Advances in Alzheimer's Disease
Vol.2 No.1(2013), Article ID:28634,9 pages DOI:10.4236/aad.2013.21004
Leptin rescues neurons from alzheimer’s disease-related pathways triggered by lipid burden
![]()
1Neurotez, Inc., Bridgewater, USA; *Corresponding Author: ntezapsidis@neurotez.com
2University of Texas, San Antonio, USA
3Stanford/VA Medical Center, Palo Alto, USA
Received 15 November 2012; revised 22 December 2012; accepted 2 January 2013
Keywords: Leptin; Cholesterol; Ceramide; AMPK; SIRT; Alzheimer’s; Tau; Amyloid-β
ABSTRACT
Adipocyte-derived leptin is a pleiotropic hormone implicated in control of lipid storage and mobilization, bone homeostasis, immune function and neuronal plasticity. Leptin has been shown to prevent accumulation of extracellular Aβ and hyperphosphorylation of tau in both cell culture and animal models. Herein an investigation was undertaken to test leptin’s ability to prevent the exacerbation/activation of AD-related pathways in neurons following their exposure to a high concentration of a variety of lipids. Specifically, cholesterol, oleic acid and/or ceramide were added to the media of cells resulting in decreased cellular viability and energy metabolism, and in increased tau phosphorylation and extracellular Aβ. Leptin increased viability, boosted cellular metabolism by activating AMPactivated protein kinase (AMPK) and the sirtuins (SIRT) and reduced tau phosphorylation and Aβ accumulation in a dose-dependent manner in response to select challenges. These findings demonstrate that leptin can attenuate the harmful effect of certain lipids that lead to exacerbation or activation of AD pathways. The study herein also provides the basis for a novel screening platform to define and identify a novel class of “metabolic” compounds addressing Alzheimer’s disease, based on a biological profile similar to leptin.
1. INTRODUCTION
A number of clinical and epidemiological studies have suggested that lifestyle factors, particularly nutritionmental and physical activities are intricately linked to the etiology of Alzheimer’s disease (AD) [1]. These studies are supported by the observation that a large percentage of AD patients present with some form of insulin resistance, impaired glucose tolerance or hyperinsulinemia, or are type II diabetic [2]. Additionally, a number of traits characteristic of the metabolic syndrome, particularly obesity, dyslipidemia, hypertension, reduced HDL cholesterol and metabolic inflammation, are also AD risk factors [1]. Indeed, ‘westernized’ high-caloric diets laden with trans and saturated fatty acids, carbohydrates and cholesterol, along with a sedentary lifestyle, promote brain dysfunction in transgenic animal models of AD [3]-illustrating the important connection between caloric regulation and mental health. This has led to the suggestion of AD as type 3 diabetes [4].
Obesity in middle age, particularly central adiposity, has been correlated with increased risk of dementia in later life independent of cardiovascular co morbidities [5,6]. Adipose tissue is the production site of leptin, a hormone that physiologically functions to regulate lipid storage and mobilization. High concentrations of leptin receptors have been found in the brain, including within the hippocampus, attesting to the hormone’s central as well as peripheral sites of action beyond regulating adiposity [7]. Direct injection of leptin into the hippocampus of rodents can improve memory processing and modulate long term potentiation and synaptic plasticity [8]. Circulating leptin is transported into the brain by binding to the lipoprotein receptor megalin at the choroid plexus or via a natural saturable leptin transporter [9]. Obesity in midlife leads to elevated circulating leptin levels, which can potentially saturate its endogenous transporter across the brain and produce a central leptin resistance-like state [10] analogous to the situation with insulin during metabolic syndrome. Furthermore, leptin resistance due to desensitization of signaling pathways or prevention of transport due to high triglycerides has been suggested.
It is speculated that dysregulation of leptin availability or sensitivity at the hippocampal region over a number of years may contribute to cognitive impairment. For individuals who are obese at midlife, studies have suggested that leptin’s transport efficiency across the blood brain barrier (BBB) is not completely restored even after weight loss by caloric restriction, despite a reduction in circulating leptin levels [10]. Weight loss is frequently observed in AD patients prior to the onset of dementia [11,12], thus obese individuals may be particularly vulnerable to cognitive dysfunction later in life considering the potential for hypoleptinemia due to adipocyte loss and inefficient transport of leptin across the BBB. To date a number of reports have addressed the correlation between reduced levels of circulating leptin and AD risk [13-16], severity of dementia [17] and cognitive decline [18,19]. In particular, a study of 785 dementia-free, older individuals followed for a median of 8.3 years identified those with plasma leptin levels in the lowest quartile of the study as being at four times greater risk for developing AD than those in the highest quartile [20].
Lipids have been reported to play an important role in activating AD-related pathways [21-23]. In cell culture models, excess levels of cholesterol [22,24], ceramide [25,26] and oleic acid [23,27] have been shown to stimulate both Aβ production and hyperphosphorylation of tau. Further, hypercholesterolemia has been reported as an AD risk factor [21,28] and genetic studies indicate that carriers of one copy of the APOE4 gene, involved in lipid metabolism, are threeto four-fold more likely to develop AD than those lacking APOE4 [29]. It is unknown whether increased AD susceptibility for ε4 carriers is due in part to an impaired ability of the lipoprotein to transport lipids and cholesterol across the BBB into the blood, or solely that its function as a transporter and scavenger of Aβ is compromised, as has been reported [30].
