Advances in Alzheimer's Disease
Vol.2 No.2(2013), Article ID:33190,7 pages DOI:10.4236/aad.2013.22009
Evaluation of the inhibitory effect of docosahexaenoic acid and arachidonic acid on the initial stage of amyloid β1-42 polymerization by fluorescence correlation spectroscopy
![]()
Department of Environmental Physiology, Shimane University Faculty of Medicine, Shimane, Japan; *Corresponding Author: michio1@med.shimane-u.ac.jp
Copyright © 2013 Koji Miwa et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 20 February 2013; revised 25 March 2013; accepted 10 April 2013
Keywords: Docosahexaenoic Acid; Arachidonic Acid; Fluorescence Correlation Spectroscopy; Amyloid β Peptide; Fibrillation
ABSTRACT
Amyloid β (Aβ)1-42 fibrillation is a crucial step in the development of pathological hallmarks, such as neuritic plaques and neurofibrillary tangles, of Alzheimer’s disease (AD). In this study, we evaluated the effects of free docosahexaenoic acid (DHA), an essential brain polyunsaturated fatty acid (PUFA), on the inhibition of Aβ1-42 fibrillation by fluorescence correlation spectroscopy (FCS), a technique capable of detecting molecular movements and interactions in solution. We also examined whether free arachidonic acid (AA), eicosapentaenoic acid (EPA), and metabolites of DHA, including neuroprotectin D1 (NPD1, 10S, 17S-dihydroxy-DHA), resolvin D1 (RvD1, 7S, 8R, 17S-trihydroxy-DHA), and didocosahexaenoyl glycerol (diDHA), affect Aβ1-42 polymerization. The results of the FCS study reveal that DHA and AA significantly reduced the diffusion time of TAMRA (5-carboxytetramethylrhodamine)-Aβ1-42 by 28% and 31%, respectively, while EPA, NPD1, RvD1, and diDHA had no effects on diffusion time. These results indicate that DHA and AA inhibited Aβ1-42 polymerization and that their inhibitory effects occurred at the initial stage of Aβ1-42 polymerization. This study will advance the research on PUFAs in preventing AD progression.
1. INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by the deposition of amyloid β (Aβ) peptides in neuritic plaques and neurofibrillar tangles in the affected brain regions [1]. Aβ1-42, which is proteolytically released from membrane-bound amyloid precursor proteins [2], constitutes the foremost component of neuritic plaques and tangles of the affected brains [3] and plays an important role in neurobehavioral impairments in AD [4]. Formation of fibers is thus central to AD pathogenesis, and a great deal of work using various techniques, including transmission electron microscopy [5], atomic force microscopy [6-8], circular dichcoism [9], polyacrylamide gel electrophoresis (PAGE) [10-12], size-exclusion chromatography [13,14], and quantitative fluorimetry [5,15], has been performed to delineate the mechanism. Consistent with the findings of other studies [16-19], we have previously reported that Aβ1-42 fibrillation involves conformational changes from α helix to β sheet and passes through various phases of fibrillation, including the formation of dimers, trimers, tetramers, oligomers, and finally matured fibrils, using thioflavin T fluorospectroscopy, PAGE, western blot, fluorescence microscopy, and transmission electron microscopy [20-23]. The natural plant compounds includeing curcumin, epigallocatechin-3-gallate and/or Ginkgo biloba extract and also fish oil components such as docosahexaenoic acid (DHA) were reported to inhibit amyloid formation [24-26]. Among these compounds, DHA is the most abundant n-3 polyunsaturated fatty acid (PUFA) in the mammalian brain [27-29], and deficiency of DHA is associated with memory impairment in AD model rats [30] and AD patients [31]. Oral administration of DHA decreases the amyloid burden in the brains of AD model rats [30] and mice [32], with a concomitant in vitro inhibition of the amyloid fibril formation, by acting at various stages of polymerization [20-23]. As one of the mechanism(s) of DHA action, we have previously shown that DHA inhibits in vitro Aβ1-42 fibrillation at the trimer/tetramer level, and thereby inhibits further progression of lateral stacking of these intermediates and finally prevents mature amyloid fibril formation [20,21]. Thus, DHA is suggested to be a potent therapeutic and preventive agent against Aβ-induced AD. However, the exact mechanisms of action of DHA remain unclear. Thus, in the present investigation, we have used fluorescence correlation spectroscopy (FCS) to delineate the temporal resolution of DHA-induced mechanisms of inhibition of amyloid fibrillation.
