Neuroscience & Medicine
Vol.3 No.2(2012), Article ID:19645,13 pages DOI:10.4236/nm.2012.32020
Pharmacological Assessment of γ-Secretase Activity from Rodent and Human Brain
![]()
1Neuroscience Research Unit, Pfizer Inc., Groton, Connecticut, USA; 2Primary Pharmacology Group, Pfizer Inc., Groton, Connecticut, USA; 3Neuroscience Medicinal Chemistry, Pfizer Inc., Groton, Connecticut, USA.
Email: *Kelly.Bales@Pfizer.com
Received February 11th, 2012; revised March 16th, 2012; accepted April 20th, 2012
Keywords: Alzheimer’s Disease; Amyloid Precursor Protein; Aβ; γ-Secretase; Pharmacology
ABSTRACT
γ-secretase is involved in the final processing of the amyloid precursor protein into a heterogeneous pool of β-amyloid (Aβ) peptides. Current Alzheimer’s disease drug discovery efforts include targeting γ-secretase activity in brain to attenuate production of the neurotoxic Aβ species. The resulting pharmacology may be affected by species-specific differences in the γ-secretase core complex or its associated proteins. Therefore, we utilized partially purified γ-secretase membranes derived from the brains of different species, including human cortex, to quantitatively assess the de novo production of both Aβ42 and Aβ40 following treatment with known γ-secretase inhibitors and modulators. We determined that the inhibitory activity of a Notch-1 sparing γ-secretase inhibitor and the modulatory activity of two classes of γ-secretase modulators were equipotent at affecting the production of Aβ across rodent and human brain membrane preparations. Additionally, the observed modulator-specific Aβ profile in isolated brain membranes across species was similar to that observed in HeLa cell membranes, and the brain and CSF of guinea pigs following oral administration. By utilizing rapidly purified γ-secretase, we were able to probe and compare the complex pharmacology of γ-secretase in the brain across common rodent species and human cortex.
1. Introduction
According to the amyloid cascade hypothesis of Alzheimer’s disease (AD) pathogenesis, deposition of Aβ peptides in brain regions critical for learning and memory is causative to disease progression [1,2]. The nearly concurrent clinical and neuropathological presentation between patients with familial forms of AD and those with the more common and sporadic late onset forms of the disease have led to the understanding that relatively small changes in the ratio of the Aβ peptides (primarily Aβ42 and Aβ40) in brain parenchyma lead to deposition of these peptides into pathological lesions [2]. The sequential cleavage of the amyloid precursor protein (APP) that liberates neurotoxic Aβ peptides is a complex process. APP undergoes regulated intra-membrane proteolysis (RIP) initiated by the β-site APP cleavage enzyme (BACE), leaving a C-terminal membrane bound stub that undergoes further proteolysis by γ-secretase [3]. These cleavage events generate of a pool of Aβ peptides of various lengths, the predominant species of which is Aβ40. Although Aβ42 represents only a minor (~5%) proportion of the Aβ peptides generated via this “amyloidogenic pathway”, the hydrophobic nature of this particular peptide is such that even small increases in relative amounts can drive fibrillization and deposition in brain parenchyma [4,5]. Thus, the reduction of Aβ in the brain by either direct inhibition or modulation of the γ- secretase enzyme is an attractive therapeutic target and is actively being pursued as a potential strategy to delay or prevent AD.
The γ-secretase enzyme is a complex consisting of four core proteins: presenilin (PS), nicastrin (NIC), anterior pharynx defective-1 (Aph-1), and presenilin enhancer-2 (PEN-2), of which PS and Aph-1 exist in several isoforms [6-8]. Additionally, several different glycosylation states of NIC have been reported to exist [9,10]. When incorporated into liposomes, catalytically activated PS1 alone contains the intrinsic activity required to cleave an exogenous C-terminal fragment of APP [11]. However, the additional members of the γ-secretase enzyme complex contribute to maximal enzyme activity by assisting with complex maturation and stability as it assembles and traffics through the endoplasmic reticulum [12-14]. More recently, several accessory proteins that are not essential for γ-secretase catalytic activity but instead influence the overall activity profile of the γ-secretase complex have been identified, suggesting yet another level of heterogeneity and complexity perhaps associated with cell and region-dependent γ-secretase activity [15-17].
Two different classes of pharmacological agents, γ- secretase inhibitors (GSIs) and γ-secretase modulators (GSMs), affect γ-secretase activity in mechanistically distinct ways [18,19]. GSIs inhibit the production of all forms of the Aβ peptides with roughly equivalent potencies, although in vivo and at low doses some GSIs have been reported to increase the Aβ42:Aβ40 ratio subtly through a poorly understood mechanism [20,21]. GSIs are thought to bind directly to PS either at the active site or at an allosteric site leading to the inhibition of γ-secretase. Regardless of the exact mechanism of action, the GSI class of compounds are burdened with the liability of inhibiting the processing of additional substrates, including but not limited to Notch-1 [22]. Moreover, the mechanism related to non-selective “pan-γ-secretase” inhibition is widely thought to have contributed to the adverse events associated with a recently halted clinical trial in mild to moderate AD patients [23]. Newer classes of GSIs that have greater in vitro potency for lowering Aβ peptides with limited activity towards Notch-1 have been developed. These “Notch-1” sparing inhibitors, however, still appear to inhibit other γ-secretase substrates as documented by the appearance of skin tumors and B-cell depletion [20,24,25]. GSI-1 (sulfonamide) [26,27] is one of these Notch-1-sparing compounds currently under investigation in the clinic. In contrast, GSMs have been identified that selectively modulate γ-secretase processing of APP to effectively inhibit the production of Aβ42 without affecting the overall production of Aβtotal [28]. These GSMs appear to be devoid of activity against other γ-secretase substrates thereby selectively reducing pathological Aβ peptides only [29,30]. Neither the mechanism of action nor the target binding site(s) of GSMs are fully understood at this time. Two distinct series of GSMs, represented by GSM-1 (NSAID-derived carboxylic acid) and GSM-2 (aryl imidazole), have been widely described [31]. The degree of selectivity between Aβ42 and Aβ40 inhibition, as well as the effects on the production of other shorter Aβ peptides (such as Aβ38), varies between these series. In vitro, these GSM compounds are capable of lowering Aβ42 while having little or no effect on the cleavage of Notch-1. Therefore, GSMs are assumed to have a more tolerable safety profile than GSIs.
Given the probability for tissueand species-specific molecular subunit heterogeneity, substrate promiscuity, as well as the presence of accessory proteins to potentially influence the pharmacological profile of γ-secretase, we developed a method to rapidly isolate partially purified γ-secretase activity derived from brain tissue from a variety of rodent species commonly used in pre-clinical drug discovery studies. We then applied this same methodology to isolate partially purified γ-secretase activity from non-Alzheimer’s diseased human brain cortex as well as from immortalized HeLa cells. Remarkably, the γ-secretase activity and the pharmacological profile of de novo Aβ42 and Aβ40 production following the addition of an exogenous C-terminal fragment of APP (C100-F) and either GSI-1, GSM-1 or GSM-2 was nearly identical across isolated brain tissue from rodent species or human frontal cortex. Additionally, the pharmacological profile for inhibiting or modulating the de novo production of Aβ following the addition these compounds to HeLa membranes prepared in an identical fashion was virtually indistinguishable from that obtained when the same assay was conducted using γ-secretase derived from brain membranes. As expected, GSI-1 very potently inhibited the de novo production of Aβ42 and Aβ40 across all of the membrane sources that we utilized, including human brain tissue, while the GSMs preferentially inhibited the de novo production of Aβ42. An increased selectivity for Aβ42 inhibition over Aβ40 with GSM-1, and the diminished selectivity observed ex vivo with GSM-2, translated in vivo where we observed a similar but less robust trend following oral administration to guinea pigs. True to their GSM nomenclature, both compounds produce robust Aβ42 selectivity over Aβtotal in vivo.