Our laboratory has previously demonstrated that leptin facilitates the uptake of Aβ by apoE [30]. Further, we have shown that leptin reduces the amount of Aβ secreted into the medium in a timeand dose-dependent fashion, which is co-incident with composition changes in lipid rafts and redistribution of β-secretase (BACE) and APP within membranes in neurons leading to suboptimum BACE activity [30]. We have also observed that leptin reduces both tau phosphorylation and Aβ production, endpoints that are mediated through pathways that are known to affect metabolism (AMPK, SIRT, GSK3β, PPRγ) [31-33] and improve cognitive performance in a transgenic model of AD [34]. To correlate these findings with the metabolic stresses associated with AD risk, we investigated the ability of leptin to suppress AD-related pathway activation following lipid challenges in neurons.
2. MATERIALS AND METHODS
2.1. Reagents
Minimum essential medium (MEM) was purchased from ATCC (Manassas, VA). Trypsin-EDTA and penicillin solution were purchased from MP Biomedicals (Solon, Ohio). Fetal bovine serum (FBS), all-trans retinoic acid (ATRA), G418, nicotinamide, methyl-β-cyclodextrin (MβCD), water-soluble cholesterol, water-soluble oleic acid and recombinant human leptin were purchased from Sigma-Aldrich (St. Louis, MO). The AMPK inhibitor, Compound C, and C2 ceramide were purchased from EMD Biosciences (San Diego, CA).
2.2. Culture and Stable Transfection of Cell Lines
The human neuroblastoma cell line, SH-SY5Y, was purchased from ATCC. Cell culture was performed according to manufacturer’s specific guidelines. Cells were propagated in MEM containing 10% FBS. Neuronal differentiation was performed as described previously [35].
To generate SY5Y stably over-expressing amyloid precursor protein (APP), cells were transfected with a mammalian expression vector encoding the 751 amino acid isoform of human APP (APP751-Accession # NM_201413) (Origene Technologies; Rockville, MD) using the FuGENE HD transfection reagent, according to manufacturer’s specific instructions (Promega; Madison, WI). Briefly, cells were transiently transfected with APP751 or vehicle for 48 h and then switched into selection medium containing a concentration range of the antibiotic G418 (100 - 600 µg/mL) to determine the optimal dose for stable selection. Selection media was changed every 3 days to remove non-viable cells. After 3 weeks, 200 µg/mL G418 yielded distinct colonies while all vehicle-transfected cells were non-viable. Cells were maintained in 10% FBS media containing 200 µg/mL G418 for expansion.
2.3. Lipid Insults
The human neuroblastoma cell line, SH-SY5Y, was purchased from ATCC. Cell culture was performed according to manufacturer’s specific guidelines. Cells were propagated in MEM containing 10% FBS. Neuronal differentiation was performed as described previously [35]. To generate SY5Y stably over-expressing amyloid precursor protein (APP), cells were transfected with a mammalian expression vector encoding the 751 amino acid isoform of human APP (APP751-Accession # NM_201413) (Origene Technologies; Rockville, MD) using the FuGENE HD transfection reagent, according to manufacturer’s specific instructions (Promega; Madison, WI). Briefly, cells were transiently transfected with APP751 or vehicle for 48 h and then switched into selection medium containing a concentration range of the antibiotic G418 (100 - 600 µg/mL) to determine the optimal dose for stable selection. Selection media was changed every 3 days to remove non-viable cells. After 3 weeks, 200 µg/mL G418 yielded distinct colonies while all vehicle-transfected cells were non-viable. Cells were maintained in 10% FBS media containing 200 µg/mL G418 for expansion.
2.4. Cell Viability Assay
RA-SY5Y, at 2 × 104 cells/well, were seeded in 96 - well microplates and treated for 18 h with a range of concentrations of MβCD, DMSO, ceramide, cholesterol or oleic acid to determine effective doses for 50% - 70% viability (data not shown), or in the presence of a range of concentrations of Leptin to determine effective doses to prevent cell death. Viability was assessed using the Cell-Titer Blue Viability Assay (Promega) by adding 20 μL of the reagent to each well for 4 h, and plates read by a microplate reader with fluorescence capabilities at Ex530 - 570 nm/Em580 - 620 nm. Viability was determined using a standard curve of known cell number and plotted as a percent of non-treated or vehicle control.
2.5. Preparation of Cell Lysates
RA-SY5Y were treated with leptin (100 ng/ml) in the presence of ceramide (25 µM), cholesterol (27.5 µg/mL), oleic acid (30 µg/mL) or vehicle (MβCD-675 µg/mL; or DMSO–0.125%) for 6 h, and then harvested by scraping. Preparation of whole cell lysates from cell pellets was performed as described previously [35].
