Advances in Microbiology
Vol.07 No.02(2017), Article ID:74394,17 pages
10.4236/aim.2017.72013
Metagenomic rDNA Profiles of the Oral Microbiome in Healthy Subjects Are Distinctive Compared to That in Smokers and Disease Subjects Despite High Variations between Subjects
Shreyasee Chakraborty, Nwadiuto Esiobu*
Microbial Biotech Laboratory, Department of Biological Sciences, Florida Atlantic University, Boca Raton, FL, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 7, 2017; Accepted: February 24, 2017; Published: February 27, 2017
ABSTRACT
The vital role of oral microbiome in the well-being of humans is only beginning to be unraveled. Employing a rigorous analysis of PCR-restriction fragment length polymorphisms (PCR-RFLP) and DNA fingerprints from denatured gradient gel electrophoresis (PCR-DGGE) of the 16S rDNA gene in metagenomic samples, this study evaluated the stability of the oral microbiome and contrasted the PCR-DGGE profiles of subjects belonging to three groups ―healthy, smokers and oral diseases; in search of distinctive patterns predictive of each group. The DNA band size, intensity and profile generated by three restriction enzymes from a 1500 bp amplicon showed a fairly stable microbial community structure (P < 0.05) in PCR-RFLP samples collected from each individual over a 3-month period. Microbial diversity indices and cluster analysis of amplicons from the V4 region of the 16S gene from the three study groups were consistent with a stable core of bacterial DNA fingerprint within and between subjects despite the enormous beta variations. Statistical analysis including multi-dimensional scaling of the DGGE fingerprints in smokers and oral disease subjects aligned both banding patterns (P-value 0.08, Student’s t-test), suggesting some similarity between the microbial consortia of smokers and subjects with dental caries and gingivitis. There was a significant difference between bacterial genomic profiles in healthy mouth and smokers/ disease (P = 0.002; paired-sample Student’s t-test). Operational taxonomic unit diversity and species richness determined by the GelCompare II software were higher in smokers (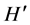 = 0.99 ± 0.12; S = 2.87 ± 0.75) and oral disease mouths (
= 0.99 ± 0.12; S = 2.87 ± 0.75) and oral disease mouths (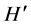 = 1.06 ± 0.085; S = 2.86 ± 0.31), than in healthy subjects (
= 1.06 ± 0.085; S = 2.86 ± 0.31), than in healthy subjects (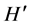 = 0.926 ± 0.07; S = 1.79 ± 0.56) suggesting that smoking is associated with a microbial community shift towards the structure found in poor oral health. It is clear that the human oral bacteria symbionts are not all random colonizers. Rather some of them constitute a fragile stable dynamic community whose disturbance could lead to disease or be indicative of disease. Understanding the dynamics of the bacterial community structure in health and disease states is a prerequisite to developing effective preventive healthcare and rapid diagnosis of diseases.
= 0.926 ± 0.07; S = 1.79 ± 0.56) suggesting that smoking is associated with a microbial community shift towards the structure found in poor oral health. It is clear that the human oral bacteria symbionts are not all random colonizers. Rather some of them constitute a fragile stable dynamic community whose disturbance could lead to disease or be indicative of disease. Understanding the dynamics of the bacterial community structure in health and disease states is a prerequisite to developing effective preventive healthcare and rapid diagnosis of diseases.
