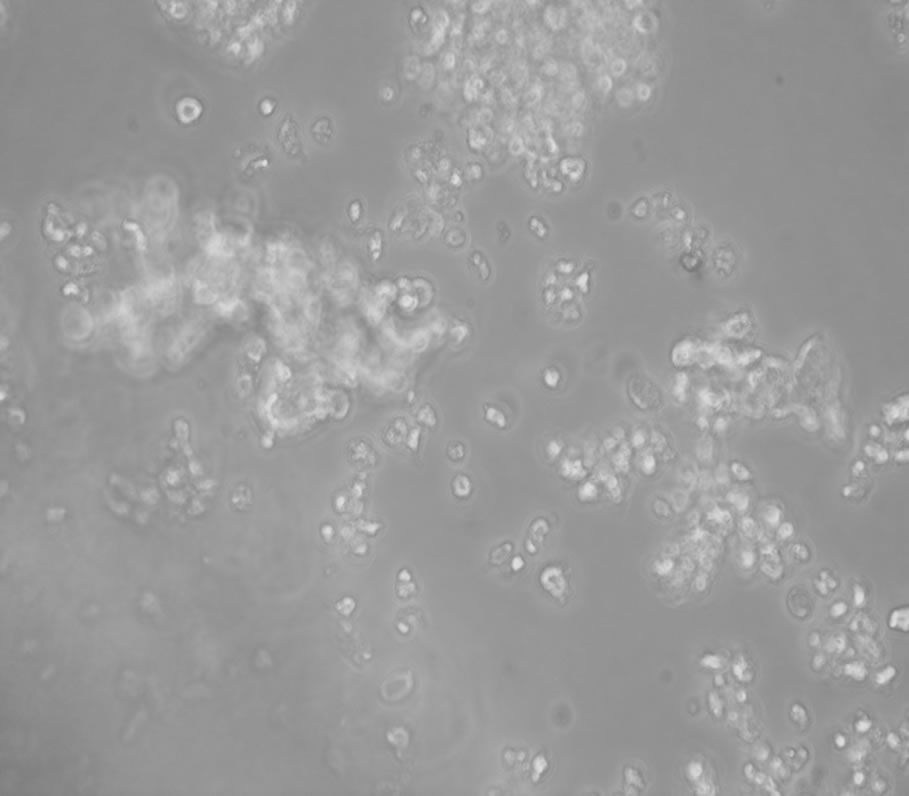 (d)
(d)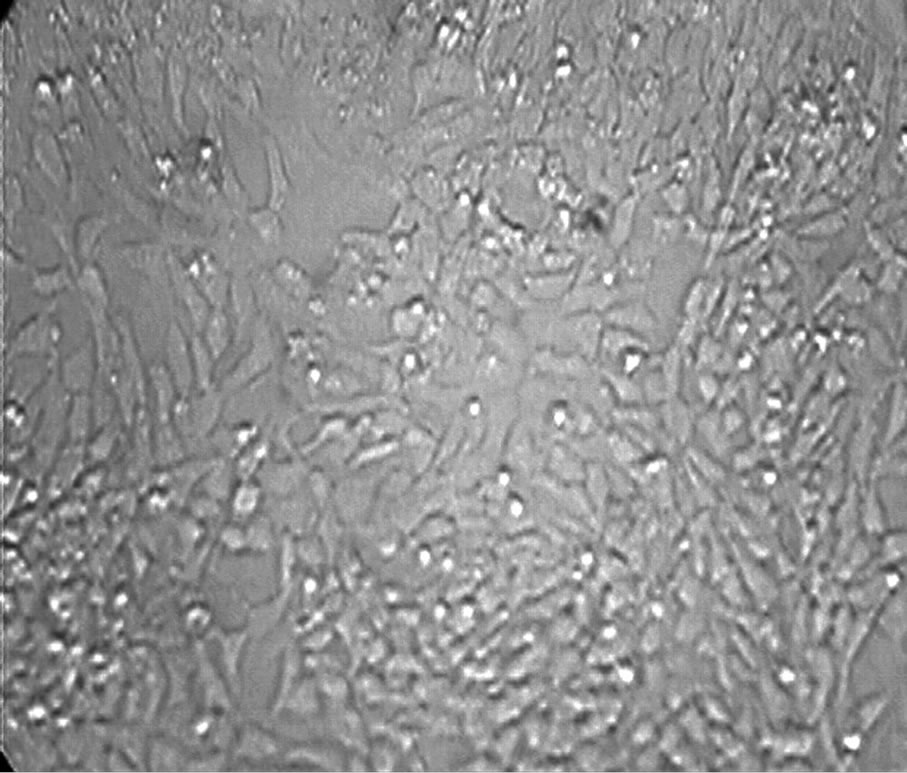 (e)
(e) (f)
(f)
Figure 2. Phase-contrast micrography shows IMR-32 cells in medium (panel (a)); treated with 5 nM S100b (panel (b)); with 10 μM Aβ with reduced (panel (c)) and oxidative 35-Methionine (panel (d)) and cells incubated simultaneously with S100b in presence of Aβ (panel (e)) and Aβ-Met-35OX (panel (f)). All treatments were performed for 48 h.
 (a)
(a)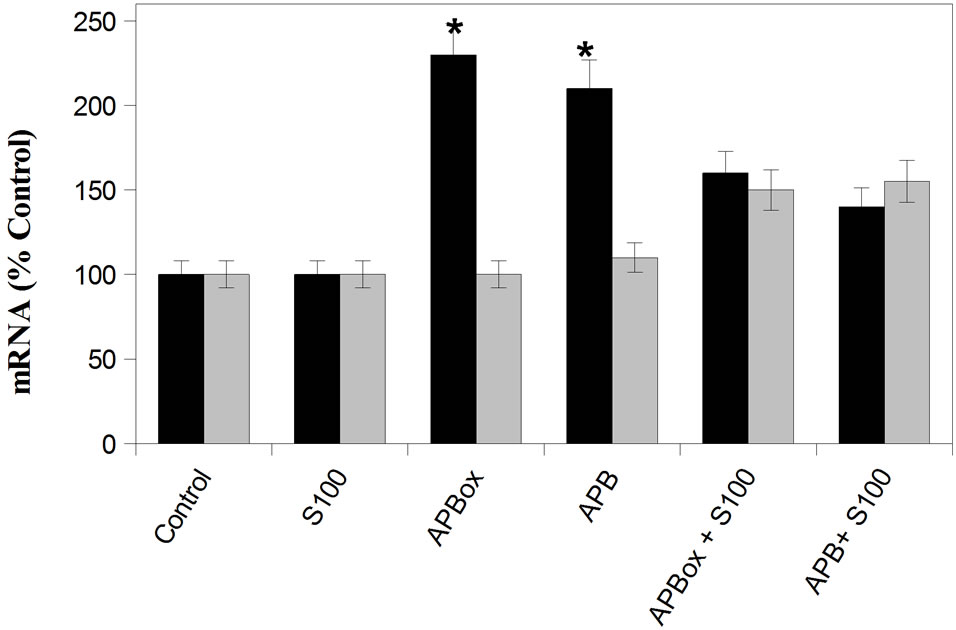 (b)
(b)
Figure 3. Panel (a): Expression of Bax and Bcl-2 genes in IMR-32 cells. Cells were incubated for 48 h with medium (control), with 5 nM S100, with 10 μM Aβ with Methionine 35 oxidated (APBox) and reduced (APB) and and with the simultaneous presence of S100 and Aβ peptides. GAPDH gene expression was used as housekeeping. Panel (b): The intensities of the bands were quantized by densitometric scanning of agarose gel bands and expressed as percent of Bax (black bars) and Bcl-2 (grey bars) mRNA expression respect to the control. Results are from eight independent experiments. Significantly different from controls *P < 0.01.
residue is mostly susceptible to oxidation in vivo [20, 36,37], and Aβ bearing oxidized Met-35 is found in large quantities in post-mortem AD plaques [38,39], while oxidative stress does not occur in the brain in vivo if the Methionine 35 residue of Aβ(1-42) is not present [40, 41].