FCS is a correlation analysis of fluctuations in the fluorescence intensity of fluorescent compounds excited by a sharply focused laser beam in a very tiny space, i.e., the so-called confocal volume. The fluorescence intensity fluctuates because of Brownian motion of the fluorescent particles. In other words, the number of particles in the confocal volume is randomly changing around the average number. This analysis gives the average number of fluorescent particles and average diffusion time when particles are passing through the tiny confocal volume. In practice, the fluorescence of dye-labeled amyloid Aβ1-42 changes because of diffusion in the confocal volume, thus the diffusion time in the presence or absence of DHA might provide greater insight into the effects of DHA on the molecular interactions of amyloid species undergoing fibrillogenesis. In addition, the effects of other PUFAs such as eicosapentaenoic acid (EPA), a precursor for DHA, and arachidonic acid (AA), the abundant n-6 PUFA in the brain, on amyloid polymerization are also unknown and thus might be studied using this technique. The DHA/AA ratio has been shown to have a significantly negative correlation with long-term memory in Aβ peptide-infused AD model rats [30] and normal rats [33]. Recently, inflammation was also shown to contribute to the amyloid pathogenesis of AD, and metabolites of DHA including neuroprotectin D1 (NPD1) and resolvin D1 (RvD1) were reported to promote anti-inflammation and provide beneficial effects [34]. didocosahexaenoyl phosphopilid species [35-37] are abundant in the brain, and thus, whether the bulky diDHA inhibits Aβ1-42 polymerization was also tested in the present experiment. Finally, the appearance of Aβ aggregates in solution [38] and the cerebrospinal fluid of AD patients [39] on FCS has been reported. Therefore, the present investigation could be considered of significant interest because it involves use of FCS, an ultrasensitive and non-invasive detection method capable of single-molecule and real-time resolution, for determining whether DHA, AA, EPA, DHA metabolites NPD1 and RvD1, and diDHA inhibit Aβ polymerization in a single experimental setting.
2. MATERIALS AND METHODS
2.1. Materials
The chemical structures of the compounds used in this experiments are indicated in Figure 1. Aβ1-42 was purchased from the Peptide Institute Inc. (Osaka, Japan). DHA (4Z, 7Z, 10Z, 13Z. 16Z, 19Z-Docosahexaenoic acid), EPA (Icosapentaenoic acid), AA (5, 8, 11, 14-icosatetraenoic acid), NPD1 [Neuroprotectin D1; 10, 17 (S)- dihydro(pero)xydocosahexa-4Z, 7Z, 11E, 13Z, 15E, 19Zenoic acid], and RvD1 [17 (S)-Resolvin D1; 7S, 8R, 17S-trihydroxy-4Z, 9E, 11E, 13Z, 15E, 19Z-docosahexaenoic acid] were purchased from Cayman Chemical Company (MI, USA). diDHA [didocosahexaenoyl glycerol; Didocosahexaenoin (4, 7, 10, 13, 16, 19, -all cis)] was purchased from Larodan Fine Chemicals AB (Malmö, Sweden). Fluorescently labeled Aβ1-42 [TAM RA (5-carboxytetramethylrhodamine)-Aβ1-42; TAMRA-la beled β-amyloid1-42] was purchased from AnaSpec Inc. (CA, USA). All other chemicals were of analytical grade.
2.2. Aβ1-42 Peptide Preparation for Analysis by FCS
Aβ1-42 peptide was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 100 μM to produce uniform, non-aggregated Aβ and stored at −30˚C until use. On the day of use, the HFIP-dissolved amyloid was blown with N2 gas at ice cold temperature and redissolved in the assembly buffer [phosphate buffered saline (pH 7.4) containing 0.05% Tween 20].
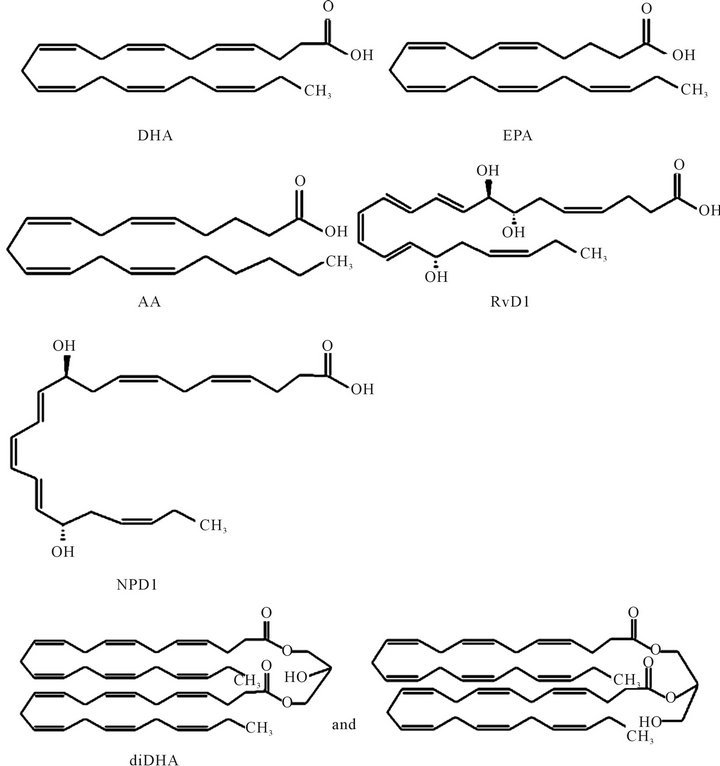
Figure 1. The chemical structures of the compounds used in the experiments.