Keywords:
Oral Microbiome, Metagenomic

1. Introduction
Over the past decade, significant progress has been achieved in cataloguing and identifying the prokaryotic symbionts of humans. For example, the sequences and identity of nearly half of the 700 prokaryotic species present in the human mouth are known, while 34% are listed as unknown phylotypes in the highly resourceful human oral database―www.homd.org [1] . The HOMD links sequence data with phenotypic, phylogenetic, clinical, and bibliographic information, allowing scientists to employ multiple technologies including next generation sequencing to create algorithms and profiles that could be predictive of health or disease in humans. The consensus that human microbiomes are important gatekeepers of good health has put analysis of microbiomes squarely at the center of healthcare. One thing however, that is sorely needed is the interpretation of the dynamics and succession of these bacteria communities. Using next generation sequencing Al Hebshi et al. found that the oral bacteria associated with oral squamous cell carcinoma were taxonomically more diverse than those found in healthy controls [2] . Similarly, dental caries, periodontitis [3] [4] , cardiovascular disease [5] , diabetes [6] and other cancers (pancreatic and gastrointestinal) [7] [8] [9] have been linked to bacteria in the mouth. While it remains unclear in some cases whether the bacteria profiles are directly responsible for the etiologies or whether they are a product of the disease pathologies; research has shown that personal habits like tobacco smoking, drinking alcohol, and diet change could contribute to poor oral health. Subgingival biofilm in smokers is enriched for pathogenic bacteria, resulting in alteration of the microbial signatures of the oral cavity with a decrease in the commensal population and a concomitant increase in pathogens [10] [11] . How these community alterations play out in several diseases of the mouth, especially those involving mixed flora or a cascade of factors triggered by microbial or other signals, is poorly understood. The recent discovery of a novel bacterial co-prevalence pattern in dental carries ? a supposedly previously well-characterized disease illustrates the dynamics of this phenomenon [12] . On the other hand, microbiome fingerprints in healthy persons are being examined for potential use in identity based on individuals’ unique body microbes [13] .
The need to begin to properly apply all these rapidly accumulating knowledge underscores the importance of research aimed at understanding microbial community changes that accompany the inception of disease. For the oral microbiome, this involves quantitative analysis of microbial signatures in search of potential biomarkers in not only the healthy state, but also that of transition to disease and actual disease states. Such discoveries, if found to be idiotypic could nicely dovetail into the generation of future personalized healthcare strategies. The oral cavity represents an important niche for studying the genomic and functional variations/profiles of microbiomes that could lead to better understanding and applications of the accumulating microbiome data to early disease diagnosis and preventive medicine because of easy access to samples, personal enclosure (unlike the skin) and its connection to other parts of the host.
This study used relatively simple techniques viz denaturing gradient gel electrophoresis (DGGE) and restriction fragment length polymorphism (PCR- RFLP) of 16S rRNA sequences to measure the stability of the oral microbiome of volunteer subjects and to contrast the genetic diversity/fingerprints of healthy non-smokers, smokers and subjects with oral diseases. These techniques have been shown to detect even single nucleotide changes in a DNA fragment [14] [15] [16] . Metagenomic samples as well as cultured communities were included to compare outcome using inexpensive and accessible protocols for the microbiome assessment.
2. Materials and Methods
2.1. Human Oral Wash Sample Collection
Oral wash samples were obtained from human subjects who self-identified as healthy (n = 5), smokers (n = 5), and subjects with oral diseases (n = 5 - Gingivitis n = 3, Dental caries n = 2) above the age of 18 years. The demographic information, periodontal parameters and smoking status are summarized in Table 1. Samples were collected in accordance with Florida Atlantic University’s approved IRB and IBC protocol (IRBNET ID #388951-2). All participants were antibiotic free. Healthy non-smokers did not smoke for at least 3 years preceding the study and all subjects adhered to their normal oral hygiene for 2 weeks before enrollment in the study. Furthermore, health and oral hygiene data were

Table 1. Demographic information, periodontal and smoking status of participants.
collected by means of a questionnaire survey. To minimize background DNA variation, participants collected 25 - 50 ml sample using the same type of bottled drinking water for a vigorous gargle early in the morning before washing their mouth. Samples were kept at −20˚C upon collection and analyzed within 12 hours.