2.6. AMPK Activity Assay
AMPK activity in RA-SY5Y cell lysates was determined using the CycLex AMPK Kinase Assay Kit (MBL International; Woburn, MA), as previously described [36, 37]. Briefly, “relative AMPK activity”, hereafter referred to as “AMPK activity”, is defined as Compound C-sensitive protein kinase activity in cell lysates. Titration of various Compound C doses identified 10 µM as the dose in which there was no further reduction in kinase activity upon increasing concentration (data not shown). Lysates were incubated in the presence or absence of 10 µM Compound C, and protein kinase activity determined by measuring phosphorylation of the insulin receptor substrate-1 (IRS-1) through immunoassay and conversion of a chromogenic substrate at an absorbance of 450 nm (A450). Normalized AMPK activity was defined as:

2.7. SIRT Activity Assay
“Total sirtuin”, hereafter termed “SIRT”, activity in cell lysates was determined using the HDAC Fluorimetric Cellular Activity Assay Kit (Enzo Life Sciences; Plymouth Meeting, PA), according to manufacturer’s specified guidelines. Briefly, SIRT activity is defined as nicotinamide-sensitive deacetylase (class III HDAC) activity in cell lysates. 5 mM nicotinamide was identified by the manufacturer as a dose in which there was no further reduction in deacetylase activity upon increasing concentration. RA-SY5Y cell lysates were incubated in the presence or absence of 5 mM nicotinamide, and SIRT activity determined by adding the Flour de Lys Substrate for deacetylation followed by exposure to the Fluor de Lys Developer to generate a fluorescent signal for detection using a fluorimeter (Ex–350 - 380 nm/Em440 - 460 nm). Normalized SIRT activity was measured in units of fluorescence intensity (Fi) and defined as:
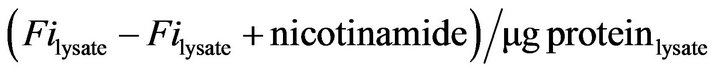
2.8. ELISAs
Aβ(1 - 40) levels in cell culture media from SY5YAPP751 treated for 18 h with the aforementioned metabolic insults in the presence or absence of leptin, were determined using the human amyloidβ 1 - 40 ELISA kit (Invitrogen; Carlsbad, CA), and phosphoand total tau levels in RA-SY5Y lysates were determined using the human Tau pSer396, pThr231 and total tau ELISA kits (Invitrogen) according to manufacturer’s specific instructions. Aβ(1 - 40), phoshoand total tau levels were calculated from a standard curve developed with OD at 450 nm using 8 serial dilutions of known concentration.
2.9. Statistical Analyses
Statistical data tests were performed by using analysis of variance and Tukey-Kramer multiple comparisons with p < 0.05 considered statistically significant.
3. RESULTS
3.1. Effect of Metabolic Challenges on Neuronal Cell Viability
Doses of the lipids which decreased neuronal viability by 25% - 50% (ceramide-25 µM; cholesterol-27.5 µg/mL and oleic Acid-30 µg/mL) after 18 h treatment (data not shown) were used to determine whether co-treatment with leptin, an important modulator of lipid homeostasis, could attenuate the toxic effects of the insults (Figure 1). RA-SY5Y were treated for 18 h with either a low (10 ng/mL; gray bars) or moderate (100 ng/mL; black bars) dose of leptin or control (white bars) in the presence or
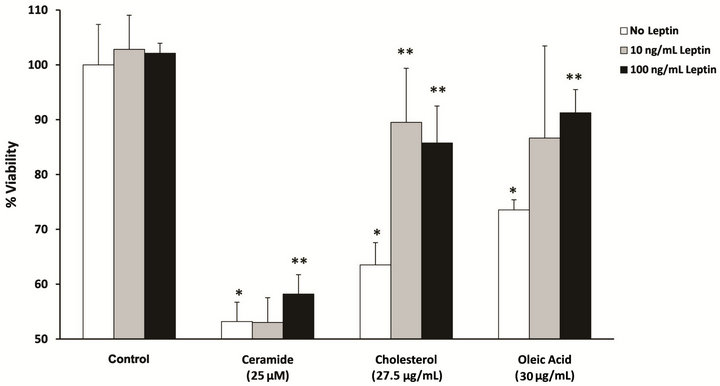
Figure 1. Leptin improves RA-SY5Y viability during metabolic insult. Cells were treated for 18 h with ceramide (25 µM), cholesterol (27.5 µg/mL), oleic acid (30 µg/mL) or control (0.125% DMSO or 675 µg/mL MβCD) in the presence or absence of leptin (10 or 100 ng/mL) and cell viability was measured. Viability was normalized to non-treated cells which were assigned a value of 100%, n=10. *p < 0.05 vs. control without insult. **p < 0.05 vs. insult without leptin.
absence of ceramide, cholesterol or oleic acid, and cell viability measured. All lipid insults induced a significant (p < 0.05) decrease in cell viability in the range of 35% ± 15% when treated with control alone. Low dose leptin was able to significantly (p < 0.05) improve viability only in response to the cholesterol insult, while a moderate dose significantly (p < 0.05) improved viability in response to all challenges. A third treatment group utilizing a high dose (1000 ng/mL) of leptin was not significantly (p > 0.05) different from the moderate group’s viability (data not shown). Of all the lipid insults, leptin most significantly attenuated the toxic effects of cholesterol (second group from right).