The use of partially purified γ-secretase activity isolated from brain (and potentially other tissue sources) will enable further characterization of cell, region, and disease-dependent requirements of this multipart atypical aspartyl protease enzyme complex. Our results also enable the refinement of drug discovery testing flow schemes to include assessment of Aβ-lowering efficacy in a more relevant and native tissue source, the human brain.
2. Materials and Methods
2.1. Compounds
GSI-1 (BMS-708163) [26,27], GSM-1 [32,33], and GSM- 2 [34] were synthesized according to literature procedures. All compounds were reconstituted in dimethyl sulfoxide (DMSO) for ex vivo studies or in 20% sulfobutyl ether-beta cyclodextrin (wt/vol) with 1.5 molar equivalents of 1M HCl for in vivo studies.
2.2. Amyloid Precursor Protein C-Terminal Substrate
DNA encoding the C-terminal 99 amino acids of the human APP gene with the addition of a methionine residue at the N-terminus as well as a C-terminal Flag tag (APP C100-F) [35] was cloned into the pET-21A expression vector (EMD Biosciences, San Diego, CA). Following transformation into BL-21(DE3) E. coli (Invitrogen, Carlsbad, CA), cells were induced for expression with 1 mM isopropyl-β-D-thiogalactoside at 20˚C for 17 h, then lysed in TNN buffer (10 mM Tris-HCl (pH 7), 200 mM NaCl, and 1% NP40 (vol/vol)) by 3 passages through a M-110L Microfluidizer (18,000 psi; Microfluidics Corp., Newton, MA). Following centrifugation (140,000 × g, 1 h), the supernatant was bound to anti-Flag M2 resin (Sigma-Aldrich, St. Louis, MO) for 2 h at 4˚C with gentle agitation, transferred to an XK16 column (GE Healthcare, UK) and allowed to flow through using an AKTAexplorer (GE Healthcare). The column was washed with TNN buffer until the baseline was stable and the bound protein was then eluted with 0.1 M glycine (pH 2.7) and 1% NP40 [36]. Each 3 ml fraction was neutralized by the addition of 1 M Tris (pH 8). Fractions that contained maximal levels of APP C100-F as identified by Western blot analysis using an antibody selective for the Flag tag (Stratagene, Santa Clara, CA) were pooled and dialyzed versus 1% NP40 for 6 h at 4˚C.
2.3. Membrane Isolation
Membranes containing γ-secretase were prepared from frozen whole brain minus cerebellum (20 - 25 g total) from 9 week old male Sprague Dawley rats (Pel Freeze Biologicals, Rogers, AR), 5 week old male Hartley guinea pigs (Lampire Biological Laboratories, Pipersville, PA), 5 month old male 129/SvE mice (Charles River Laboratories, Wilmington, MA), frontal cortex from a human brain donor that did not have Alzheimer’s disease (46 year-old female, 4 h post-mortem tissue collection; Analytical Biological Services Inc., Wilmington, DE), or frozen HeLa cell pellet (25 g). Briefly, brain tissue was crudely homogenized using a polytron in 5 ml/g buffer A (50 mM MES (2-(N-morpholino)ethanesulfonic acid, pH 6), 150 mM KCl, 5 mM CaCl2, 5 mM MgCl2, and 1x Complete Protease Inhibitor Cocktail Tablets without EDTA (ethylenediaminetetraacetic acid) (Roche, Switzerland)). Following centrifugation (800 × g, 10 min, 4˚C), the supernatant (S1) was transferred to a fresh tube and centrifuged again (100,000 × g, 1 h, 4˚C). The resulting pellet (P2) was resuspended in buffer A by dounce homogenization, using a tight-fitting pestle, to a final concentration of approximately 2.5 mg/ml total protein (w/v). HeLa cell membrane fractions were prepared in the same manner, except cells were initially broken by microfluidising (2 × 10,000 psi, M-110L Microfluidizer; Microfluidics Corp.).
2.4. Presenilin1 Quantification
PS1-NTF was quantified in serial human frontal cortex and HeLa membrane fractions following the manufacturer suggested protocol (DY-149; R & D Systems, Inc., Minneapolis, MN). Briefly, membrane fractions were subjected to sandwich ELISA using two different antihuman PS1-NTF antibodies with colorimetric horseradish-peroxidase readout.
2.5. Analysis of γ-Secretase Core Components
Proteins from P2 membrane fractions across all sources were size fractionated on a NuPage 4% - 12% Bis-Tris gel with MES SDS (sodium dodecyl sulfate) running buffer (Invitrogen) via gel electrophoresis. The proteins were then transferred onto PVDF (polyvinylidene fluoride) or nitrocellulose and hybridized with antibodies directed against PS1-CTF (PRB-354P, Covance), PS1- NTF (a gift from Dr. Yue-Ming Li, MSKCC, New York), PS2-CTF (NB110-57435, Novus Biologicals, Littleton, CO), NIC (N1660, Sigma-Aldrich), PEN-2 (3981, ProSci Inc., Poway, CA), Aph-1A (PRB-551P, Covance), Aph- 1B (ab24614, Abcam, Cambridge, UK) or actin (MAB- 1501, Millipore, Billerica, MA). Following hybridization, the blots were washed and incubated with the appropriate tertiary IRDye 800CW antibody (Li-Cor, Lincoln, NE) prior to visualization (Odyssey, Li-Cor).
2.6. γ-Secretase Activity Assay
γ-Secretase activity was assessed by incubating P2 membrane (300 μg/ml rat, guinea pig, mouse or HeLa P2 membranes (225 μg/ml human membranes)) at 37˚C for 75 min with 0.3 μM APP C100-F substrate in the presence of 10 mM Pipes (pH 7), 1 mM MgCl2, 1 mM CaCl2, 30 mM KCl, and 0.25% CHAPSO (vol/vol), followed by measurement of de novo produced Aβ42 and Aβ40 (see below for protocol). As needed, compound in a final concentration of 1% DMSO (vol/vol) was included. The reaction was stopped following the addition of RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 4 mM Tris-HCl, pH 8.0, vol/vol). Samples were then diluted 10-fold in 5M Guanidine HCl and 50 mM Tris-HCl (pH 8), and incubated for an additional 30 min at 50˚C. Each reaction was subjected to solid phase extraction using an Oasis 60 μm HLB plate (60 mg; Waters Corp, Milford, MA), eluted in 2% NH4OH and 90% MeOH (vol/vol), and evaporated to dryness.
2.7. In Vivo Assessment of γ-Secretase Modulators
All animal treatment protocols were approved by Pfizer’s Institutional Animal Care and Use Committee and were compliant with Animal Welfare Act regulations. 2 - 3 week old male Hartley guinea pigs (180 - 210 g; Charles River Laboratories, Inc.) were dosed orally with GSM-1 (32 mg/kg), GSM-2 (30 mg/kg), or vehicle (n = 5 per group). Six hours following compound administration, CSF and hemibrains were collected following CO2 euthanasia and quickly frozen on dry ice. Briefly, hemibrains were homogenized in 0.4% DEA (diethylamine) (vol/vol) and 50 mM NaCl, incubated overnight at 4˚C, and centrifuged (135,000 × g, 1 h), prior to Aβ extraction from the supernatant (Oasis 60 μm HLB plate, 60 mg; Waters Corp), as above. CSF was collected via cisterna magna puncture and assayed directly for Aβ.