2.2. Direct Metagenomic DNA Extraction and Plate Wash DNA Extraction
About 20 mL of each sample was dispensed into 15 micro-centrifuge tubes and spun at 15,000 rpm for 10 min in an aerosol-tight microcentrifuge to harvest bacteria cells. The pellets were then spooled into one tube and used for direct extraction of the metagenomes. To contrast the bacterial community structure of directly extracted genomes with cultivable community, a sub-set of bacteria pellets was washed in 1.5 mL of the Phosphate Buffered Saline, top-spread (100 µL) on Brain Heart Infusion Agar (BHIA) and incubated for 48 h at 37˚C. Thereafter, the community of cultivatable bacteria was harvested, washed in PBS and collected by centrifugation at 15,000 rpm for 10 min. The subsequent steps for genomic DNA extraction from direct and plate wash bacterial communities were done using the Qiagen Dneasy Blood and Tissue DNA purification kit (Qiagen, Valencia, CA) following the manufacturer’s instruction.
2.3. PCR Amplification for Restriction Fragment Length Polymorphism (PCR-RFLP)
The extracted metagenomes of samples intended for the PCR-RFLP analysis were amplified using the universal primer 1492 Reverse (5’GGTTACCTTGTT ACGACTT-3’) and the primer 27 Forward (5’-AGAGTTTGATCCTGGCTC AG-3’) [17] that targets the V1-V9 region of the 16S rRNA gene. PCR was performed using the Mastercycler Nexus PCR Systems (Eppendorf North America, NY, USA). Each PCR reaction mixture (a total volume of 25 μl) contained: a standardized 100 ng (1 - 3 μl depending on the initial concentration) of total genomic DNA; 0.5 micromol of each primers and 12.5 μl of 2X PCR Master Mix which contains Tag DNA polymerase, dNTP’s, PCR buffer and Mgcl2 (Promega Inc,WI, USA). Reaction mixtures were incubated for 4 min at 94˚C for denaturation, followed by 35 cycles of 1 min at 94˚C, annealing for 30 sec at 45˚C, and extension for 2 min at 72˚C. PCR product (approximately 1500 bp) was confirmed by electrophoresis in 1% agarose gel at 120 V for 30 min. 10 µL of the amplified PCR product was utilized for restriction digest.
2.4. PCR Amplification for DGGE Fingerprinting
A set of universal primers for bacterial 16S rRNA gene―Bac1 (‘5-CGCCCGCCG CGCCCCGCGCCCGTCCCGCCGCCCCCGCCCG-CCTACGGGAGGCAGCAG-3’) and Bac2 (5-CCGTCAATTCCTTTRAGTTT-3’) [18] which targets the hypervariable region V4-V5 of the Escherichia coli 16S rDNA ribosomal locus was used to generate a 300 bp amplicon. A 40-nucleotide GC clamp was added to the 5’ end of Bac1 to normalize the different amplicons and increase resolution. PCR conditions were: initial denaturation at 95˚C for 3 min and 35 cycles consisting of 1 min at 95˚C, 1 min at 56˚C, 2 min at 72˚C and an additional cycle of 5 min at 72˚C for chain elongation. The PCR products were evaluated by electrophoresis in 3.0% agarose gel at 60 V for 60 min and the size of amplicons (300 bp) were confirmed according to a 1 kb molecular size standard. A standard 20 µL (approximately 300 ng) of PCR amplified product was then employed for the DGGE/ TGGE analysis.
3. Microbial DNA Fingerprinting Techniques
3.1. Restriction Fragment Length Polymorphism (RFLP)
The 1500 bp PCR-amplified bacterial 16S rDNA segment was digested using 3 restriction enzymes: HaeIII, Alu1 and Sau3A (1U), according to the manufacturer’s instructions (New England Biolabs, Beverly, Mass.). The DNA fragments were separated by gel electrophoresis on a 3% low melting agarose gel run in 1X TAE (Tris Acetate-EDTA, pH-8) buffer. The RFLP or ribopattern profile were digitally captured and recorded by means of the FOTO/Analyst® Investigator/FX System using the PC Image Acquistion Software (Fotodyne Incorporated, Hartland, WI).