3.2. Leptin Increases Cellular Metabolism in Response to Metabolic Challenges
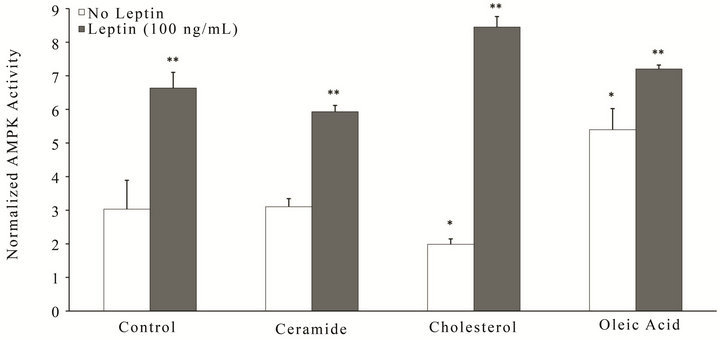
We have previously shown that leptin regulates AD pathways via activation of the cellular energy sensors, AMP-activated protein kinase (AMPK) and the sirtuins (SIRT) in neuronal cells [31,33]. To this end, we determined whether leptin has the ability to attenuate these effects on cellular energetics in an acute model of metabolic stress (Figure 2). Utilizing similar lipid doses as for the viability experiments, RA-SY5Y were treated for 6 h with leptin (100 ng/mL) or vehicle in the presence or absence of ceramide, cholesterol or oleic acid, and cellular energy status measured by AMPK (Figure 2(a)) or SIRT activity (Figure 2(b)). All insults were able to significantly (p < 0.05) suppress SIRT activity (Figure 2(b); white bars), while only cholesterol showed a similar effect on AMPK activity at the specified dose (Figure 2(a); second white bar from right). Treatment with leptin (Figures 2(a) and (b); gray bars) was able to signifycantly (p < 0.05) boost both AMPK and SIRT activity in response to all lipid insults, with the exception of ceramide for SIRT (Figure 2(b); second gray bar from left). Interestingly, as was observed for the viability experiments, leptin most significantly attenuated the suppressive effects of cholesterol (second group from right) on cellular energy metabolism.
3.3. Leptin Suppresses AD-Related Pathway Activation Following Exposure to Exogenous Lipids
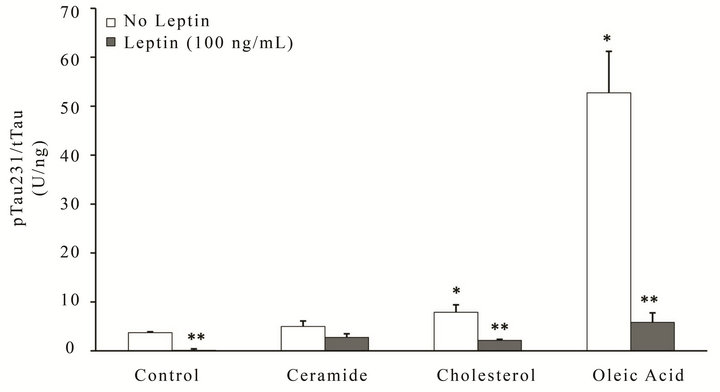
Metabolic stress has been linked to the contribution/activation of AD pathological pathways, with lipids known to play an important role [21-23]. We therefore investigated the extent to which extracellular lipids can increase the phosphorylation of tau (Figure 3) and Aβ production (Figure 4) in neuronal cells, and determined whether leptin could prevent these effects. Utilizing similar lipid doses as previous experiments, RA-SY5Y were treated for 6 h with leptin (100 ng/mL) or control in the presence or absence of ceramide, cholesterol or oleic acid, and phosphorylation of tau at two different epitopes, pTau231 (Figure 3(a)) or pTau396 (Figure 3(b)), as well as total tau measured by ELISA. Challenging cells with either cholesterol (second group from right) or oleic acid
 (a)
(a)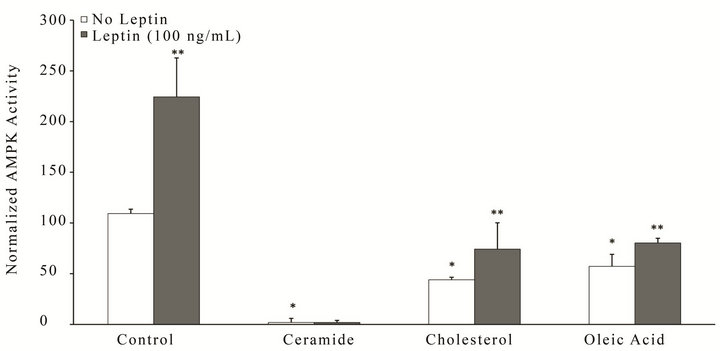 (b)
(b)
Figure 2. Leptin boosts AMPK and SIRT activity in RA-SY5Y during metabolic insult. Cells were treated for 6 h with ceramide, cholesterol, oleic acid or control in the presence or absence of leptin (100 ng/mL) and (a) AMPK or (b) SIRT activity measured. All activity values were normalized to total protein, n = 3. *p < 0.05 vs. control without insult. **p < 0.05 vs. insult without leptin.