In this complex pathogenetic network, numerous exogenous and endogenous substances are been tested to individuate possible molecules able to oppose the neurodegenerative mechanisms generated by Aβ(1-42) [42- 44]. In this context the observation that during the early stages of AD, microglia produces numerous proinflammatory molecules such as S100b which expression is highly modulated by reactive astrocytes in close vicinity of beta-amyloid deposits, induced us to investigate theinterplay between Aβ, both with Met-35 oxidate and
 (a)
(a) (b)
(b)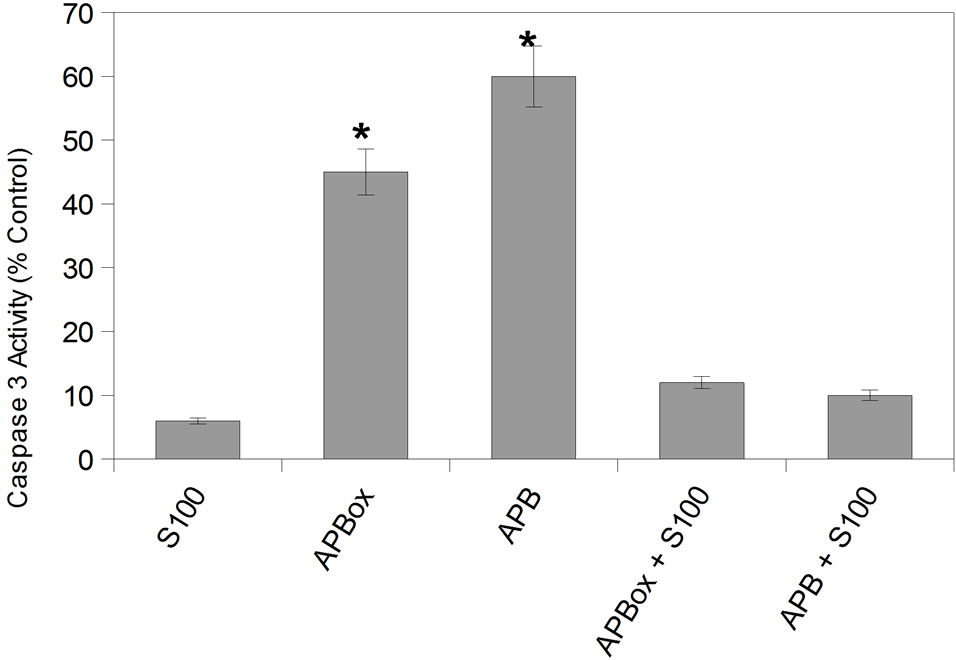 (c)
(c)
Figure 4. Panel (a): Expression of caspase 3 gene in IMR-32 cells. Cells were incubated for 48 h with medium (control), with 5 nM S100b, with 10 μM Aβ with Methionine 35 oxidated (APBox) and reduced (APB) and and with the simultaneous presence of S100 and Aβ peptides. GAPDH gene expression was used as housekeeping (data not shown). Panel (b): The intensities of the bands were quantized by densitometric scanning of agarose gel bands and expressed as percent of caspase 3 mRNA expression respect to the control. Panel (c) Caspase-3 activity (expressed as percent of control) in IMR-32 cells incubated for 48 h with medium (control), with 5 nM S100b, with 10 μM Aβ with Methionine 35 oxidated (APBox) and reduced (APB) and and with the simultaneous presence of S100b and Aβ peptides. Results are from eight independent experiments. Significantly different from controls *P < 0.01.
reduced, and S100b. In particular the S100b is carefully modulated during AD evolution, coming by nanomolar values, during the first phases of pathology, to micromolar concentration [11-13]. In this study we examined the possible participation of this protein, at different concentrations, to contrast the AD progression.