3.2. Denaturing Gradient Gel Electrophoresis (DGGE)
DGGE is a powerful technique that can separate nucleic acid fragments based on their nucleotide sequence, distinguishing between sequences with single nucleotide polymorphisms [19] . The diversity and richness of bacteria from the study groups were determined by separating the 16S rDNA gene amplicons (V3-V4) to create a profile based on their nucleotide sequence in the denatured gradient gels (DGGE). DGGE was performed using DGGE system from CBS scientific company (San Diego, CA, USA) for all DGGE experiments. A 30% - 70% linear DNA denaturing gradient (100% denaturant is equivalent to 7 mol/L urea and 40% deionized formamide) was formed in 8% (w/v) polyacrylamide gels. PCR products were loaded directly in each lane, and electrophoresis was performed at a constant 60 V at 58˚C for 16 h in 1X Tris-acetate-EDTA (TAE) buffer at pH 8.5. The image of the DGGE profile was captured, after staining with Ethidium Bromide for 30 min, using the FOTO/Analyst® Investigator/FX System using the PC Image Acquistion Software (Fotodyne Incorporated, Hartland, WI). in the same manner as explained above. The different banding patterns, which reflected the variations in nucleotide sequence and hence diversity; were noted and analyzed using the GelCompare software.
3.3. Analysis of the 16S rDNA Fingerprint Generated by PCR-RFLP and PCR-DGGE
An image of the RFLP gel was captured by Fotodyne Imager as mentioned above. The banding patterns were examined to identify the commonalities and differences among the different study groups. The GelCompare II (Applied Maths, Kortrijk, Belgium) software was employed in the analysis of the 16S rDNA fingerprint. This software searches for discriminative bands between selected groups of patterns and also searches for unique and common bands within selections. To obtain an objective comparison and normalization of the bands on different slab gels, a molecular weight marker was included in each gel at least two times. Digitized images were normalized and combined [20] In addition, experimental conditions were kept exactly the same through-out the DGGE experiments. A similarity matrix was created using the Dice similarity coefficient [21] [22] . For the same slab, gels similarity matrix was created using the Pearson correlation coefficient which employs the densitometry curve created by the fingerprints. Similarities were displayed graphically as a dendrogram. Cluster analysis was performed for the fingerprint data based on the binary presence or absence of bands (presence = 1, absence = 0). For band comparison, a band position tolerance value of 0.8% was allowed to compensate for misalignment of homologous bands due to technical imperfections. The unweighted pair group method using average linkages (UPGMA) [20] was used to cluster the patterns. Diversity indices were also calculated for PCR-DGGE analysis: richness (S) was determined from the number of bands in each lane, and the Shannon-Wiener index (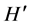 ) was calculated from
) was calculated from 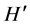 = −ΣPilnPi [23] , where Pi is the importance probability of the bands in a lane, calculated from ni/N, where ni is the peak height of a band and N is the sum of all peak heights in the densitometric curve. Evenness (E) was calculated as E =
= −ΣPilnPi [23] , where Pi is the importance probability of the bands in a lane, calculated from ni/N, where ni is the peak height of a band and N is the sum of all peak heights in the densitometric curve. Evenness (E) was calculated as E =  /
/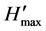 , where
, where 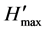 = ln S [24] . Significant differences in the number of detected PCR amplicons in the DGGE gels were determined using analysis of variance (anova) and the paired-samples Student’s t-test. Further statistical analyses were performed using Microsoft Excel 2010. All P-values < 0.05 were two-tailed and considered significant.
= ln S [24] . Significant differences in the number of detected PCR amplicons in the DGGE gels were determined using analysis of variance (anova) and the paired-samples Student’s t-test. Further statistical analyses were performed using Microsoft Excel 2010. All P-values < 0.05 were two-tailed and considered significant.