2.8. Measurement of Aβ Peptides
Aβ peptides were measured by ELISA-time resolved fluorescence. Briefly, following extraction each pellet was reconstituted in blocking solution (phosphate buffered saline + 0.05% tween 20 (vol/vol) + 1% bovine serum albumin (wt/vol)) at half the initial reaction volume. De novo Aβ40 and Aβ42 were captured using the cleavage specific antibodies RN1219 and 10G3 (Rinat, Pfizer Inc., San Francisco, CA), respectively, with 4G8-biotin (Covance, Princeton, NJ) and Streptavidin-Europium (SAEu; PerkinElmer, Waltham, MA) to report. Aβtotal (1-x), Aβ1-38, Aβx-40, and Aβx-42 extracted from guinea pig brain or CSF were detected using the following sandwich ELISA pairs, respectively: 6E10(Covance)/4G8-biotin, 6E10/R341-biotin [37], 1219/4G8-biotin, and 10G3/4G8- biotin, with SA-Eu reporter. Following a 20 min addition of Delfia enhancement solution (PerkinElmer) to the samples, plates were read by EnVision (PerkinElmer) using standard Delfia filters and mirror. The amount of Aβ in each unknown was determined by back calculation from Aβ40 and Aβ42 (American Peptide, Sunnyvale, CA) standard curves. IC50 values were generated using a nonlinear regression model for curve fitting (Y = Bottom + (Top – Bottom)/(1 + 10^((LogIC50-X) × HillSlope))) and significant differences between groups were detected by one-way ANOVA followed by Dunnett’s post hoc (GraphPad Software Inc., San Diego, CA).
2.9. Data Analysis
Data are presented as mean ± SEM of individual experiments, where each condition was performed in triplicate. Serial membrane fraction data has been normalized to the P2 fraction and represents the mean of 2 HeLa or 4 human membrane experiments. Experiments in which P2 membrane concentration, C100-flag substrate concentration, reaction time, and drug concentration were varied stem from 2 - 4 individual experiments, depending upon the membrane source and endpoint measured. In vivo guinea pig data represent 5 animals in each treatment group and are expressed as a percentage of their own vehicle group. Statistical significance was determined using one way ANOVA with Dunnett’s post hoc.
3. Results
3.1. Isolation of an Active γ-Secretase Complex
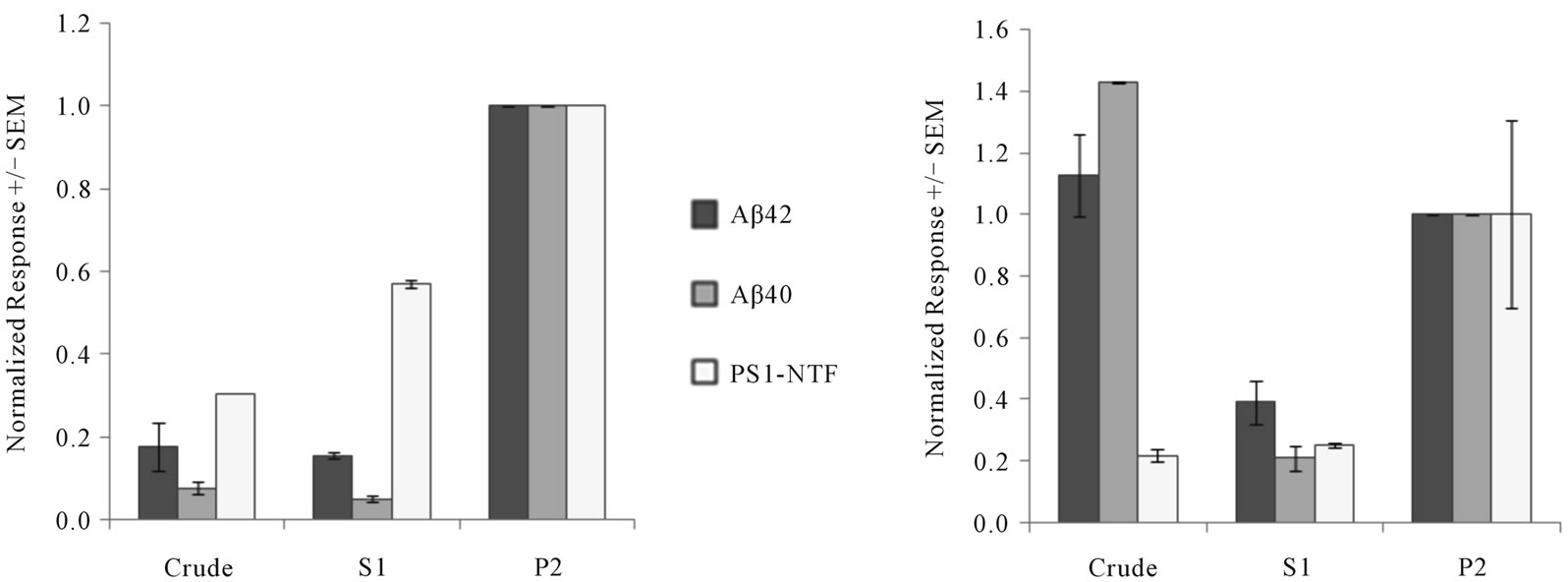
The level of subcellular fractionation required to obtain an active γ-secretase complex was assessed by measuring de novo Aβ42 and Aβ40 production following a multistep centrifugation protocol. Exogenous APP C100-F substrate was included to enable Aβ detection as there was no detectable endogenous Aβ or endogenous APPderived Aβ in the membranes. Crude homogenate as well as the S1 supernatant and P2 membranes were prepared from both immortalized HeLa cells and human brain frontal cortex. As has been previously reported [35,38], the optimal γ-secretase activity in the HeLa cell extract was obtained in the P2 membrane fraction, while little to no activity was observed in the prior fractions (Figure 1(a)). In contrast, we were able to measure de novo production of Aβ peptides from crude human brain homogenate with activity levels similar or higher than that measured in the corresponding P2 fraction (Figure 1(b)). Similar activity in crude mouse brain homogenate was observed as well (data not shown). The levels of PS1- NTF in each of the subcellular fractions isolated from HeLa cells and human brain was assessed as a surrogate of active γ-secretase complex levels, however, we did not observe a direct correlation between normalized PS1- NTF levels and Aβ production (Figures 1(a) and (b)). We then isolated partially purified γ-secretase activity in the form of P2 membrane fractions from guinea pig, rat, and mouse brain tissue. We were able to demonstrate the presence of each of the γ-secretase core components, PS1-CTF, PS1-NTF, PS2-CTF, NIC, PEN-2, Aph-1A, and Aph-1B in the brain membranes from each species as well as from membranes prepared from HeLa cells (Figure 2). The presence of distinct PS (PS1 and PS2) and Aph-1 protein (Aph-1A and Aph-1B) isoforms in membranes prepared from immortalized HeLa cells supports the notion that the γ-secretase enzyme may be composed of heterogeneous subunits even when isolated from a relatively homogenous cellular environment. The presence of a heterogeneous γ-secretase pool within brain tissue appears even more plausible since we observed differences in the migration pattern of both PEN-2 and Aph-1 across the various species that we studied. This latter observation may be attributed to differences in
 (a) (b)
(a) (b)
Figure 1. Activity of serial membrane fractions in HeLa cells and human brain tissue. The de novo production of Aβ42 or Aβ40 in serial membrane preparations from (a) HeLa cells or (b) human cortical brain was compared to levels of PS1 NTF in each. 300 μg of total protein from crude, S1, or P2 preparations were incubated with C100-F substrate for 75 min at 37˚C, followed by Aβ detection. PS1 NTF levels were measured directly in each preparation by sandwich ELISA. All data was normalized to the P2 fraction and represent mean ± SEM.

Figure 2. Identification of the core components of the γ- secretase complex in P2 membranes isolated from mouse (ms), rat, guinea pig (gp), and human (hu) brain. γ-secretase expression in HeLa cell P2 membranes was included as a comparator. For each sample, 30 μg of total protein was size fractionated by gel electrophoresis on a NuPage 4% - 12% Bis-Tris gel. Proteins were then visualized using specific antibodies. Visualization of actin was included for normalization of total protein loading.
protein-protein interaction which resist the electrophoresis denaturing conditions and/or post-translational modifications to these core components.