(far right group) resulted in significant (p < 0.05) increases in tau phosphorylation at either epitope, with oleic acid incurring the greatest effect (–15 to 20-fold increase). Simultaneous treatment with leptin signifycantly (p < 0.05) prevented hyperphosphorylation (far right group, gray bars) more dramatically than the other insults.
In parallel to the tau studies, we investigated whether exposure of neuronal cells to high lipids could exacerbate the extracellular production of Aβ, and likewise determined whether leptin could prevent these effects (Figure 4). SY5Y stably over-expressing human APP751 were treated for 18 h with leptin (100 ng/mL) or control in the presence or absence of ceramide, cholesterol or oleic acid, and the amount of extracellular Aβ(1 − 40) measured by ELISA. All lipid insults induced a signifycant (p < 0.05) increase in the amount of Aβ(1 − 40) produced (white bars), with ceramide (second group from left, white bar) incurring the greatest effect (–3 - fold increase). Treatment with leptin (gray bars) signifycantly (p < 0.05) reduced Aβ(1 − 40) levels as similar to control (first group from right, white bar) for all insults. Leptin’s ability to reduce Aβ(1 − 40) production following ceramide challenge (second group from left, gray bar) was the most remarkable of all insults.
4. DISCUSSION
Excess buildup of lipids exerts toxic effects on the brain, which could exacerbate or even trigger Alzheimer’s disease pathological cascades [38]. Lipids are a broad group of molecules which encompass fatty acids, glycero-, phospho-, sphingoand sterol lipids, among others. Our studies began by investigating the relation-
 (a)
(a)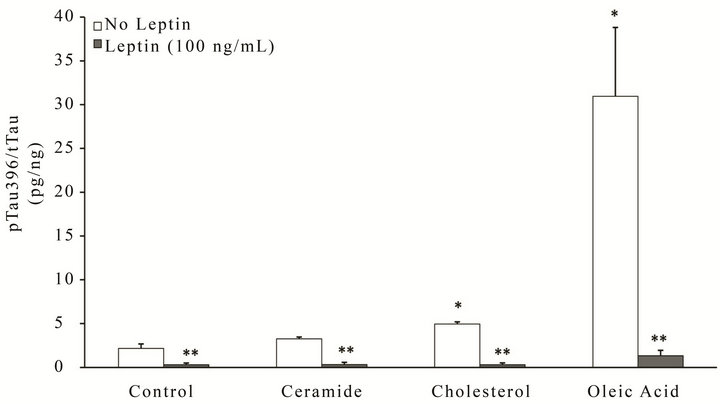 (b)
(b)
Figure 3. Leptin reverses metabolic insult-induced hyperphosphorylation of tau in RA-SY5Y. Cells were treated for 6h with ceramide, cholesterol, oleic acid or control in the presence or absence of Leptin (100 ng/mL) and (a) pTau231, (b) pTau396 or total tau measured by ELISA. All concentration values were normalized to total tau, n = 3. *p < 0.05 vs. control without insult
**p < 0.05 vs. insult without leptin.
ship between several types of lipids and viability of SY5Y neuroblastoma cells differentiated with retinoic acid (RA-SY5Y). We specifically utilized: a) ceramide, a lipid composed of sphingosine and a fatty acid, typically found in cell membraines, b) cholesterol, a waxy steroid of fat, also found in cell membranes; and c) oleic acid, a monounsaturated omega-9 fatty acid found in vegetable and animal fats. Prolonged states of positive energy balance due to high levels of fatty acids, triglycerides and/or cholesterol, as is often the case in obesity, can disrupt cellular energy metabolism and may have similar negative effects on neurons [39]. Metabolic diseases, particularly obesity and diabetes, have been associated with increased risk of cognitive impairment and AD [39]. In support, the contribution of diet and nutrition to AD incidence has been extensively documented [1,40]. Leptin, an adipocyte-derived hormone which physiologically functions in the control of lipid storage and mobilization, impedes production of Aβ and hyperphosphorylation of tau [30,34,35]. The epidemiological findings combined with leptin’s ability to regulate both lipid metabolism and AD pathobiology led us to investigate the relationship between lipids, leptin and activation of AD-related pathways.