In this regard, it has been evidenced that S100b at nanomolar concentrations is able to protect human neuroblastoma cells (IMR-32) against Aβ-mediated toxicity. Since Aβ-mediated mechanism of toxicity seems to be mediated by an apoptotic pathway [45], we investigated the expression of Bax, Bcl-2 and caspase 3 genes, three of the main important apoptosis-related genes [46,47], in Aβ-treated cells both with oxidated and reduced methionine 35 with and without S100b.
Bax is a gene encoding for a protein which accelerates programmed cell death by its translocation to the mitochondrion membrane, leading to the release of cytochrome c. When translocated to the mitochondrial membrane, Bax can in fact homodimerize and triggers the activation of terminal caspases by alteration of mitochondrial functions, which results in the release of apoptosis-promoting factors into the cytoplasm [46].
Bcl-2 product is instead a protein localized in the cytoplasmic face of the mitochondrial outer membrane, endoplasmic reticulum, and nuclear envelope [45,46]. Bcl-2 has been shown to prevent cytochrome c release, caspase activation, and cell death.
The results of present study evidenced that Aβ treatments resulted in increased Bax expression in IMR-32 cells: but when Aβ-treated cells were incubated in the presence of S100b, Bax expression results up-regulated. On the other hand, our results show that the expression of the anti-apoptotic gene Bcl-2 is very low in control and in IMR-32 cells treated with Aβ peptides while treatment with S100b considerably increases the expression of this gene. This finding indicates that a significant shift of Bax/Bcl-2 ratio in favour of Bcl-2 occurs in Aβ/S100b- treated cells with respect to that showed by Aβ-treated cells. Another important apoptotic event is characterized by the caspase 3 activation. In particular, mRNA accumulation of Bax may be sufficient to initiate the intrinsic pathway of caspase activation and therefore to lead to cell death [47,48]. Indeed, Bax can form homodimers/ heterodimers with other members of the Bcl-2 family upon translocation to the mitochondrial membrane, and activate a cascade of events including increased mitochondrial membrane permeability, release of cytochrome c into the cytoplasm and activation of caspases. Our results, evidenced that the treatment of IMR-32 with Aβ peptides increases the expression of caspase 3 and its enzymatic activity: also in this case, the simultaneous presence of S100b determines a protective effect, inhibiting the production and activity of this pro-apoptotic enzyme. As expected and described previously in literature [18], the reduced peptide was far more effective than the oxidized form in determining the apoptotic cascade but the protective effect of S100b is equally effective demonstrating a common interplay with the Aβ peptides, independently by the oxidative state of methionine 35. To summarize, our results indicate that Aβ-mediated degeneration in IMR-32 cells is due to a large shift of Bax/Bcl-2 ratio in favour of the pro-apoptotic Bax with subsequent cell death. The presence of nanomolar concentrations of S100b significantly lowers the toxicity induced by Aβ peptides through the reduction of the pro-apoptotic Bax/Bax homodimers and the increase of antiapoptotic Bcl-2/Bcl-2 homodimers. Moreover Bcl-2 has been shown to prevent caspase activation and cell death; therefore, the effect of S100b on caspase 3 is mediated both by a direct effect on RNAm expression and indirectly through enzyme activity post-transcriptionally regulated by Bcl-2/Bcl-2 homodimers.
Another important question is the increase of S100b observed in late phases of Alzheimer: the levels of S100 beta protein and mRNA, were elevated 10 - 20-fold in extracts of temporal lobe from autopsy samples of Alzheimer’s disease patients compared to those of aged control patients. Our data evidenced that the treatment with S100b at micromolar concentration on IMR-32 results strongly toxic potentiating the neurotoxicity by beta amyloide.