3.2. Optimization of Assay Conditions
We next examined the effect of increasing concentrations of P2 membranes on γ-secretase activity (Figure 3(a)) and observed a consistent and concentration-dependent increase in the level of γ-secretase activity as measured by the de novo production of Aβ42 and Aβ40 regardless of species. Similarly, we also observed an increase in γ- secretase activity with increasing concentrations of HeLa cell P2 membranes as the source of γ-secretase activity. All subsequent studies that probed the pharmacological profile of γ-secretase activity using guinea pig, rat, and mouse were conducted with 300 μg/ml of total P2 membrane protein. Studies utilizing human brain membranes were conducted using 225 μg/ml total protein to conserve this resource.
We also examined the effect of varying the concentration of exogenous APP C100-F substrate and reaction incubation time on de novo Aβ42 and Aβ40 production (Figures 3(b) and (c)). We found Aβ production to be linear with increasing substrate concentrations up to 0.3 μM and reaction times up to 2 h at 37˚C. Additionally, the amount of Aβ production measured from an exogenous substrate was consistent across the species suggesting the use of an equivalent amount of γ-secretase activity in our assays. We chose a reaction time point of 75 min and a final APP C100-F concentration of 0.3 μM for all subsequent experiments where we probed the pharmacological profile of the GSI and GSMs.

Figure 3. Brain-derived γ-secretase activity is similar across species and is dependent upon (a) the concentration of membrane protein and (b) the addition of exogenous APP C100-F substrate as well as (c) duration of the reaction time. A 75 min reaction at 37˚C with 0.3 μM APP C100-F and 300 μg/ml rodent or HeLa membrane protein (225 μg/ml human membrane protein) was chosen to produce consistent and measurable production of Aβ40 and Aβ42 across all species. Data points represent mean ± SEM.
3.3. Pharmacological Evaluation of γ-Secretase Inhibition and γ-Secretase Modulation
To assess the pharmacological profile of γ-secretase activity in brain tissue derived from multiple species, we measured the de novo production of both Aβ42 and Aβ40 in our optimized membrane assay, with and without the addition of γ-secretase interacting compounds. We chose to investigate compounds that had been previously reported to inhibit (GSM-1) or modulate (GSM-1 or GSM- 2) γ-secretase activity via differing mechanisms. Preincubation of γ-secretase membranes with these GSMs prior to APP C100-F substrate addition produced no significant change to de novo Aβ production, suggesting the GSM binding kinetics are not rate-limiting (data not shown).
Interestingly, GSI-1 inhibited equally the de novo production of Aβ42 and Aβ40 (IC50 ~ 0.3 - 0.9 nM) regardless of the species source of γ-secretase activity (Figure 4, Table 1). Additionally, the de novo production of Aβ42 or Aβ40 was inhibited to a similar level when γ-secretase activity was derived from HeLa membranes. The inhibitory potency in our γ-secretase membrane assay was in good agreement with the whole cell lowering activity that has been previously reported for this compound [26].
Following the addition of GSM-1 or GSM-2 to partially purified γ-secretase derived from various membrane sources, we observed consistent yet differential effects on the ability of these two compounds to inhibit the de novo synthesis of Aβ42 or Aβ40 (Figure 4). Importantly, the pharmacological profile that we observed when utilizing γ-secretase derived from human brain was nearly identical to that which we observed when utilizing γ-secretase activity derived from rodent brain or HeLa cells. The ability of GSM-1 to selectively inhibit the de novo production of Aβ42 (IC50 205 - 431 nM) was approximately 200-fold greater than the effect measured on Aβ40 (IC50 55 - 84 μM), regardless of the source of γ- secretase enzyme (Table 1). Although GSM-2 was very potent at inhibiting the de novo synthesis of Aβ42 (IC50 8 - 33 nM), the potency at blocking the de novo production of Aβ40 was also retained (IC50 82 - 653 nM). The selectivity profile of GSMs that we observed when using

Figure 4. The Aβ profile following the addition of pharmacological tools GSI-1, GSM-1, and GSM-2 to P2 membranes prepared from the brains of guinea pig (blue circle), rat (red square), mouse (black diamond), and human (green triangle). γ- Secretase P2 membranes derived from rodent and human brain as well as the peripheral HeLa cell line (purple inverted triangle) produce a similar Aβ profile in response to GSI and GSM treatment. Data points represent mean ± SEM.
γ-secretase derived from HeLa cells was very similar to that which we observed when assessing γ-secretase activity derived from brain.
3.4. In Vivo Modulation of γ-Secretase
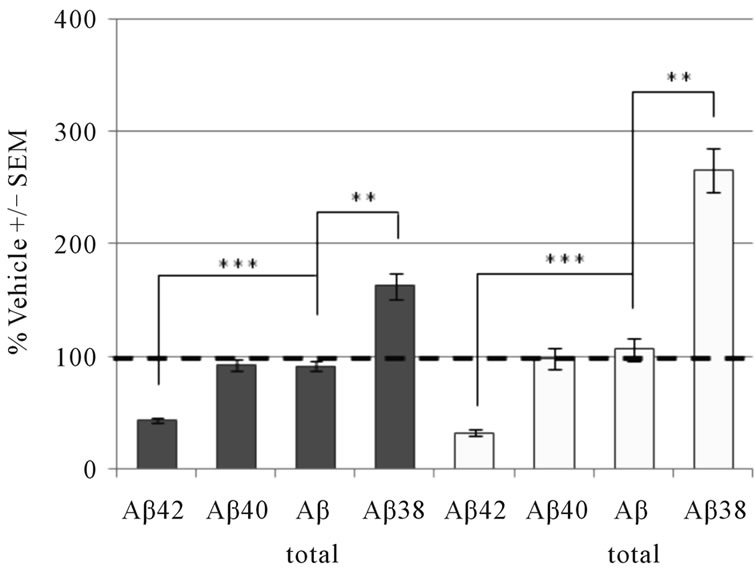
To compare our brain membrane results with in vivo efficacy, we administered GSM-1 and GSM-2 to guinea pigs and measured Aβ levels in CSF as well as crude brain homogenates (Figure 5). Following a single oral dose of GSM-1 (32 mg/kg), we observed a significant decrease in the levels of Aβ42 in brain (56% decrease) as well as CSF (67% decrease). The levels of Aβ40 as well as Aβtotal (1-x) remained unchanged in both brain and CSF. Similar to previous reports, we also observed a significant increase (>100%) in Aβ1-38 levels in brain and CSF [39,40]. A single acute administration of GSM-2 (30 mg/kg) to guinea pigs also resulted in a significant decrease Aβ42 and Aβ40 (~80% decrease) in both brain homogenates as well as CSF while the levels of Aβtotal were slightly lower or unchanged. We measured a significant increase (>200%) in Aβ1-38 in brain and CSF [29,41].
4. Discussion
An in vitro assay to measure γ-secretase activity by measuring the de novo production of Aβ40 has been described previously [35,36] using γ-secretase enriched membranes derived from HeLa cells following the addition of a C-terminal fragment of APP as an exogenous source of substrate. Although several investigators have reported the use of γ-secretase derived from rodent or human brain [38,42-44], to our knowledge there are no reports that compare the pharmacological activity of γ-secretase inhibitors and modulators in brain membranes that are prepared from multiple species, including human brain tissue, nor do these reports measure Aβ42 generation. Here we describe a rapid and facile protocol for the partial purification of a γ-secretase complex from brain


Table 1. IC50 values calculated from the de novo production of Aβ42 and Aβ40 in P2 brain membranes isolated from examined species. Distinct Aβ profiles generated by a heterogeneous pool of partially purified brain γ-secretase in response to varying γ-secretase pharmacological modulation reveal similar Aβ IC50 values across the examined species. Aβ IC50 values generated by the HeLa cell line also correlate with brain membrane results. IC50 values were generated by non-linear regression curve fitting and represent mean ± SEM.
tissue that retains enzymatic cleavage activity towards an exogenous APP substrate. By quantitatively measuring the de novo production of Aβ42 and Aβ40 we were able to investigate the ex vivo pharmacological profile of brain γ-secretase activity following the inhibition or modulation of this enzyme complex. Furthermore, the use of a partially purified enzyme complex enables the direct comparison of γ-secretase activity across multiple species, including humans, which may not otherwise be possible since pharmacokinetic properties of diverse compounds may limit exposure at the relevant target site.