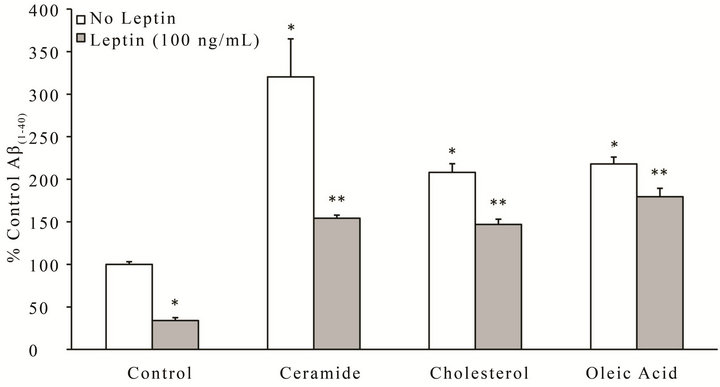
Figure 4. Leptin reverses metabolic insult-induced accumulation of extracellular Aβ(1 - 40) in SY5YAPP-751. Cells were treated for 18 h with ceramide, cholesterol, oleic acid or control in the presence or absence of leptin (100 ng/mL), and culture media collected for determination of Aβ(1 - 40) levels by ELISA. Results were normalized to total protein and presented as a percentage relative to control, which was assigned a value of 100%, n = 3. *p < 0.05 vs. control without insult. **p < 0.05 vs. insult without leptin.
We utilized three classes of lipids with known effects on AD pathways to serve as metabolic challenges in testing leptin’s protective functions. We first determined the optimal dose that would decrease neuronal viability by 25% - 50% for each insult. The insults significantly increased tau phosphorylation (Figure 3) and extracellular production of Aβ (Figure 4), and depressed cellular metabolism through deactivation of the energy sensors, AMPK and SIRT (Figure 2).
For most lipid insults, leptin co-administration negated the deleterious effects; however, each assay identified a unique insult that leptin was most able to abrogate. For viability and cellular metabolism studies leptin was most successful in preventing the effects of cholesterol, while for tau and Aβ, leptin was most successful in preventing the effects of oleic acid and ceramide, respectively. This indicates that heterologous deleterious metabolic challenges on neurons may be responsible in preferentially triggering either the amyloidogenic or tau hyperphosphorylation pathways. However, most importantly, leptin can correct the metabolic imbalances that high concentration of extracellular lipids may induce to neurons preventing AD-like cascades.
These results have been the core of our novel cellbased screening platform that utilizes an array of assay-specific lipid challenges to identify both novel and existing compounds which behave similar to leptin in modulating viability, energy metabolism and AD pathways. The platform utilizes cell viability as the preliminary screen to demonstrate feasibility as well as determine the effective dose for the subsequent readouts of the assay. Discovery of lead compounds will initiate testing in animal models of AD, and further drug development efforts will determine therapeutic significance.
Some of the categories that we are exploring include: a) high-affinity activators of the leptin receptor, which could entail leptin-like peptides, such as muteins or fusion proteins, or small molecule leptin receptor agonists; b) modulators of components of the leptin signaling pathway; c) modulators of AMPK and SIRT [31, 33]; d) novel insulin-like or insulin-sensitizing compounds, such as the PPARγ agonist thioglitazones; e) inhibitors to factors involved in lipid biosynthesis, such as sterol regulatory-element binding proteins (SREBPs).
Attenuation of the detrimental effects that exposure of neurons to excess lipid levels imparts is clinically significant. The progressive deterioration of brain lipid homeostasis in AD patients can elicit locally increased cholesterol [41], ceramide [26] and oleic acid [42] levels, further suggesting that assessment of the molecular and cell biology of lipids in the context of Alzheimer’s disease and the impact of lipid-related changes on neurometabolism should be of interest to basic and applied scientists alike [43]. Our present report provides evidence that leptin can suppress activation of AD pathological pathways in response to high doses of these lipids. These findings further support leptin as a potential AD therapeutic and provide a framework to screen novel and existing compounds which act similar or complementary to leptin.
5. ACKNOWLEDGEMENTS
This work was supported by the National Institute on Aging (SBIR
-1R43AG029670).
REFERENCES
- Bhat, N.R. (2010) Linking cardiometabolic disorders to sporadic Alzheimer’s disease: A perspective on potential mechanisms and mediators. Journal of neurochemistry 115, 551-562. doi:10.1111/j.1471-4159.2010.06978.x
- Watson, G.S. and Craft, S, (2003) The role of insulin resistance in the pathogenesis of Alzheimer’s disease: Implications for treatment. CNS Drugs, 17, 27-45. doi:10.2165/00023210-200317010-00003
- Hooijmans, C.R., Van der Zee, C.E., Dederen, P.J., Brouwer, K.M., Reijmer, Y.D., Van Groen, T., Broersen, L.M., Lutjohann, D., Heerschap, A. and Kiliaan, A.J. (2009) DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiology of Disease, 33, 482-498. doi:10.1016/j.nbd.2008.12.002
- Moroz, N., Tong, M., Longato, L., Xu, H. and De la Monte, S.M. (2008) Limited Alzheimer-type neurodegeneration in experimental obesity and type 2 diabetes mellitus. Journal of Alzheimer’s disease: JAD, 15, 29-44.