4. Results
The 16S rDNA gene segment from oral bacteria community for each subject generated a profile of DNA gel bands after restriction (PCR-RFLP). These banding patterns represented their individual oral microbiome structure or identity, band profile of composite dataset including healthy non-smokers, smokers and disease subjects using PCR-RFLP and DGGE were compared and contrasted. The discriminatory as well as uniform/core bands were detected using the GelCompare II version 6.6 software.
4.1. Restriction Fragment Length Polymorphisms of Ribotypes Generated by HaeIII Restriction
PCR-RFLP ribopattern analysis of the complete data-set for the three different groups using HaeIII showed the existence of well-resolved stable bands of 700 bp, 500 bp and 350 bp across all samples in both cultured and metagenomic subsets (Figure 1(a), Figure 1(b)). A distinctive 300 bp fragment in the pool that was conspicuous in smokers was also present in subjects with oral disease. Cluster analysis generated UPGMA dendrograms (Figure 2) for the respective

Figure 1. RFLP of HaeIII digest of the PCR product containing V1-V9 hypervariable region from oral metagenomic samples (a) and culture plate wash samples (b) separated on 3% agarose gel. ((a), (b)) Lane 1: Molecular weight marker (100 bp; Promega, Madison, WI), Lane 2-6: Healthy Non-smokers, Lane 7-11: Smokers, Lane 12-16 Oral Disease.
subsets with distinct clustering patterns. In the cultivated subset there was aclade (denoted by solid outline) that surprisingly included Smoker#3 and Healthy#3 (r = 91%, Cophenetic coefficient, CPCC = 79%) (Figure 2(a)). Overall however, disease group and the smokers demonstrated tight clustering. No discernable clear-cut clades/clustering were observed in the metagenomic samples (Figure 2(b)). However both dendrograms in Figure 2(a) and Figure 2(b) positioned Healthy #3 in close proximity with smokers (r > 95%), similar to the results from cultured bacteria community.
4.2. Restriction Fragment Length Polymorphisms of Ribotypes Generated by Sau3AI Restriction
Figure 3 shows the restriction fragment length polymorphisms generated by Sau3 A1 from the 16S rDNA gene product of oral bacteria community of healthy, smoker and oral disease subjects.
Although there were very little differences between the three study groups, with all showing 4 stable visible bands at 1000 bp/380bp/180bp/150bp; cluster analysis of the cultured community branched into two distinct groups I and II in Figure 4(b). Cluster II is predominantly disease group. Interestingly, Healthy #3 and Smoker #4 were all grouped in cluster II similar to results obtained from HaeIII restriction digest. Periodontal disease group were tightly clustered in the metagenomic subset. However, the smokers and healthy non-smokers were scattered generating no extrapolative cluster in Figure 4(a).
4.3. Restriction Fragment Length Polymorphisms of Ribotypes Generated by AluI Restriction
In both cultured plate wash and metagenomic extracts,bands of 1200 bp/900 bp/

Figure 2. UPGMA Cluster Analysis of PCR-RFLP DNA of cultured (a) and metagenomic (b) oral bacteria using HaeIII. Similarity matrices were calculated using Pearson correlation coefficient based on the densitometric curve. (a) HP1-5 Culture from Healthy non-smokers HPS1-5―Culture from Smokers, HPD1-5―Culture from oral disease subjects. (b) HH1-5―Metagenomic samples from Healthy non-smokers, HS 1-5 Metagenomic samples from Smokers, and HD 1-5 Metagenomic samples from disease subjects.

Figure 3. RFLP from Sau3A1 digest of the PCR product containing V1-V9 hypervariable regions from cultured oral bacteria separated on 3% agarose gel. Lanes1and 18: Molecular weight marker (100 bp; Promega Inc, Madison, WI), Lanes 3-7: Healthy Non-smokers, Lane 8-12: Smokers, Lanes 13-17 Disease subjects.