The assessment of de novo production of Aβ42 from γ-secretase containing membranes has proven challenging due to the low levels of Aβ42 generated and, therefore, many reports have focused solely on measuring the de novo synthesis of Aβ40. The few reports that have measured the de novo production of Aβ42 have relied on
 (a)
(a)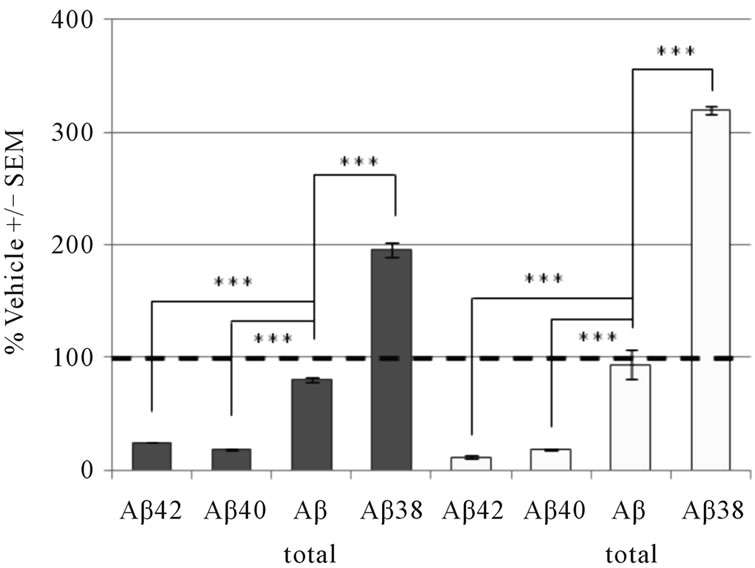 (b)
(b)
Figure 5. Modulation of brain and CSF Aβ peptide levels in guinea pig following acute administration of (a) 32 mg/kg GSM-1 or (b) 30 mg/kg GSM-2. These two distinct chemical series differ in their effects on Aβ40 inhibition, but both decrease Aβ42 and increase Aβ38 in brain (dark bars) and CSF (light bars) without affecting the production of total Aβ. Data points represent mean ± SEM from n = 5 animals per treatment group. **p < 0.005; ***p < 0.0005, one way ANOVA with Dunnett’s post hoc.
gel electrophoresis and subsequent western blot analysis to detect and quantify various peptides. Since the deposition of Aβ42 in brain regions critical for learning and memory is thought to be correlative to the progression of AD, we modified our original assay parameters to enable the detection of Aβ42. By incorporating guanidine salt denaturation, solid-phase extraction, and a highly sensitive and selective Aβ42 capture antibody we were able to follow the de novo synthesis of Aβ42, even after the addition of a very potent GSI when Aβ42 levels were significantly reduced (Figures 4 and 5).
We prepared serial subcellular fractionations from HeLa cells and human brain cortex for assessment of γ-secretase activity as measured by de novo Aβ42 and Aβ40 production. Since HeLa cell membranes have historically served as the enzyme source in publications that utilize partially purified γ-secretase, we wanted to understand how γ-secretase activity that was measured from brain subcellular fractions compared to that measured in HeLa membranes. Interestingly, we found significant differences in the de novo production of Aβ when assessed in the crude homogenate from these two membrane sources. Our findings confirm the need to isolate the P2 fraction in HeLa cells in order to obtain γ-secretase activity; however, we found that de novo production of Aβ could be measured from crude human brain homogenates at levels that were equivalent to that measured from the P2 fraction (Figure 1). We also measured a significant level of de novo Aβ production in crude homogenates from mouse brain (data not shown). Although we could detect PS1-NTF in all of the fractions that we isolated, the relative level of PS1-NTF did not correlate with the level of γ-secretase activity as measured by de novo Aβ production in either the HeLa cell or human brain fractions. As expected, while PS is necessary for γ-secretase activity, the level of PS1-NTF was not a good predictor of activity since PS must first undergo endoproteolysis for the γ-secretase complex to become enzymatically active [45].
The four core γ-secretase complex proteins, PS, NIC, Aph-1, and PEN-2 were identified in the P2 membrane preparations from rodent brains, human brain, and HeLa cells (Figure 2). The presence of different Aph-1 isoforms as well as protein from both PS family members (PS1 and PS2) was not surprising due to the cellular heterogeneity of the brain tissue that we utilized. The presence of both Aph-1 isoforms and PS proteins in the HeLa cell membrane preparations indicates the heterogeneous nature of the γ-secretase complex even within this rather uniform cell line. Our experiments were not able to clarify the exact composition of the γ-secretase complex that was responsible for the activity that we measured in any of the membrane preparations that we utilized. However, given the ubiquitous and promiscuous nature of γ-secretase activity it is highly likely that even within a single cell type, the γ-secretase complex is composed of multiple protein isoforms. While we observed some differences in the relative levels of each of the γ-secretase core protein components as well as potential post-translational effects, these differences did not appear to impact either γ-secretase activity or the pharmacological profile.
The native brain membrane assay for the assessment of γ-secretase activity that we describe here is dependent upon the concentration of membrane and substrate, as well as the length of time the reaction is allowed to incubate (Figure 3). The absence of C100-F substrate in the reaction resulted in background levels of Aβ detection suggesting there is no endogenous membrane Aβ or Aβ generated from endogenous APP contributing to the overall Aβ signal. The reaction components were optimized to ensure that a maximal level of de novo Aβ42 synthesis could be measured even under conditions where pharmacological inhibition was observed (Figure 4). The amount of de novo Aβ42 and Aβ40 that we measured was nearly identical regardless of the membrane source of γ-secretase activity confirming that we had utilized a similar level of active γ-secretase enzyme complex for studies where we probed the pharmacological effects of inhibiting or modulating the enzyme complex. When tested at high concentrations in our assay, both the GSI and GSMs that we investigated resulted in levels of Aβ that were at background level, thus excluding the possibility that a PS-independent or non-specific cleavage artifact had contributed to the overall de novo synthesis of Aβ42 or Aβ40.
As expected, the GSI that we tested in our membrane assay was able to very potently inhibit the de novo synthesis of both Aβ42 and Aβ40 regardless of the species source of γ-secretase activity. The whole cell potency of GSI-1 (IC50 = 0.3 nM) was very congruent with the inhibitory activity that we measured when assessing the de novo production of these Aβ peptides from partially purified γ-secretase derived from brain membranes (IC50 = 0.3 - 0.9 nM). Aβ lowering effects in brain of treated dogs following administration of this same GSI have also been reported [26]. In contrast, GSM-1, a compound that has previously been reported to preferentially inhibit Aβ42 in cells, was very effective at preferentially inhibiting the de novo synthesis of Aβ42 in our native brain membrane assay. Following addition of GSM-1 to partially purified γ-secretase from rodent or human brain, we observed an approximate 200-fold inhibition in de novo Aβ42 synthesis when compared to Aβ40. GSM-2 also preferentially inhibited the de novo production of Aβ42 although to a more limited degree. The differential Aβ selectivity displayed by these two classes of GSMs may be explained by reports that GSM compounds have different γ-secretase binding sites and therefore may differentially alter the interaction between the γ-secretase active site and APP [46,47]. Most interestingly, we observed very similar pharmacology for GSI-1, GSM-1, and GSM-2 in membranes prepared from common preclinical rodent species and membranes prepared from human cortical tissue.