2.3. Preparation of DHA, EPA, AA, NPD1, RvD1 and diDHA
DHA, EPA, AA, NPD1, and RvD1 dissolved in ethanol were stored at −80˚C, and diDHA dissolved in chloroform was stored at −30˚C until use. On the day of use, DHA, EPA, AA, and diDHA were mixed with assembly buffer at a final concentration of 20 μM, and NPD1 and RvD1 were mixed at a final concentration of 50 nM. Only freshly prepared DHA, EPA, AA, NPD1, RvD1, and diDHA were used.
2.4. FCS Measurement
In the present experiment, the FCS measurements were performed on a Fluoro Point Light (Olympus, Tokyo, Japan) at room temperature using the on-board 543- nm helium/neon laser at a laser power of 100 μW for excitation. TAMRA-Aβ1-42 dissolved in 1% NH4OH was stored at −30˚C. On the day of use, it was re-dissolved in assembly buffer at 1 nM, with or without DHA, EPA, AA, NPD1, RvD1, and diDHA, and quickly mixed with non-labeled Aβ1-42. Free rhodamine was used as a reference dye. The measurements were performed in a sample volume of 50 μL in a 384-well glass-bottomed microplate. The samples were sequentially and automatically loaded into the device, the optical system of which was also automatically adjusted for each measurement. Initially, the samples were subjected to FCS measurement at zero time. Afterward, the samples were incubated at 37˚C for 1 h, followed by a second reading using the Fluoro Point Light device. All experiments were performed under identical conditions, with a data acquisition time of 10 s per measurement, and measurements were repeated five times per sample. Only freshly prepared TAMRA-Aβ1-42 was used.
2.5. Statistical Analysis
Results are expressed as means ± S.E. The data were analyzed by unpaired Student’s t-test and one-way ANOVA. ANOVA followed by Dunnett’s test was used for post hoc comparisons. The statistical program used was PASW Statistics 18.0 (IBM-SPSS, Inc., USA). Statistical significance was set at P < 0.05.
3. RESULTS
3.1. Effect of Various DHA Concentrations on Diffusion Time of Aβ1-42
Figure 2 shows the results of FCS studies of the dosedependent effect of DHA on Aβ polymerization using rhodamine-labeled Aβ1-42 (TAMRA-Aβ1-42). One-way analysis of diffusion time of Aβ1-42 showed that DHA inhibited Aβ1-42 polymerization in a concentration-dependent manner. DHA (10 and 20 μM) significantly inhibited Aβ1-42 polymerization (Figure 2), as indicated by the decreased diffusion times, compared with the control (Aβ1-42 alone) without DHA.
3.2. Effect of DHA, AA, and EPA on Diffusion Time of Aβ1-42
After observing an inhibitory effect with 20 μM DHA, which decreased the diffusion time of Aβ1-42 by 28%, we decided to use 20 μM concentrations of AA and EPA for our next experiments. Addition of 20 μM AA decreased the diffusion time of Aβ1-42 by 31%, but the addition of 20 μM EPA did not significantly affect the diffusion time, demonstrating that AA inhibited Aβ1-42 polymerization but EPA did not (Figure 3).
3.3. Effect of DHA Metabolites NPD1 and RvD1 on Diffusion Time of Aβ1-42
In our experiment, 50 - 500 nM NPD1 and 50 - 500
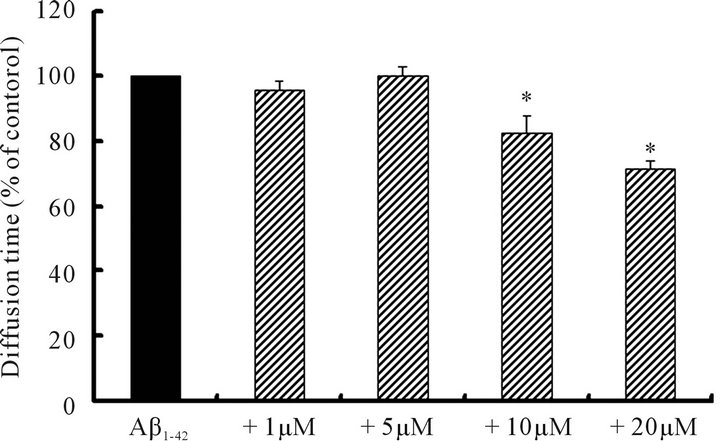
Figure 2. The effect of DHA concentrations on Aβ1-42 polymerization examined by FCS. TAMRA-Aβ1-42 was dissolved in assembly buffer at 1 nM (final concentration) with (1 - 20 μM) (n = 5) or without (control) (n = 5) DHA and quickly mixed with 10 μM non-labeled Aβ1-42. Data were analyzed by ANOVA followed by Dunnett’s test. Asterisks * indicate a significant difference (P < 0.05).