- Whitmer, R.A., Gunderson, E.P,, Barrett-Connor, E., Quesenberry, C.P. Jr. and Yaffe, K. (2005) Obesity in middle age and future risk of dementia: A 27-year longitudinal population based study. BMJ, 330, 1360. doi:10.1136/bmj.38446.466238.E0
- Whitmer, R.A,, Gustafson, D.R., Barrett-Connor, E., Haan, M.N., Gunderson, E.P., Yaffe, K. (2008) Central obesity and increased risk of dementia more than three decades later. Neurology, 71, 1057-1064. doi:10.1212/01.wnl.0000306313.89165.ef
- Huang, X.F., Koutcherov, I., Lin, S., Wang, H.Q. and Storlien, L. (1996) Localization of leptin receptor mRNA expression in mouse brain. Neuroreport, 7, 2635-2638. doi:10.1097/00001756-199611040-00045
- Harvey, J., Shanley, L.J., O’Malley, D. and Irving, A.J. (2005) Leptin: A potential cognitive enhancer? Biochemical Society Transactions, 33, 1029-1032. doi:10.1042/BST20051029
- Dietrich, M.O., Spuch, C., Antequera, D., Rodal, I., De Yebenes, J.G., Molina, J.A., Bermejo, F. and Carro, E. (2008) Megalin mediates the transport of leptin across the blood-CSF barrier. Neurobiology of Aging, 29, 902-912. doi:10.1016/j.neurobiolaging.2007.01.008
- Adam, C.L. and Findlay, P.A. (2010) Decreased bloodbrain leptin transfer in an ovine model of obesity and weight loss: Resolving the cause of leptin resistance. International Journal of Obesity, 34, 980-988. doi:10.1038/ijo.2010.28
- Barrett-Connor, E., Edelstein, S.L., Corey-Bloom, J., Wiederholt, W.C. (1996) Weight loss precedes dementia in community-dwelling older adults. Journal of American Geriatrics Society, 44, 1147-1152.
- Mazzali, G., Bissoli, L., Gambina, S., Residori, L., Pagliari, P., Guariento, S., Sun, M., Broggio, E., Bosello, O. and Zamboni, M. (2002) Energy balance in Alzheimer’s disease. The Journal of Nutrition Health and Aging, 6, 247- 253.
- Olsson, T., Nasman, B., Rasmuson, S. and Ahren, B. (1998) Dual relation between leptin and cortisol in humans is disturbed in Alzheimer’s disease. Biological Psychiatry, 44, 374-376.
- Power, D.A., Noel, J., Collins, R. and O’Neill, D. (2001) Circulating leptin levels and weight loss in Alzheimer's disease patients. Dement Geriatr Cogn Disord 12, 167-170. doi:10.1159/000051252
- Zhou, R., Deng, J., Zhang, M., Zhou, H.D. and Wang, Y.J. (2011) Association between bone mineral density and the risk of Alzheimer’s disease. Journal of Alzheimer’s Disease: JAD, 24, 101-108.
- Bigalke, B., Schreitmuller, B., Sopova, K., Paul, A., Stransky, E., Gawaz, M., Stellos, K. and Laske, C. (2011) Adipocytokines and CD34 progenitor cells in Alzheimer’s disease. PloS One, 6, e20286. doi:10.1371/journal.pone.0020286
- Ray, S. and Wyss-Coray, A. (2005) Methods for diagnosis, stratification, and monitoring of Alzheimer’s disease. World Intellectual Property Organization, WO 2005/ 052592 A2, Satoris, Inc., Menlo Park.
- Holden, K.F., Lindquist, K., Rosano, C., Tylavsky, F.A., Harris, T.B. and Yaffe, K. (2006) Low serum leptin is associated with poor cognitive performance in the elderly. 58th Annual Meeting of American Academy of Neurology, San Diego, 1-8 April 2006, p. S41.006.
- Holden, K.F., Lindquist, K., Tylavsky, F.A., Rosano, C., Harris, T.B. and Yaffe, K. (2008) Serum leptin level and cognition in the elderly: Findings from the health ABC study. Neurobiology of Aging, 30, 1483-1489.
- Lieb, W., Beiser, A.S., Vasan, R.S., Tan, Z.S., Au, R., Harris, T.B., Roubenoff, R., Auerbach, S., DeCarli, C., Wolf, P.A. and Seshadri, S. (2009) Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA: The Journal of the American Medical Association, 302, 2565-2572. doi:10.1001/jama.2009.1836
- Puglielli, L., Tanzi, R.E. and Kovacs, D.M. (2003) Alzheimer’s disease: the cholesterol connection. Nature Neuroscience, 6, 345-351. doi:10.1038/nn0403-345
- Glockner, F., Meske, V., Lutjohann, D. and Ohm, T.G. (2011) Dietary cholesterol and its effect on tau protein: a study in apolipoprotein E-deficient and P301L human tau mice. Journal of Neuropathology and Experimental Neurology, 70, 292-301. doi:10.1097/NEN.0b013e318212f185
- Wilson, D.M. and Binder, L.I. (1997) Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. The American Journal of Pathology, 150, 2181-2195.
- Hooff, G.P., Peters, I., Wood, W.G., Muller, W.E. and Eckert, G.P. (2010) Modulation of cholesterol, farnesylpyrophosphate, and geranylgeranylpyrophosphate in neuroblastoma SH-SY5Y-APP695 cells: Impact on amyloid beta-protein production. Molecular Neurobiology, 41, 341-350. doi:10.1007/s12035-010-8117-5
- Darios, F., Muriel, M.P., Khondiker, M.E., Brice, A. and Ruberg, M. (2005) Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of tau. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 25, 4159-4168.