Acute oral administration of GSM-1 or GSM-2 to guinea pigs resulted in a similar Aβ40 and Aβ42 modulation profile in brain and CSF. Following administration of GSM-1 to guinea pigs there was a 56% reduction in the levels of brain Aβ42 measured and an even greater reduction in the level of CSF Aβ42 (Figure 5). The levels of Aβ40 or Aβtotal were unchanged and there was a significant elevation in Aβ38 in both brain and CSF. In contrast, acute oral administration of GSM-2 resulted in a significant decrease in both Aβ42 and Aβ40 in brain and CSF with little to no effect on total Aβtotal and an elevation in Aβ38 levels. Although we did not measure the de novo synthesis of Aβ38 or Aβtotal in our γ-secretase membrane assay, the overall pharmacological profile for modulating the de novo synthesis of Aβ42 or Aβ40 observed in vivo following oral administration of GSM-1 or GSM-2 to guinea pigs was very similar to that observed in our native brain membrane assay across multiple species. For example, the 180-fold selectivity for Aβ42 over Aβ40 inhibition in guinea pig brain membranes with GSM-1 is apparent as a significant preference for in vivo Aβ42 inhibition in both guinea pig brain and CSF. In contrast, the greatly reduced 28-fold Aβ42 to Aβ40 selectivity observed in guinea pig brain membranes with GSM-2 is apparently not a strong enough effect to observe in vivo. Additionally, the pharmacological profile for inhibiting or modulating de novo Aβ production following the addition of the GSI or GSMs to HeLa P2 membranes was virtually indistinguishable from that obtained using partially purified γ-secretase derived from rodent or human brain, thus confirming the use of this assay as a reasonable surrogate with which to probe various γ-secretase pharmacological profiles prior to in vivo testing.
The very similar γ-secretase activity and pharmacology that we observed across species with this set of compounds may be explained by the high degree of sequence homology amongst the core protein members of the γ-secretase complex. Emerging evidence suggests that additional γ-secretase accessory proteins may influence the cell, region, and perhaps even stimulus-dependent processing of APP resulting in the generation of pathological Aβ peptides [48,49]. By isolating and characterizing γ-secretase activity from human brain we were able to probe the pharmacological profile of various compounds, thus confirming the potential to directly inhibit or selectively modulate the de novo production of pathologically relevant Aβ species. Our assay now also offers the potential of investigating γ-secretase activity in selective brain regions like the hippocampus or cerebellum, or, with slight substrate modifications, in brain tissue isolated from subjects with Alzheimer’s disease. Moreover, the native brain membrane assay that we have described here should also help to inform differing mechanisms of γ-secretase pharmacology as more novel compounds with different modes of action are identified.
5. Acknowledgements
We would like to thank Drs. Philip Iredale and Sarah Grimwood for critical review of our manuscript. No financial conflicts of interest exist. This work was supported by Pfizer Inc.
REFERENCES
- J. Hardy and D. J. Selkoe, “The Amyloid Hypothesis of Alzheimer’s Disease: Progress and problems on the road to therapeutics,” Science, Vol. 297, No. 5580, 2002, pp. 353-356. doi:10.1126/science.1072994
- E. Karran, M. Mercken and B. D. Strooper, “The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the development of therapeutics,” Nature Reviews Drug Discovery, Vol. 10, No. 9, 2011, pp. 698- 712. doi:10.1038/nrd3505
- S. F. Lichtenthaler, C. Haass and H. Steiner, “Regulated Intramembrane Proteolysis-Lessons from Amyloid Precursor Protein Processing,” Journal of Neurochemistry, Vol. 117, No. 5, 2011, pp. 779-796. doi:10.1111/j.1471-4159.2011.07248.x
- T. Iwatsubo, A. Odaka, N. Suzuki, H. Mizusawa, N. Nukina and Y. Ihara, “Visualization of Aβ42(43) and Aβ40 in Senile Plaques with End-Specific Aβ Monoclonals: Evidence That an Initially Deposited Species Is Aβ42(43),” Neuron, Vol. 13, No. 1, 1994, pp. 45-53. doi:10.1016/0896-6273(94)90458-8
- J. T. Jarrett, E. P. Berger and P. T. Lansbury, Jr., “The Carboxy Terminus of the β Amyloid Protein Is Critical for the seeding of Amyloid Formation: Implications for the pathogenesis of Alzheimer’s disease,” Biochemistry, Vol. 32, No. 18, 1993, pp. 4693-4697. doi:10.1021/bi00069a001
- D. M. Kovacs, H. J. Fausett, K. J. Page, T. W. Kim, R. D. Moir, D. E. Merriam, R. D. Hollister, O. G. Hallmark, R. Mancini, K. M. Felsenstein, B. T. Hyman, R. E. Tanzi and W. Wasco, “Alzheimer-Associated Presenilins 1 and 2: Neuronal Expression in brain and localization to intracellular membranes in Mammalian Cells,” Nature Medicine, Vol. 2, No. 2, 1996, pp. 224-229. doi:10.1038/nm0296-224
- K. Shirotani, D. Edbauer, S. Prokop, C. Haass and H. Steiner, “Identification of distinct Gamma-Secretase Complexes with different APH-1 variants,” Journal of Biological Chemistry, Vol. 279, No. 40, 2004, pp. 41340- 41345. doi:10.1074/jbc.M405768200
- L. Serneels, T. Dejaegere, K. Craessaerts, K. Horre, E. Jorissen, T. Tousseyn, S. Hebert, M. Coolen, G. Martens, A. Zwijsen, W. Annaert, D. Hartmann and B. De Strooper, “Differential contribution of the three Aph1 genes to Gamma-Secretase Activity in vivo,” Proceedings of the National Academy of Sciences of USA, Vol. 102, No. 5, 2005, pp. 1719-1724. doi:10.1073/pnas.0408901102
- S. Arawaka, H. Hasegawa, A. Tandon, C. Janus, F. Chen, G. Yu, K. Kikuchi, S. Koyama, T. Kato, P. E. Fraser and P. St George-Hyslop, “The levels of Mature Glycosylated Nicastrin Are Regulated and correlate with gammaSecretase Processing of amyloid β-Precursor Protein,” Journal of Neurochemistry, Vol. 83, No. 5, 2002, pp. 1065-1071. doi:10.1046/j.1471-4159.2002.01207.x
- F. Chen, A. Tandon, N. Sanjo, Y. J. Gu, H. Hasegawa, S. Arawaka, F. J. Lee, X. Ruan, P. Mastrangelo, S. Erdebil, L. Wang, D. Westaway, H. T. Mount, B. Yankner, P. E. Fraser and P. St George-Hyslop, “Presenilin 1 and presenilin 2 Have Differential Effects on the stability and maturation of nicastrin in Mammalian brain,” Journal of Biological Chemistry, Vol. 278, No. 22, 2003, pp. 19974- 19979. doi:10.1074/jbc.M210049200