Figure 4. UPGMA Cluster Analysis of metagenomes and cultured oral bacterial ribotype using Sau3AI. Similarity matrices were calculated using Pearson correlation coefficient based on the densitometric curve. (a) SH―Healthy Metagenomic non-smokers, SS― Smokers Metagenomic, and SD―Disease Metagenomic from periodontal disease. On the scale r values are expressed as percentages (b) SP―cultured bacteria from healthy subjects, SPS―cultured smokers bacteria, SPD―cultured bacteria from disease subjects.
580 bp were present. Remarkably, the 450 bp length fragment which was distict in healthy and some smokers in the cultured plate wash category disappeared in 80% disease subjects (Figure 5(a)). The tight clustering of the healthy group in resultant dendrogram is noteworthy (Figure 5(b)). The smokers and disease samples were closely aligned sharing similarity as high as 95%.
4.4. PCR-DGGE Fingerprints of Ribotypes from Oral Bacteria of Healthy Subjects, Smokers and Disease Subjects
Gel Slab 1 contained 16S rDNA (V4) PCR products of Healthy non-smokers and Smokers while Gel Slab 2 contained amplicons from Smokers and Disease subjects. The culture independent method generated a more complex fingerprint compared to the culture dependent profile (figure not shown). UPGMA cluster analysis of Gel slab 1 (Healthy/Smokers) of the metagenomic subset did not demonstrate discernable clustering and was highly diverse (Figure 6(a)). In gel slab 2 (Smokers/Disease), a higher similarity was observed between groups with as high as 90% complementarity (Figure 6(b)).
On the other hand, the profile of the cultured set showed three sets of inimitable bands in smokers (Figure 7(a) Lane 6,7; Bands 3,4,5) and 4 bands in disease (Figure 7(a) Lanes 11,12; Bands 1,2,6,7). Culture dependent communities showed similar cluster alignments (Figure 7(b) & Figure 7(c)). There was widespread variations in smokers and healthy (Figure 7(b)). On the other hand close alignment as reported in the case of metagenomic community were noted

Figure 5. (a) RFLP analysis of AluI digest of the PCR product containing V1-V9 hypervariable region of cultured oral bacterial separated on 3% agarose gel. Lane 1,2: Molecular weight marker (100 bp; Promega Inc, Madison, WI), Lane 3-7: Healthy Non-smokers, Lane 8-12: Smokers, Lane 13-17 5 (b) UPGMA Cluster Analysis of composite dataset for cultured oral bacteria using AluI. Similarity matrices were calculated using Pearson correlation coefficient based on the densitometric curve. APH―Cultured from healthy subset, APS―Cultured from smoker subset, APD―from disease subset.

Figure 6. Pearson-UPGMA Cluster Analysis of metagenomic isolates of (a) healthy and smokers in Gel slab 1 and (b) Smokers and oral disease in Gel slab 2. H―Metagenomic isolates from healthy non-smokers, S―Metagenomic isolates from smokers, S#―Smokers in gel Slab 2, D―Disease. On the scale r-values are expressed as percentage.

Figure 7. (a) DGGE profile of PCR amplified 16srDNA segments of cultivated isolates. Lane 1-5 Healthy, Lane 6-10 smokers, Lane 11-15 Disease. Pearson-UPGMA Cluster Analysis of cultured isolates of (b) healthy and smokers in Gel slab 1 and (c) Smokers and oral disease in Gel slab 2. PH―healthy non-smokers, PS―smokers, #ps―Smokers in gel Slab 2, pd―Disease in gel slab 2. On the scale r-values are expressed as percentage.
between smokers and disease (Figure 7(c)).
It is noteworthy that the DGGE gel fingerprint generated clusters that showed similarity as high as 75% of Healthy #3 with smokers and Smoker #4 with disease (r = 0.85). This DGGE data is in accord with that of PCR-RFLP analysis reported above.