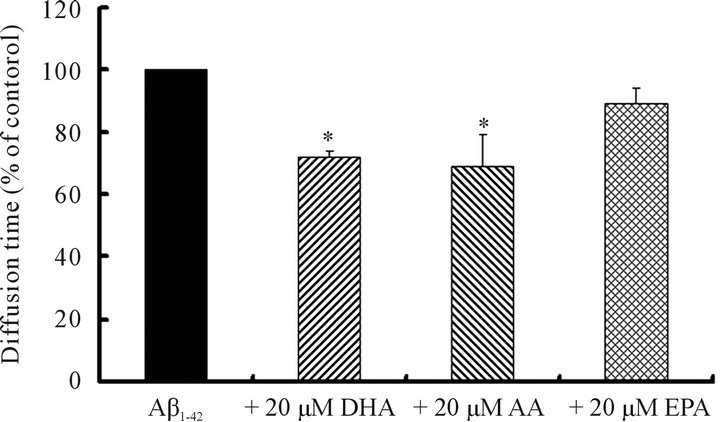
Figure 3. The effect of polyunsaturated fatty acids DHA, EPA, and AA on Aβ1-42 polymerization examined by FCS. The diffusion time of 10 μM Aβ1-42 alone (n = 6 - 7) and with 20 μM DHA (n = 7), AA (n = 6), or EPA (n = 7). Data were analyzed by ANOVA followed by Dunnett’s test. Asterisks * indicate a significant difference (P < 0.05).
nM RvD1 did not affect the diffusion time of Aβ1-42 (Figure 4), indicating that DHA metabolites did not have any effect on Aβ1-42 polymerization.
3.4. Effect of diDHA on Diffusion Time of Aβ1-42
The diffusion time of Aβ1-42 was not increased by the addition of 20 μM diDHA (Figure 5), indicating that the bulky DHA did not affect Aβ1-42 polymerization within 1 h.
4. DISCUSSION
In these experiments, using FCS, we examined the effect of PUFAs, such as DHA, AA, and EPA, diDHA, and the DHA metabolites NPD1 and RvD1, on Aβ1-42 polymerization within 1 h of the start of fibrillation. In the present experimental paradigm, we mixed TAMRA-labeled Aβ1-42 with unlabeled Aβ1-42 such that the fluorescent label would be co-assembled into the fibers. The process enabled TAMRA-Aβ1-42 to continue to fluoresce and provide information on its molecular interactions in terms of the diffusion time in the confocal volume, and therefore
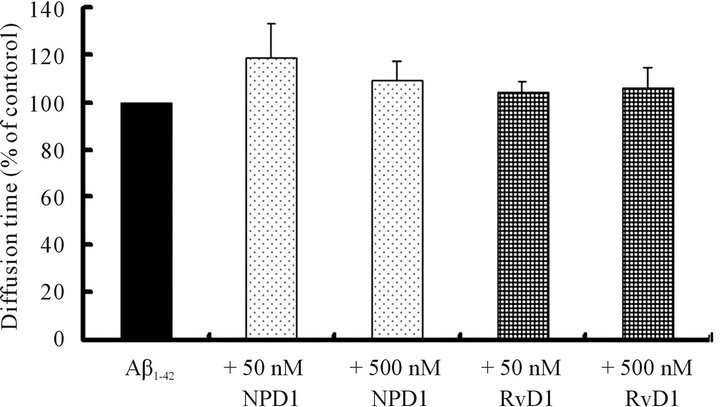
Figure 4. The effect of the DHA metabolites NPD1 and RvD1 on Aβ1-42 polymerization examined by FCS. The diffusion time of 10 μM Aβ1-42 alone (n = 6 - 7) and with 50 and 500 nM NPD1 (n = 6) or RvD1 (n = 7). Data were analyzed by ANOVA followed by Dunnett’s test.
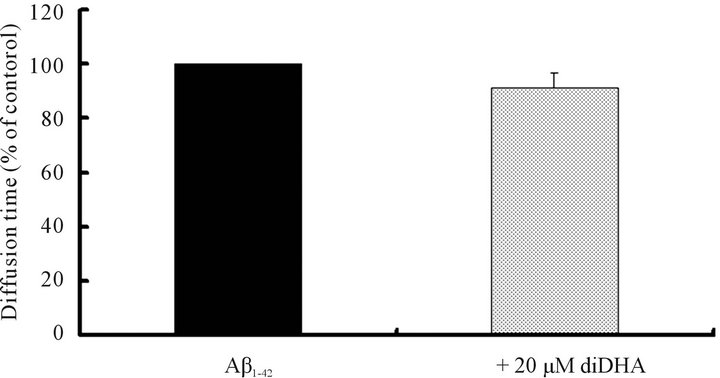
Figure 5. The effect of diDHA on Aβ1-42 polymerization examined by FCS. The diffusion time of 10 μM Aβ1-42 alone (n = 7) and with 20 μM diDHA (n = 7). Data were analyzed by unpaired Student’s t-test.