- K. Ahn, C. C. Shelton, Y. Tian, X. Zhang, M. L. Gilchrist, S. S. Sisodia and Y. M. Li, “Activation and Intrinsic Gamma-Secretase Activity of presenilin 1,” Proceedings of the National Academy of Sciences of USA, Vol. 107, No. 50, 2010, pp. 21435-21440. doi:10.1073/pnas.1013246107
- N. Takasugi, T. Tomita, I. Hayashi, M. Tsuruoka, M. Niimura, Y. Takahashi, G. Thinakaran and T. Iwatsubo, “The role of presenilin cofactors in the Gamma-Secretase Complex,” Nature, Vol. 422, No. 6930, 2003, pp. 438-441. doi:10.1038/nature01506
- Y. W. Zhang, W. J. Luo, H. Wang, P. Lin, K. S. Vetrivel, F. Liao, F. Li, P. C. Wong, M. G. Farquhar, G. Thinakaran and H. Xu, “Nicastrin is critical for stability and trafficking but Not Association of Other Presenilin/ Gamma-Secretase Components,” Journal of Biological Chemistry, Vol. 280, No. 17, 2005, pp. 17020-17026. doi:10.1074/jbc.M409467200
- W. J. Luo, H. Wang, H. Li, B. S. Kim, S. Shah, H. J. Lee, G. Thinakaran, T. W. Kim, G. Yu and H. Xu, “PEN-2 and APH-1 Coordinately Regulate Proteolytic Processing of presenilin 1,” Journal of Biological Chemistry, Vol. 278, No. 10, 2003, pp. 7850-7854. doi:10.1074/jbc.C200648200
- G. He, W. Luo, P. Li, C. Remmers, W. J. Netzer, J. Hendrick, K. Bettayeb, M. Flajolet, F. Gorelick, L. P. Wennogle and P. Greengard, “Gamma-Secretase Activating Protein Is a Therapeutic Target for Alzheimer’s disease,” Nature, Vol. 467, No. 7311, 2010, pp. 95-98. doi:10.1038/nature09325
- S. Zhou, H. Zhou, P. J. Walian and B. K. Jap, “CD147 is a Regulatory Subunit of the Gamma-Secretase Complex in Alzheimer’s Disease Amyloid β-Peptide Production,” Proceedings of the National Academy of Sciences of USA, Vol. 102, No. 21, 2005, pp. 7499-7504. doi:10.1073/pnas.0502768102
- F. Chen, H. Hasegawa, G. Schmitt-Ulms, T. Kawarai, C. Bohm, T. Katayama, Y. Gu, N. Sanjo, M. Glista, E. Rogaeva, Y. Wakutani, R. Pardossi-Piquard, X. Ruan, A. Tandon, F. Checler, P. Marambaud, K. Hansen, D. Westaway, P. St George-Hyslop and P. Fraser, “TMP21 is a Presenilin Complex Component That Modulates GammaSecretase But Not Epsilon-Secretase Activity,” Nature, Vol. 440, No. 7088, 2006, pp. 1208-1212. doi:10.1038/nature04667
- A. F. Kreft, R. Martone and A. Porte, “Recent advances in the identification of Gamma-Secretase Inhibitors to Clinically Test the Aβ Oligomer Hypothesis of Alzheimer’s disease,” Journal of Medicinal Chemistry, Vol. 52, No. 20, 2009, pp. 6169-6188. doi:10.1021/jm900188z
- M. Pettersson, G. W. Kauffman, C. W. am Ende, N. C. Patel, C. Stiff, T. P. Tran and D. S. Johnson, “Novel Gamma-Secretase Modulators: A Review of patents from 2008 to 2010,” Expert Opinion on Therapeutic Patents, Vol. 21, No. 2, 2011, pp. 205-226.
- T. A. Lanz, M. J. Karmilowicz, K. M. Wood, N. Pozdnyakov, P. Du, M. A. Piotrowski, T. M. Brown, C. E. Nolan, K. E. Richter, J. E. Finley, Q. Fei, C. F. Ebbinghaus, Y. L. Chen, D. K. Spracklin, B. Tate, K. F. Geoghegan, L. F. Lau, D. D. Auperin and J. B. Schachter, “ConcentrationDependent Modulation of amyloid-β in vivo and in vitro using the Gamma-Secretase Inhibitor, LY-450139,” Journal of Pharmacology and Experimental Therapeutics, Vol. 319, No. 2, 2006, pp. 924-933. doi:10.1124/jpet.106.110700
- E. Siemers, M. Skinner, R. A. Dean, C. Gonzales, J. Satterwhite, M. Farlow, D. Ness and P. C. May, “Safety, tolerability, and changes in amyloid β concentrations after administration of a Gamma-Secretase Inhibitor in volunteers,” Clinical Neuropharmacology, Vol. 28, No. 3, 2005, pp. 126-132. doi:10.1097/01.wnf.0000167360.27670.29
- A. Haapasalo and D. M. Kovacs, “The Many Substrates of Presenilin/Gamma-Secretase,” Journal of Alzheimer’s Disease, Vol. 25, No. 1, 2011, pp. 3-28.
- B. P. Imbimbo, F. Panza, V. Frisardi, V. Solfrizzi, G. D’Onofrio, G. Logroscino, D. Seripa and A. Pilotto, “Therapeutic intervention for Alzheimer’s disease with Gamma-Secretase Inhibitors: Still a Viable Option?” Expert Opinion on Investigational Drugs, Vol. 20, No. 3, 2011, pp. 325-341.
- F. Panza, V. Frisardi, B. P. Imbimbo, C. Capurso, G. Logroscino, D. Sancarlo, D. Seripa, G. Vendemiale, A. Pilotto and V. Solfrizzi, “Review: gamma-Secretase inhibitors for the treatment of Alzheimer’s disease: The Current State,” CNS Neuroscience & Therapeutics, Vol. 16, No. 5, 2010, pp. 272-284. doi:10.1111/j.1755-5949.2010.00164.x
- S. Demehri, A. Turkoz and R. Kopan, “Epidermal Notch1 Loss Promotes Skin Tumorigenesis by impacting the Stromal Microenvironment,” Cancer Cell, Vol. 16, No. 1, 2009, pp. 55-66. doi:10.1016/j.ccr.2009.05.016
- K. W. Gillman, J. E. Starrett Jr., M. F. Parker, K. Xie, J. J. Bronson, L. R. Marcin, K. E. McElhone, C. P. Bergstrom, R. A. Mate, R. Williams, J. E. Meredith Jr., C. R. Burton, D. M. Barten, J. H. Toyn, S. B. Roberts, K. A. Lentz, J. G. Houston, R. Zaczek, C. F. Albright, C. P. Decicco, J. E. Macor and R. E. Olson, “Discovery and evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable gamma-Secretase Inhibitor,” ACS Medicinal Chemistry Letters, Vol. 1, No. 3, 2010, pp. 120-124. doi:10.1021/ml1000239
- J. Starrett, J. E., K. W. Gillman and R. E. Olson, “Novel Alpha-(N-Sulfonamido)Acetamide Compound as an inhibitor of Beta Amyloid Peptide Production,” U.S. Patent No. 2009/0227642 A0227641, 2009.
- S. Weggen, J. L. Eriksen, P. Das, S. A. Sagi, R. Wang, C. U. Pietrzik, K. A. Findlay, T. E. Smith, M. P. Murphy, T. Bulter, D. E. Kang, N. Marquez-Sterling, T. E. Golde and E. H. Koo, “A subset of NSAIDs Lower Amyloidogenic Aβ42 independently of Cyclooxygenase Activity,” Nature, Vol. 414, No. 6860, 2001, pp. 212-216. doi:10.1038/35102591
- M. Z. Kounnas, A. M. Danks, S. Cheng, C. Tyree, E. Ackerman, X. Zhang, K. Ahn, P. Nguyen, D. Comer, L. Mao, C. Yu, D. Pleynet, P. J. Digregorio, G. Velicelebi, K. A. Stauderman, W. T. Comer, W. C. Mobley, Y. M. Li, S. S. Sisodia, R. E. Tanzi and S. L. Wagner, “Modulation of Gamma-Secretase Reduces β-Amyloid Deposition in a Transgenic Mouse Model of Alzheimer’s disease,” Neuron, Vol. 67, No. 5, 2010, pp. 769-780. doi:10.1016/j.neuron.2010.08.018
- S. Weggen, J. L. Eriksen, S. A. Sagi, C. U. Pietrzik, T. E. Golde and E. H. Koo, “Aβ42-Lowering Nonsteroidal AntiInflammatory Drugs Preserve Intramembrane Cleavage of the Amyloid Precursor Protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain,” Journal of Biological Chemistry, Vol. 278, No. 33, 2003, pp. 30748-30754. doi:10.1074/jbc.M304824200
- D. Oehlrich, D. J. Berthelot and H. J. Gijsen, “gammaSecretase Modulators as Potential Disease Modifying Anti-Alzheimer’s Drugs,” Journal of Medicinal Chemistry, Vol. 54, No. 2011, pp. 669-698.