The synergical toxic effect exerted by S100 and amyloide peptide could be discussed on the basis of RAGE (advanced glycation end-product receptor) expression. RAGE is an inducible receptor, which exists on neurons and microglial cells, responsible of extracellular elimination of neurotoxic advanced glycation end-products (AGEs) [49]. Indeed, RAGE is multi-ligand receptors: able to accept more than one ligand concomitantly including Aβ peptides, amphoterins, and also S100b [49-51]. Hence, S100b concentration, modulating also the expression of RAGE, could underlie the progression of AD and of a range of other chronic disorders [25,52-53].
5. CONCLUSION
In conclusion, our results evidenced that during early AD pathogenesis, a network between astrocytes and neurons could govern the Aβ toxicity through apoptotic signal modulation. Instead, in advanced phases of pathology, the (over)-expression of S100b is so high to accelerate AD-like pathology. In this light the biosynthesis modulation of pro-inflammatory cytokines such as S100b, may be a promising therapeutic strategy to delay AD progression.
6. ACKNOWLEDGEMENTS
This work was supported by grants from the Italian Ministry of Education, University, and Research (PRIN 2010-2011 20109MXHMR-006 to B.G.) and by Italian National Research Council (CNR).
REFERENCES
- Donato, R., Cannon, BR., Sorci, G., Riuzzi, F., Hsu, K., Weber, D.J. and Geczy, C.L. (2012) Function of S100 proteins. Current Molecular Medicine, 13, 24-57.
- Donato, R., Sorci, G., Riuzzi, F., Arcuri, C., Bianchi, R., Brozzi, F., Tubaro, C. and Giambanco, I. (2009) S100B’s double life: Intracellular regulator and extracellular signal. Biochimica et Biophysica Acta, 1793, 1008-1022. doi:10.1016/j.bbamcr.2008.11.009
- Adami, C., Sorci, G., Blasi, E., Agneletti, A.L., Bistoni, F. and Donato, R. (2001) S100B expression in and effects on microglia. Glia, 33, 131-142. doi:10.1002/1098-1136(200102)33:2<131::AID-GLIA1012>3.0.CO;2-D
- Donato, R. (2003) Intracellular and extracellular roles of S100 proteins. Microscopy Research and Technique, 60, 540-551.
- Yamaguchi, F., Umeda, Y., Shimamoto, S., Tsuchiya, M., Tokumitsu, H., Tokuda, M. and Kobayashi, R. (2012) S100 proteins modulate protein phosphatase 5 function: A link between CA2+ signal transduction and protein dephosphorylation. The Journal of Biological Chemistry. 287, 13787-13798.
- Ciccarelli, R., Di Iorio, P., Bruno, V., Battaglia, G., D’ Alimonte, I., D’Onofrio, M., Nicoletti, F. and Caciagli, F. (1999) Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia, 27, 275-281. doi:10.1002/(SICI)1098-1136(199909)27:3<275::AID-GLIA9>3.0.CO;2-0
- Ahlemeyer, B., Beier, H., Semkova, I., Schaper, C., Krieglstein, J. (2000) S-100beta protects cultured neurons against glutamateand staurosporine-induced damage and is involved in the antiapoptotic action of the 5 HT(1A)- receptor agonist, Bay x 3702. Journal of Brain Research, 858, 121-128.
- Pinto, S.S., Gottfried, C., Mendez, A., Gonçalves, D., Karl, J., Gonçalves, C.A., Wofchuk, S. and Rodnight, R. (2000) Immunocontent and secretion of S100B in astrocyte cultures from different brain regions in relation to morphology. FEBS Letters, 486, 203-207.
- Tort, A.B., Portela, L.V., da Purificação Tavares, M., Gonçalves, C.A., Netto, C., Giugliani, R. and Souza DO. (2004) Specificity and sensitivity of S100B levels in amniotic fluid for Down syndrome diagnosis. Life Sciences, 76, 379-384.