on the effect(s) of DHA on their dynamic interactions. This means that Aβ1-42 molecules, before being intercalated into the amyloid fibers, existed in a different conformation, and this conformation is most probably a monomer with a smaller diffusion time. However, after 1 h of incubation of the amyloid samples, the monomeric conformation of Aβ1-42 was changed to a higher molecular species such as Aβ1-42 fibers. The speculation is based on the fact that after 1 h of incubation, the samples showed a longer diffusion time, thus again indicating that amyloid peptides were transformed into fibers and/or other molecular species such as dimers/trimmers etc, which exhibit a different fluorescence fluctuation on FCS. However, transformation of the monomers was disturbed by DHA, implying that the monomer to fibril transformation was inhibited. DHA inhibited Aβ1-42 polymerization by 28% within 1 h, suggesting that the inhibitory effect of DHA on Aβ1-42 polymerization occurred at the initial stage, earlier than the trimer/tetramer level that was suggested by the thioflavin T fluorescence spectroscopy experiment [21]. In AD model rats, DHA prevented learning deficits and the impairment of spatial cognition ability. DHA also inhibited Aβ1-42 polymerization in vitro [21,30,40]. These results further support the inhibitory effect of DHA on Aβ1-42 polymerization and indicate the possibility that the DHA might exert the inhibitory effect at the primary steps of fibrillation during the initial onset of AD.
AA also reduced the Aβ1-42 diffusion time by 31% within 1 hour of fibrillation, suggesting that AA also had an inhibitory effect on Aβ1-42 polymerization. Kotani et al. (2006) suggested that DHA and AA supplementation improves cognitive dysfunction by improving membrane fluidity that affects the neurogenesis and/or synaptogenesis [41]. Hashimoto et al. (2002) showed that the pre-administration of DHA to AD model rats suppressed the increase in lipid peroxide and reactive oxygen species levels that accompanied a decrease in the amounts of AA in the cerebral cortex and hippocampus [30]. We showed the inhibitory effect of AA on Aβ1-42 polymerization for the first time. Further investigations will be needed to explore the role of AA in AD. On the other hand, the EPA did not affect the Aβ1-42 polymerization, as indicated by the lack of effect on the diffusion time of amyloid peptides in the FCS. As reported previously, EPA attenuated the IL-1β induced-impairment of spatial memory by the modulation of noradrenergic and serotonergic systems, and EPA protected against Aβ-induced memory deficit in AD model rat by decreasing oxidative stress and increasing the expression of synaptic plasticity-related proteins after being converted into DHA [42,43]. When ethyl-EPA was administered to rats and dogs, DHA concentrations in the brain increased in a time-dependent manner, with a concomitant decrease in EPA concentrations [44]. These results suggested that the beneficial effects of EPA on AD occurred after EPA was transformed into DHA.
Being the member of the same n-3 PUFA family, EPA and DHA have recently been claimed to influence various biological functions, including depression [45], which are initiated ultimately by the brain. The differenttial effect of DHA versus EPA on Aβ1-42 polymerization also remains to be clarified. According to molecular dynamics simulations, DHA exhibits high conformational flexibility and undergoes rapid interconversion between torsional states, including extended and bent conformation [46]. According to computational analyses of fatty acids containing 20 and 22 carbons, DHA, AA, and EPA form ball-shaped curves [47,48]. DHA and AA have six and four double bonds, respectively, in their carbon skeleton, and these double bonds may face each other. EPA, which has five (an odd number) double bonds, may have double bonds that do not face each other while it interacts with amyloid peptides. These results and possibilities might relate to the differences between the inhibitory effects of DHA, AA, and EPA on Aβ1-42 polymerization. Previous studies have shown that NPD1, a DHA metabolite that is generated during the resolution phase of acute inflammatory actions, has anti-apoptotic and anti-inflammatory effects and represses Aβ1-42-triggered activation of proinflammatory genes [49-51]. RvD1, another DHA metabolite, is also produced during inflammation [52]. Inflammation also contributes to AD pathogenesis [34]. We examined whether DHA metabolites had an inhibitory effect on Aβ1-42 polymerization. NPD1 and RvD1 did not affect Aβ1-42 polymerization. These results suggest that DHA itself inhibited Aβ1-42 polymerization before being converted to its metabolites, including NPD1 and RvD1. DHA has been shown to be rapidly incorporated into various cells, primarily into phospholipids [46]. High concentration of DHA is found in retinal rod outer segment membranes [53], sperm [54], and synaptosomes [55], and didocosahexaenoyl phospholipid species have been isolated from several tissues [35-37]. In this experiment, diDHA did not affect Aβ1-42 polymerization, suggesting that free DHA, but not didocosahexaenoyl glycerol, exerted the inhibitory effect on Aβ1-42 polymerization.
5. CONCLUSION
Using FCS, we showed that DHA and AA inhibit Aβ1-42 polymerization within short period of time. This provides new insight into the function of DHA and AA at the initial stage of Aβ1-42 polymerization. Finally, the results of the present study demonstrate that continuous intake of DHA might delay the onset and progression of AD initiated by the earlier deposition of Aβ1-42 fibers.
6. ACKNOWLEDGEMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research (C) from the Ministry of Education Culture, Sports, Science and Technology, Japan (23500955, M.H.).