- R. M. Page, K. Baumann, M. Tomioka, B. I. Perez-Revuelta, A. Fukumori, H. Jacobsen, A. Flohr, T. Luebbers, L. Ozmen, H. Steiner and C. Haass, “Generation of Aβ38 and Aβ42 is independently and Differentially Affected by familial Alzheimer Disease-Associated Presenilin Mutations and Gamma-Secretase Modulation,” Journal of Biological Chemistry, Vol. 283, No. 2, 2008, pp. 677-683. doi:10.1074/jbc.M708754200
- J. C. Hannam, J. J. Kulagowski, A. Madin, M. P. Ridgill and E. M. Steward, “Preparation of Piperidines and related compounds for treatment of Alzheimer’s disease,” International Patent No. WO2006/043064, 2006.
- T. Kimura, N. Kitazawa, T. Kaneko, N. Sato, K. Kawano, K. Ito, M. Takaishi, T. Sasaki, Y. Yoshida, T. Uemura, T. Doko, D. Shinmyo, D. Hasegawa, T. Miyagawa and H. Hagiwara, “Multi-cyclic compounds,” U.S. Patent No. 2009/0062529 A0062521, 2009.
- Y. M. Li, M. T. Lai, M. Xu, Q. Huang, J. DiMuzioMower, M. K. Sardana, X. P. Shi, K. C. Yin, J. A. Shafer and S. J. Gardell, “Presenilin 1 is linked with gammasecretase activity in the Detergent Solubilized State,” Proceedings of the National Academy of Sciences of USA, Vol. 97, No. 11, 2000, pp. 6138-6143. doi:10.1073/pnas.110126897
- W. T. Kimberly, W. P. Esler, W. Ye, B. L. Ostaszewski, J. Gao, T. Diehl, D. J. Selkoe and M. S. Wolfe, “Notch and the Amyloid Precursor Protein Are Cleaved by Similar Gamma-Secretase(s),” Biochemistry, Vol. 42, No. 1, 2003, pp. 137-144. doi:10.1021/bi026888g
- D. L. Miller, A. Potempska and P. D. Mehta, “Humoral Immune Responses to Peptides Derived from the β- amyloid Peptide C-Terminal Sequence,” Amyloid, Vol. 14, No. 1, 2007, pp. 39-50.
- J. Franberg, H. Welander, M. Aoki, B. Winblad, L. O. Tjernberg and S. Frykman, “Rat Brain Gamma-Secretase Activity Is Highly Influenced By Detergents,” Biochemistry, Vol. 46, No. 25, 2007, pp. 7647-7654. doi:10.1021/bi0621258
- J. Hawkins, D. C. Harrison, S. Ahmed, R. P. Davis, T. Chapman, I. Marshall, B. Smith, T. L. Mead, A. Medhurst, G. M. Giblin, A. Hall, M. I. Gonzalez, J. Richardson and I. Hussain, “Dynamics of Aβ42 reduction in plasma, CSF and brain of Rats Treated with the GammaSecretase Modulator, GSM-10h,” Neurodegenerative Diseases, Vol. 8, No. 6, 2011, pp. 455-464. doi:10.1159/000324511
- B. Van Broeck, J. M. Chen, G. Treton, M. Desmidt, C. Hopf, N. Ramsden, E. Karran, M. Mercken and A. Rowley, “Chronic treatment with a Novel Gamma-Secretase Modulator, JNJ-40418677, Inhibits Amyloid Plaque Formation in a Mouse Model of Alzheimer’s disease,” British Journal of Pharmacology, Vol. 163, No. 2, 2011, pp. 375-389. doi:10.1111/j.1476-5381.2011.01207.x
- E. Portelius, B. Van Broeck, U. Andreasson, M. K. Gustavsson, M. Mercken, H. Zetterberg, H. Borghys and K. Blennow, “Acute effect on the Aβ Isoform Pattern in CSF in response to Gamma-Secretase Modulator and inhibitor treatment in dogs,” Journal of Alzheimer’s Disease, Vol. 21, No. 3, 2010, pp. 1005-1012.
- R. Wang, B. Wang, W. He and H. Zheng, “Wild-Type Presenilin 1 Protects against Alzheimer Disease Mutation-Induced Amyloid Pathology,” Journal of Biological Chemistry, Vol. 281, No. 22, 2006, pp. 15330-15336. doi:10.1074/jbc.M512574200
- J. L. Goggi, H. D. Lewis, J. Mok, T. Harrison, M. S. Shearman, J. R. Atack and J. D. Best, “A Comparative Assessment of Gamma-Secretase Activity in transgenic and Non-Transgenic Rodent Brain,” Journal of Neuroscience Methods, Vol. 157, No. 2, 2006, pp. 246-252. doi:10.1016/j.jneumeth.2006.05.001
- J. Y. Hur, H. Welander, H. Behbahani, M. Aoki, J. Franberg, B. Winblad, S. Frykman and L. O. Tjernberg, “Active Gamma-Secretase Is Localized to Detergent-Resistant Membranes in Human Brain,” FEBS Journal, Vol. 275, No. 6, 2008, pp. 1174-1187. doi:10.1111/j.1742-4658.2008.06278.x
- G. Thinakaran, D. R. Borchelt, M. K. Lee, H. H. Slunt, L. Spitzer, G. Kim, T. Ratovitsky, F. Davenport, C. Nordstedt, M. Seeger, J. Hardy, A. I. Levey, S. E. Gandy, N. A. Jenkins, N. G. Copeland, D. L. Price and S. S. Sisodia, “Endoproteolysis of Presenilin 1 and Accumulation of Processed Derivatives in Vivo,” Neuron, Vol. 17, No. 1, 1996, pp. 181-190. doi:10.1016/S0896-6273(00)80291-3
- A. Ebke, T. Luebbers, A. Fukumori, K. Shirotani, C. Haass, K. Baumann and H. Steiner, “Novel Gamma-secretase Modulators Directly Target Presenilin,” Journal of Biological Chemistry, Vol. 286, 2011, pp. 37181- 37186.
- C. J. Crump, B. A. Fish, S. V. Castro, D.-M. Chau, N. Gertsik, K. Ahn, C. Stiff, N. Pozdnyakov, K. R. Bales, D. S. Johnson and Y.-M. Li, “Piperidine Acetic Acid Based Gamma-Secretase Modulators Directly Bind to Presenilin-1,” ACS Chemical Neuroscience, Vol. 2, No. 12, 2011, pp. 705-710.
- B. De Strooper and W. Annaert, “Novel Research Horizons for presenilins and Gamma-Secretases in Cell Biology and disease,” Annual Review of Cell and Developmental Biology, Vol. 26, 2010, pp. 235-260. doi:10.1146/annurev-cellbio-100109-104117
- T. Wakabayashi, K. Craessaerts, L. Bammens, M. Bentahir, F. Borgions, P. Herdewijn, A. Staes, E. Timmerman, J. Vandekerckhove, E. Rubinstein, C. Boucheix, K. Gevaert and B. De Strooper, “Analysis of the gamma-secretase interactome and validation of Its Association with Tetraspanin-Enriched Microdomains,” Nature Cell Biology, Vol. 11, No. 11, 2009, pp. 1340-1346. doi:10.1038/ncb1978
Abbreviations
NOTES
*Corresponding author.