- Netto, C.B., Portela, L.V., Ferreira, C.T., Kieling, C., Matte, U., Felix, T., da Silveira, T.R., Souza, DO., Gonçalves, C.A. and Giugliani, R. (2005) Ontogenetic changes in serum S100B in down syndrome patients. Clinical Biochemistry, 38, 433-435. doi:10.1016/j.clinbiochem.2004.12.014
- Jesse, S., Steinacker, P., Cepek, L., von Arnim, C.A., Tumani, H., Lehnert, S., Kretzschmar, H.A., Baier, M. and Otto, M. (2009) Glial fibrillary acidic protein and protein S-100B: Different concentration pattern of glial proteins in cerebrospinal fluid of patients with Alzheimer’s disease and Creutzfeldt-Jakob disease. Journal of Alzheimer’s Disease, 17, 541-51.
- Chaves, M.L., Camozzato, A.L., Ferreira, E.D., Piazenski, I., Kochhann, R., Dall’Igna, O., Mazzini, G.S., Souza, DO. and Portela, L.V. (2010) Serum levels of S100B and NSE proteins in Alzheimer’s disease patients. Journal of Neuroinflammation, 27, 6.
- Li, C., Zhao, R., Gao, K., Wei, Z., Yin, M.Y., Lau, L.T., Chui, D. and Hoi Yu. A.C. (2011) Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Current Alzheimer Research, 8, 67-80. doi:10.2174/156720511794604543
- Medeiros, R. and LaFerla, F.M. (2013) Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Experimental Neurology, 239, 133-138.
- Marshak, D.R., Pesce, S.A., Stanley, L.C. and Griffin, W.S. (1992) Increased S100 beta neurotrophic activity in Alzheimer’s disease temporal lobe. Neurobiology of Aging, 13, 1-7. doi:10.1016/0197-4580(92)90002-F
- Mori, T., Koyama, N., Arendash, G.W., Horikoshi-Sakuraba, Y., Tan, J. and Town, T. (2010) Overexpression of human S100B exacerbates cerebral amyloidosis and gliosis in the Tg2576 mouse model of Alzheimer’s disease. Glia, 58, 300-314.
- Castaño, E.M., Maarouf, C.L., Wu, T., Leal, M.C., Whiteside, C.M., Lue, L.F., Kokjohn, T.A., Sabbagh, M.N., Beach, T.G. and Roher, A.E. (2012) Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochemistry International, 62, 145-156.
- Clementi, M.E., Pezzotti, M., Orsini, F., Sampaolese, B., Mezzogori, D., Grassi, C., Giardina, B. and Misiti, F. (2006) Alzheimer’s amyloid beta-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: An intriguing role for methionine 35. Biochemical and Biophysical Research Communications, 342, 206- 213.
- Butterfield, D.A., Galvan, V., Lange, M.B., Tang, H., Sowell, R.A., Spilman, P., Fombonne, J., Gorostiza, O., Zhang, J., Sultana, R. and Bredesen, D.E. (2010) In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid betapeptide of APP. Free Radical Biology & Medicine, 48, 136-144.
- Butterfield, D.A. and Sultana, R. (2011) Methionine-35 of aβ(1-42): Importance for oxidative stress in Alzheimer disease. Journal of Amino Acids, 2011, 1-10. doi:10.4061/2011/198430
- Pike, C.J., Burdick, D., Welencewicz, A.J., Glabe, C.G. and Cotman C.W. (1993) Neurodegeneration induced by β-amyloid peptides in vitro: The role of peptide assembly state. The Journal of Neuroscience, 13, 1676-1678.
- Boland, K., Behrens, M., Choi, D., Manias, K. and Perlmutter, D.H. (1996) The serpin-enzyme complex receptor recognizes soluble, nontoxic amyloid-beta peptide but not aggregated, cytotoxic amyloid-beta peptide. The Journal of Biological Chemistry, 271, 18032-18044. doi:10.1074/jbc.271.30.18032
- Cory, A.H., Owen, T.C., Barltrop, J.A. and Cory, J.G. (1991) Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Communications, 3, 207-212.
- Clementi, M.E., Marini, S., Coletta, M., Orsini, F., Giardina, B. and Misiti, F. (2005) Abeta(31-35) and Abeta(25-35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Letters, 579, 2913-2918.