REFERENCES
- Selkoe, D.J. (1991) The molecular pathology of Alzheimer’s disease. Neuron, 6, 487-498. doi:10.1016/0896-6273(91)90052-2
- Selkoe, D.J. (1993) Physiological production of the β amyloid protein and the mechanism of Alzheimer’s disease. Trends in Neurosciences, 16, 403-409. doi:10.1016/0166-2236(93)90008-A
- Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N. and Ihara, Y. (1994) Visualization of A β 42(43) and A β 40 in senile plaques with end-specific A β monoclonals: Evidence that an initially deposited species is A β 42(43). Neuron, 13, 45-53. doi:10.1016/0896-6273(94)90458-8
- Berman, D.E., Dall’Armi, C., Voronov, S.V., McIntire, L.B., Zhang, H., Moore, A.Z., Staniszewski, A., Arancio, O., Kim, T.W. and Di Paolo, G. (2008) Oligomeric amyloid-β peptide disrupts phosphatidyl-inositol-4, 5-bisphophate metabolism. Nature Neuroscience, 11, 547-554. doi:10.1038/nn.2100
- Naiki, H. and Nakakuki, K. (1996) First-order kinetic model of Alzheimer’s β-amyloid fibril extension in vitro. Laboratory Investigation, 74, 374-83.
- Stine, W.B. Jr., Snyder, S.W., Ladror, U.S., Wade, W.S., Miller. M.F., Perun, T.J., Holzman, T.F. and Krafft, G.A. (1996) The nanometer-scale structure of amyloid-β visualized by atomic force microscopy. Journal of Protein Chemistry, 15, 193-203. doi:10.1007/BF01887400
- Harper, J.D., Wong, S.S., Lieber, C.M. and Lansbury, P.T. (1997) Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chemistry & Biology, 4, 119-125. doi:10.1016/S1074-5521(97)90255-6
- Harper, J.D., Lieber, C.M. and Lansbury Jr., P.T. (1997) Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chemistry & Biology, 4, 951-959. doi:10.1016/S1074-5521(97)90303-3
- Terzi, E., Hölzemann, G. and Seelig, J. (1995) Self-association of β-amyloid peptide (1-40) in solution and binding to lipid membranes. Journal of Molecular Biology, 252, 633-642. doi:10.1006/jmbi.1995.0525
- Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C.L., and Beyreuther, K. (1991) Aggregation and secondary structure of synthetic amyloid β A4 peptides of Alzheimer’s disease. Journal of Molecular Biology, 218, 149-163. doi:10.1016/0022-2836(91)90881-6
- Burdick, D., Soreghan, B., Kwon, M., Kosmoski, J., Knauer, M., Henschen, A., Yates, J., Cotman, C. and Glabe, C. (1992) Assembly and aggregation properties of synthetic Alzheimer’s A4/β amyloid peptide analogs. Journal of Biological Chemistry, 267, 546-554.
- Sweeney, P.J., Darker, J.G., Neville, W.A., Humphries, J. and Camilleri, P. (1993) Electrophoretic techniques for the analysis of synthetic amyloid β-A4-related peptides. Analytical Biochemistry, 212, 179-184. doi:10.1006/abio.1993.1310
- Garzon-Rodriguez, W., Sepulveda-Becerra, M., Milton, S. and Glabe, C.G. (1997) Soluble amyloid Aβ-(1-40) exists as a stable dimer at low concentrations. Journal of Biological Chemistry, 272, 21037-21044. doi:10.1074/jbc.272.34.21037
- Shen, C.L., Fitzgerald, M.C. and Murphy, R.M. (1994) Effect of acid predissolution on fibril size and fibril flexibility of synthetic β-amyloid peptide. Biophysical Journal, 67, 1238-1246. doi:10.1016/S0006-3495(94)80593-4
- LeVine, H. III. (1993) Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Science, 2, 404-410. doi:10.1002/pro.5560020312
- Serpell, L.C. (2000) Alzheimer’s amyloid fibrils: Structure and assembly. Biochimica et Biophysica Acta, 1502, 16-30. doi:10.1016/S0925-4439(00)00029-6
- Barrow, C.J. and Zagorski, M.G. (1991) Solution structures of β peptide and its constituent fragments: Relation to amyloid deposition. Science, 253, 179-182. doi:10.1126/science.1853202
- Shao, H., Jao, S., Ma, K. and Zagorski, M.G. (1999) Solution structures of micelle-bound amyloid β-(1-40) and β-(1-42) peptides of Alzheimer’s disease. Journal of Molecular Biology, 285, 755-773. doi:10.1006/jmbi.1998.2348
- Kirkitadze, M.D., Condron, M.M. and Teplow, D.B. (2001) Identification and characterization of key kinetics intermediates in amyloid β-protein fibrillogenesis. Journal of Molecular Biology, 312, 1103-1119. doi:10.1006/jmbi.2001.4970
- Hashimoto, M., Shahdat, H.M., Yamashita, S., Katakura, M., Tanabe, Y., Fujiwara, H., Gamoh, S., Miyazawa, T., Arai, H., Shimada, T. and Shido, O. (2008) Docosahexaenoic acid disrupts in vitro amyloid β1-40 fibrillation and concomitantly inhibits amyloid levels in cerebral cortex of Alzheimer’s disease model rats. Journal of Neurochemistry, 107, 1634-1646. doi:10.1111/j.1471-4159.2008.05731.x