- Haughey, N.J., Bandaru, V.V., Bae, M. and Mattson, M.P. (2010) Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochimica et biophysica acta, 1801, 878-886. doi:10.1016/j.bbalip.2010.05.003
- Liu, Y., Yang, L., Conde-Knape, K., Beher, D., Shearman, M.S. and Shachter, N.S. (2004) Fatty acids increase presenilin-1 levels and [gamma]-secretase activity in PSwt-1 cells. Journal of Lipid Research, 45, 2368-2376. doi:10.1194/jlr.M400317-JLR200
- Merched, A., Xia, Y., Visvikis, S., Serot, J.M. and Siest, G. (2000) Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiology of Aging, 21, 27-30. doi:10.1016/S0197-4580(99)00103-7
- Mahley, R.W. and Huang, Y. (2006) Apolipoprotein (apo) E4 and Alzheimer’s disease: Unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurologica Scandinavica. Supplementum, 185, 8-14. doi:10.1111/j.1600-0404.2006.00679.x
- Fewlass, D.C., Noboa, K., Pi-Sunyer, F.X., Johnston, J.M., Yan, S.D. and Tezapsidis, N. (2004) Obesity-related leptin regulates Alzheimer’s Aβ. FASEB Journal, 18, 1870-1878. doi:10.1096/fj.04-2572com
- Greco, S.J., Hamzelou, A., Johnston, J.M., Smith, M.A., Ashford, J.W. and Tezapsidis, N. (2011) Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochemical and Biophysical Research Communications, 414, 170-174. doi:10.1016/j.bbrc.2011.09.050
- Greco, S.J., Sarkar, S., Casadesus, G., Zhu, X., Smith, M.A., Ashford, J.W., Johnston, J.M. and Tezapsidis, N. (2009) Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neuroscience Letters, 455, 191-194. doi:10.1016/j.neulet.2009.03.066
- Greco, S.J., Sarkar, S., Johnston, J.M. and Tezapsidis, N. (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochemical and Biophysical Research Communications, 380, 98-104. doi:10.1016/j.bbrc.2009.01.041
- Greco, S.J., Bryan, K.J., Sarkar, S., Zhu, X., Smith, M.A., Ashford, J.W., Johnston, J.M., Tezapsidis, N. and Casadesus, G. (2010) Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. Journal of Alzheimer’s Disease: JAD, 19, 1155-1167.
- Greco, S.J., Sarkar, S., Johnston, J.M., Zhu, X., Su, B., Casadesus, G., Ashford, J.W., Smith, M.A. and Tezapsidis, N. (2008) Leptin reduces Alzheimer’s disease-related tau phosphorylation in neuronal cells. Biochemical and Biophysical Research Communications, 376, 536-541. doi:10.1016/j.bbrc.2008.09.026
- Wang, Y., Nishi, M., Doi, A., Shono, T., Furukawa, Y., Shimada, T., Furuta, H., Sasaki, H. and Nanjo, K. (2010) Ghrelin inhibits insulin secretion through the AMPKUCP2 pathway in beta cells. FEBS Letters, 584, 1503- 1508. doi:10.1016/j.febslet.2010.02.069
- Yeh, C.H., Chen, T.P., Wang, Y.C., Lin, Y.M. and Fang, S.W. (2010) AMP-activated protein kinase activation during cardioplegia-induced hypoxia/reoxygenation injury attenuates cardiomyocytic apoptosis via reduction of endoplasmic reticulum stress. Mediators of Inflammation, 2010, 130636. doi:10.1155/2010/130636
- Martins, I.C., Kuperstein, I., Wilkinson, H., Maes, E., Vanbrabant, M., Jonckheere, W., Van Gelder, P., Hartmann, D., D’Hooge, R., De Strooper, B., Schymkowitz, J. and Rousseau, F. (2008) Lipids revert inert Abeta amyloid fibrils to neurotoxic protofibrils that affect learning in mice. The EMBO Journal, 27, 224-233. doi:10.1038/sj.emboj.7601953
- Kapogiannis, D. and Mattson, M.P. (2011) Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurology, 10, 187-198. doi:10.1016/S1474-4422(10)70277-5
- Pasinetti, G.M. and Eberstein, J.A. (2008) Metabolic syndrome and the role of dietary lifestyles in Alzheimer’s disease. Journal of Neurochemistry, 106, 1503-1514. doi:10.1111/j.1471-4159.2008.05454.x
- Leduc, V., Jasmin-Belanger, S. and Poirier, J. (2010) APOE and cholesterol homeostasis in Alzheimer’s disease. Trends in Molecular Medicine, 16, 469-477. doi:10.1016/j.molmed.2010.07.008
- Fraser, T., Tayler, H. and Love, S. (2010) Fatty acid composition of frontal, temporal and parietal neocortex in the normal human brain and in Alzheimer's disease. Neurochemical Research, 35, 503-513. doi:10.1007/s11064-009-0087-5
- Foley, P. Lipids in Alzheimer’s disease: A century-old story. Biochimica et Biophysica Acta, 1801, 750-753.