- Businaro, R., Leone, S., Fabrizi, C., Sorci, G., Donato, R., Lauro, G.M. and Fumagalli, L. (2006) S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. Journal of Neuroscience Research, 83, 897-906. doi:10.1002/jnr.20785
- Villarreal, A., Aviles Reyes, R.X., Angelo, M.F., Reines, A.G., Ramos, A.J. (2011) S100B alters neuronal survival and dendrite extension via RAGE-mediated NF-κB signaling. Journal of Neurochemistry, 117, 321-332. doi:10.1111/j.1471-4159.2011.07207.x
- De Strooper, B. (2010) Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiological Reviews, 90, 465-494. doi:10.1152/physrev.00023.2009
- Ubhi, K. and Masliah, E. (2013) Alzheimer’s disease: Recent advances and future perspectives. Journal of Alzheimer’s Disease, 33, S185-194.
- Schweers, O., Schönbrunn-Hanebeck, E., Marx, A. and Mandelkow, E. (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. The Journal of Biological Chemistry, 269, 24290-24297,
- Wahlström, A., Hugonin, L., Perálvarez-Marín, A., Jarvet, J. and Gräslund, A. (2008) Secondary structure conversions of Alzheimer’s Abeta(1-40) peptide induced by membrane-mimicking detergents. FEBS J, 275, 117-128. doi:10.1111/j.1742-4658.2008.06643.x
- Manzoni, C., Colombo, L., Bigini, P., Diana, V., Cagnotto, A., Messa, M., Lupi, M., Bonetto, V., Pignataro, M., Airoldi, C., Sironi, E., Williams, A. and Salmona, M. (2011) The molecular assembly of amyloid aβ controls its neurotoxicity and binding to cellular proteins. PLoS One, 6, e24909. doi:10.1371/journal.pone.0024909
- Butterfield, A., Swomley, A.M. and Sultana, R. (2012) Amyloid β-peptide 1-42-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxidants & Redox Signaling, 18, 1-55.
- Butterfield, D.A. (2003) Amyloid beta-peptide [1-42]- associated free radical-induced oxidative stress and neurodegeneration in Alzheimer’s disease brain: Mechanisms and consequences. Current Medicinal Chemistry, 10, 2651-2659. doi:10.2174/0929867033456422
- Butterfield, D.A., Perluigi, M. and Sultana, R. (2006) “Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. European Journal of Pharmacology, 545, 39-50. doi:10.1016/j.ejphar.2006.06.026
- Sultana, R. and Butterfield, D.A. (2013) Oxidative modification of brain proteins in Alzheimer’s disease: Perspective on future studies based on results of redox proteomics studies. Journal of Alzheimer’s Disease, 33, 243- 251.
- Butterfield, D.A. and Bush, A.I. (2004) Alzheimer’s amyloid beta-peptide (1-42): Involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiology of Aging, 25, 563-568. doi:10.1016/j.neurobiolaging.2003.12.027
- Misiti, F., Clementi, M.E. and Giardina, B. (2010) Oxidation of methionine 35 reduces toxicity of the amyloid beta-peptide(1-42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochemistry International, 56, 597-602. doi:10.1016/j.neuint.2010.01.002
- Naslund, J., Schierhorn, A., Hellman, U., Lannfelt, L., Roses, A.D., Tjernberg, L.O., Silberring, J., Gandy, S.E., Winblad, B. and Greengard, P. (1994) Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proceedings of the National Academy of Sciences of the United States of America, 91, 8378-8382. doi:10.1073/pnas.91.18.8378
- Kuo, Y.M., Kokjohn, T.A., Beach, T.G., Sue, L.I., Brune, D., Lopez, J.C., Kalback, W.M., Abramowski, D., Sturchler-Pierrat, C., Staufenbiel, M. and Roher, A.E. (2001) Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer’s disease brains. The Journal of Biological Chemistry, 276, 12991-12998. doi:10.1074/jbc.M007859200