- Hossain, S., Hashimoto, M., Katakura, M., Miwa, K., Shimada, T. and Shido, O. (2009) Mechanism of docosahexaenoic acid-induced inhibition of in vitro Aβ1-42 fibrillation and Aβ1-42 induced toxicity in SH-SY5Y cells. Journal of Neurochemistry, 111, 568-579. doi:10.1111/j.1471-4159.2009.06336.x
- Hashimoto, M., Shahdat, H.M., Katakura, M., Tanabe, Y., Gamoh, S., Miwa, K., Shimada, T. and Shido, O. (2009) Effects of docosahexaenoic acid on in vitro amyloid beta peptide 25-35 fibrillation. Biochimica et Biophysica Acta, 1791, 289-296. doi:10.1016/j.bbalip.2009.01.012
- Hashimoto, M., Katakura, M., Hossain, S., Rahman, A., Shimada, T. and Shido, O. (2011) Docosahexaenoic acid withstands the Aβ25-35 induced neurotoxicity in SH-SY5Y cells. Journal of Nutritional Biochemistry, 22, 22-29. doi:10.1016/j.jnutbio.2009.11.005
- Ono, K., Hasegawa, K., Naiki, H. and Yamada, M. (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Journal of Neuroscience Research, 75, 742-750. doi:10.1002/jnr.20025
- Rezai-Zadeh, K., Arendash, G.W., Hou, H., Fernandez, F., Jansen, M., Runfeldt, M., Shytle, R.D. and Tan, J. (2008) Green tea epigallocatechin-3-gallate (EGCG) reduces β- amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer’s transgenic mice. Brain Research, 1214, 177-187. doi:10.1016/j.brainres.2008.02.107
- Luo, Y., Smith, J.V., Paramasivam, V., Burdick, A., Curry, K.J., Buford, J.P., Khan, I., Netzer, W.J., Xu, H. and Butko, P. (2002) Inhibition of amyloid-β aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proceedings of the National Academy of Sciences of the United States of America, 99, 12197-12202. doi:10.1073/pnas.182425199
- Lauritzen, L., Hansen, H.S., Jørgensen, M.H. and Michaelsen, K.F. (2001) The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Progress in Lipid Research, 40, 1-94. doi:10.1016/S0163-7827(00)00017-5
- Innis, S.M. (2007) Dietary (n-3) fatty acids and brain development. Journal of Nutrition, 137, 855-859.
- Tozuka, Y., Wada, E. and Wada, K. (2009) Bio-communication between mother and offspring: Lessons from animals and new perspectives for brain science. Journal of Pharmacological Sciences, 110, 127-132. doi:10.1254/jphs.09R01CP
- Hashimoto, M., Hossain, S., Shimada, T., Sugioka, K., Yamasaki, H., Fujii, Y., Ishibashi, Y., Oka, J. and Shido, O. (2002) Docosahexaenoic acid provides protection from impairment of learning ability in Alzheimer’s disease model rats. Journal of Neurochemistry, 81, 1084- 1091. doi:10.1046/j.1471-4159.2002.00905.x
- Soderberg, M., Edlund, C., Kristensson, K. and Dallner, G. (1991) Fatty acid composition of brain phospholipids in aging and in Alzheimer’s Disease. Lipids, 26, 421-425. doi:10.1007/BF02536067
- Grimm, M.O., Kuchenbecker, J., Grösgen, S., Burg, V.K., Hundsdörfer, B., Rothhaar, T.L., Friess, P., deWilde, M.C., Broersen, L.M., Penke, B., Péter, M., Vígh, L., Grimm, H.S. and Hartmann, T. (2011) Docosahexaenoic acid reduces amyloid β production via multiple pleiotropic mechanisms. Journal of Biological Chemistry, 286, 14028- 14039. doi:10.1074/jbc.M110.182329
- Gamoh, S., Hashimoto, M., Sugioka, K., Hossain, S., Hata N., Misawa, Y. and Masumura, S. (1999) Chronic administration of docosahexaenoic acid improves reference memory-related learning ability in young rats. Neuroscience, 93, 237-241. doi:10.1016/S0306-4522(99)00107-4
- Ariel, A. and Serhan, C.N. (2007) Resolvins and protectins in the terminatrion program of acute inflammation. Trends in Immunology, 28, 176-183. doi:10.1016/j.it.2007.02.007
- Miljanich, G.P., Sklar, L.A., White, D.L. and Dratz, E.A. (1979) Disaturated and dipolyunsaturated phospholipids in the bovine retinal rod outer segment disk membrane. Biochimica et Biophysica Acta, 552, 294-306. doi:10.1016/0005-2736(79)90284-0
- Bell, M.V., Dick, J.R. and Buda, C. (1997) Molecular speciation of fish sperm phospholipids: Large amounts of dipolyunsatusated phosphatidylserine. Lipids, 32, 1085- 1091. doi:10.1007/s11745-997-0140-y
- Bell, M.V. and Tocher, D.R. (1989) Molecular species composition of the major phospholipids in brain and retina from rainbow trout (Salmo gairdneri). Biochemical Journal, 264, 909-915.