- Butterfield, D.A., Galvan, V., Lange, M.B., Tang, H., Sowell, R.A., Spilman, P., Fombonne, J., Gorostiza, O., Zhang, J., Sultana, R. and Bredesen, D.E. (2010) In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid betapeptide of APP. Free Radical Biology and Medicine, 48, 136-144. doi:10.1016/j.freeradbiomed.2009.10.035
- Johansson, A.S., Bergquist, J., Volbracht, C., Päiviö, A., Leist, M., Lannfelt, L. and Westlind-Danielsson, A. (2007) Attenuated amyloid-beta aggregation and neurotoxicity owing to methionine oxidation. Neuroreport, 18, 559-563. doi:10.1097/WNR.0b013e3280b07c21
- Parihar, M.S. and Hemnani, T. (2004) Alzheimer’s disease pathogenesis and therapeutic interventions. Journal of Clinical Neuroscience, 11, 456-467. doi:10.1016/j.jocn.2003.12.007
- Kumar, S., Okello, E.J. and Harris, J.R. (2012) Experimental inhibition of fibrillogenesis and neurotoxicity by amyloid-beta (Aβ) and other disease-related peptides/ proteins by plant extracts and herbal compounds. Subcellular Biochemistry, 65, 295-326. doi:10.1007/978-94-007-5416-4_13
- Vassallo, N. and Scerri, C. (2012) Mediterranean diet and dementia of the Alzheimer type. Current Aging Science, 6, 150-162.
- Kuwana, T. and Newmeyer, D.D. (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Current Opinion in Cell Biology, 15, 691-699. doi:10.1016/j.ceb.2003.10.004
- Murphy, K.M., Ranganathan, V., Farnsworth, M.L., Kavallaris, M. and Lock, R.B. (2000) Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death & Differentiation, 7, 102-111. doi:10.1038/sj.cdd.4400597
- Budihardjo, I., Oliver, H., Lutter, M., Luo, X. and Wang, X. (1999) Biochemical pathways of caspase activation during apoptosis. Annual Review of Cell and Developmental Biology, 15, 269-290. doi:10.1146/annurev.cellbio.15.1.269
- Fesik, S.W. and Shi, Y. (2001) Controlling the caspases. Science, 294, 1477-1478. doi:10.1126/science.1062236
- Yan, S.D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., Slattery, T., Zhao, L., Nagashima, M., Morser, J., Migheli, A., Nawroth, P., Stern, D. and Schmidt, A.M. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature, 382, 685-691. doi:10.1038/382685a0
- Hofmann, M.A., Drury, S., Fu, C., Qu, W., Taguchi, A., Lu, Y., Avila, C., Kambham, N., Bierhaus, A., Nawroth, P., Neurath, M.F., Slattery, T., Beach, D., McClary, J., Nagashima, M., Morser, J., Stern, D. and Schmidt, A.M. (1999) RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell, 97, 889-901. doi:10.1016/S0092-8674(00)80801-6
- Meneghini, V., Bortolotto, V., Francese, M.T., Dellarole, A., Carraro, L., Terzieva, S. and Grilli, M. (2013) Highmobility group box-1 protein and β-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-κB axis: Relevance for Alzheimer’s disease. The Journal of Neuroscience, 33, 6047- 6059. doi:10.1523/JNEUROSCI.2052-12.2013
- Sathe, K., Maetzler, W., Lang, J.D., Mounsey, R.B., Fleckenstein, C., Martin, H.L., Schulte, C., Mustafa, S., Synofzik, M., Vukovic, Z., Itohara, S., Berg, D. and Teismann, P. (2012) S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-α pathway. Brain, 135, 3336-3347. doi:10.1093/brain/aws250
- Slowik, A., Merres, J., Elfgen, A., Jansen, S., Mohr, F., Wruck, C.J., Pufe, T. and Brandenburg, L.O. (2012) Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)—And amyloid beta 1-42-induced signal transduction in glial cells. Molecular Neurodegeneration, 7, 55.
NOTES
*All Authors of the manuscript declare to have not any direct financial relation with the commercial identities mentioned in the present paper.