- Tjernberg, L.O., Paramanik, A., Björling, S., Thyberg, P., Thyberg, J., Nordstedt, C., Berndt, K.D., Terenius, L. and Rigler, R. (1999) Amyloid β-peptide polymerization studied using fluorescence correlation spectroscopy. Chemistry & Biology, 6, 53-62. doi:10.1016/S1074-5521(99)80020-9
- Pitschke, M., Prior, R., Haupt, M. and Riesner, D. (1998) Detection of single amyloid β-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nature Medicine, 4, 832- 834. doi:10.1038/nm0798-832
- Hashimoto, M., Tanabe, Y., Fujii, Y., Kikuta, T., Shibata, H. and Shido, O. (2005) Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid β-infused rats. Journal of Nutrition, 135, 549-555.
- Kotani, S., Sakaguchi, E., Warashina, S., Matsukawa, N., Ishikura, Y., Kiso, Y., Sakakibara, M., Yoshimoto, T., Guo, J. and Yamashima, T. (2006) Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neuroscience Research, 56, 159-164. doi:10.1016/j.neures.2006.06.010
- Song, C. and Horrobin, D. (2004) Omega-3 fatty acid ethyl-eicosapentaenoate, but not soybean oil, attenuates memory impairment induced by central IL-1β administration. Journal of Lipid Research, 45, 1112-1121. doi:10.1194/jlr.M300526-JLR200
- Hashimoto, M., Hossain, S., Tanabe, Y., Kawashima, A., Harada, T., Yano, T., Mizuguchi, K. and Shido, O. (2009) The protective effect of dietary eicosapentaenoic acid against impairment of spatial cognition learning ability in rats infused with amyloid β1-40. Journal of Nutritional Biochemistry, 20, 965-973. doi:10.1016/j.jnutbio.2008.08.009
- Ishiguro, J., Tada, T., Ogihara, T., Ohzawa, N., Murakami, K. and Kosuzume, H. (1988) Metabolic disposition of ethyl eicosapentaenoate and its metabolites in rats and dogs. Journal of Pharmacobio-Dynamics, 11, 251-261. doi:10.1248/bpb1978.11.251
- Martins, J.G., Bentsen, H. and Puri, B.K. (2012) EPA in major depressive disorder: Eicosapentaenoic acid appears to be the key omega 3 fatty acid component associated with efficacy in major depressive disorder: A critique of Bloch and Hannestad and updated meta-analysis. Molecular Psychiatry, 17, 1-6.
- Stillwell, W. and Wassall, S.R. (2003) Docosahexaenoic acid: Membrane properties of a unique fatty acid. Chemistry and Physics of Lipids, 126, 1-27. doi:10.1016/S0009-3084(03)00101-4
- Yonezawa, Y., Hada, T., Uryu, K., Iijima, H., Yoshida, H. and Mizushina, Y. (2006) Inhibitory action of C22-fatty acids on DNA polymerases and DNA topoisomerases. International Journal of molecular Medicine, 18, 583-588.
- Mizushina, Y., Dairaku, I., Yanaka, N., Takeuchi, T., Ishimaru, C., Sugawara, F., Yoshida, H. and Kato, N. (2007) Inhibitory action of polyunasaturated fatty acids on IMP dehydrogenase. Biochimie, 89, 581-590. doi:10.1016/j.biochi.2007.01.009
- Lukiw, W.J. and Bazan, N.G. (2008) Docosahexaenoic acid and the aging brain. Journal of Nutritioin, 138, 2510-2514. doi:10.3945/jn.108.096016
- Serhan, C.N., Gotlinger, K., Hong, S., Lu, Y., Siegelman, J., Baer, T., Yang, R., Colgan, S.P. and Petasis, N.A. (2006) Anti-inflammatory actions of neuroprotectin D1/ Protectin D1 and its natural stereoisomers: Assignment of dihydroxy-containing docosatrienes. Journal of Immunology, 176, 1848-1859.
- Lukiw, W.J., Cui, J.G., Marcheselli, V.L., Bodker, M., Botkjaer, A., Gotlinger, K., Serhan, C.N. and Bazan, N.G. (2005) A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzhemier disease. Journal of Clinical Investigation, 115, 2774-2783. doi:10.1172/JCI25420
- Serhan, C.N. and Chiang, N. (2008) Endgenous pro-resolving and anti-inflammatory lipid mediators: A new pharmacologic genus. British Journal of Pharmacology, 153, S200-S215. doi:10.1038/sj.bjp.0707489
- Wiegand, R.D. and Anderson, R.E. (1983) Phospholipid molecular species of frog outer segment membranes. Experimental Eye Research, 37, 159-173. doi:10.1016/0014-4835(83)90075-1
- Neil, A.R. and Masters, C.J. (1973) Metabolism of fatty acids by ovine spermatozoa. Journal of the Society for Reproduction and Fertility, 34, 279-287. doi:10.1530/jrf.0.0340279
- Breckernridge, W.C., Gombos, G. and Morgan, I.G. (1972) The lipid composition of adult rat brain synaptosomal plasma membranes. Biochimica et Biophysica Acta, 266, 695-707. doi:10.1016/0005-2736(72)90365-3