The similarity matrix of the DGGE patterns was also analyzed using multi-dimensional scaling (MDS) as shown in Figure 8.
It was not surprising that the mean numbers of detected amplicons were lower (13.2 ± 3.8) in the cultivated community that in the metagenomic group (20.4 ± 2.7). The banding patterns were significantly different between the groups as determined by the analysis of variance study in both communities (p = 0.009, ANOVA). Significant differences were also found in the number of bands between Healthy non-smokers and disease groups (P = 0.002; paired-sample Student’s t-test). However, the difference between smokers and disease groups were not significant (p = 0.47; paired-sample Student’s t-test); a result that is consistent with the outcome of MDS analysis above.
4.5. Comparative Analysis of Oral Bacteria Community Structure and Diversity in Cultured and Metagenomic Samples from Healthy, Smokers and Oral Disease Subjects
The Shannon Weiner diversity index calculated with GelCompareII software showed highest diversity in periodontal disease groups with a mean value of

Figure 8. Coordinate space window, resulting from multi-dimensional scaling (MDS) analysis of the similarity matrices derived from DGGE pattern analysis of (a) Cultured oral bacteria community and (b) Metagenomic oral bacteria community. The entries are shown as dots in a cubic coordinate system. Green: Healthy, Red: Smokers and Blue: Disease. The MDS statistic tool showed widespread distribution between smokers (red dots) and healthy (green dots). However, disease (blue dots) and smokers (red dots) were closely associated and signified relatedness between the group.
Table 2. Diversity indices calculated from the DGGE banding profiles of V4 region of bacterial 16S rDNA from the mouths of healthy non-smokers, smokers and disease study groups.
 = 1.06 ± 0.085 (p = 0.014). Community diversity and species richness was considerably low in healthy non-smokers Table 2.
= 1.06 ± 0.085 (p = 0.014). Community diversity and species richness was considerably low in healthy non-smokers Table 2.
Overall, diversity in healthy subjects was significantly high (p < 0.05) in metagenomic subset compared to that of cultivated (Figure 9).
5. Discussion
The Human oral microbiome database is replete of many important research that show not only the diversity of the human mouth but actual taxonomic ranks of these oral-bionts employing next generation sequencing and shot-gun sequencing technologies. The overarching quest is to understand the ecology and potential roles of the respective oral bacteria in health and or disease and to be able to use such knowledge in advancing dental and oral health. In this study, we tested our hypothesis that the community structure of healthy subjects will be different from that of disease patients, while the smokers bacteria will represent

Figure 9. Diversity calculated using the PCR-DGGE band profile of the oral bacteria in healthy non-smokers, smokers and disease subjects from (a) metagenomic samples and (b) cultivated samples. H―Healthy, S―Smoker, D―Disease.
a disturbed niche, clustering somewhere between the former groups. Whereas most studies have focused on the analysis of bacterial genomes directly extracted from the mouth (metagenomes) because of the detailed and deeper resolution it provides, taking into account cultured and non-cultured bacteria. This presumed advantage of metagenomic approach over cultured bacteria community (not isolates) analysis has been scarcely studied in healthy non-smokers, smokers and oral disease humans. In other words, will the cultured community structure be as useful as the metagenomics community in predicting health, transition to disease or outright disease state? For practical probiotic or diagnostic purposes, the bacterial communities or isolates need to be cultivable. An important feature of the experimental design in this study is the rigorous analysis of bacteria communities of same samples cultivated on a rich medium versus direct extraction. The PCR-DGGE procedure is well-suited for the detection of DNA polymorphisms in situations where RFLP is low but DNA sequence variations are expected but fidelity of the DNA fingerprints relies on the amount of samples used and the experimental conditions. We employed two old-fashioned methods PCR-RFLP and the PCR-DGGE under meticulously controlled laboratory conditions to analyze the community profile, diversity and bacterial signatures associated with variousoral health status. Although the resolution of members (taxa) of any bacterial consortium are bound to be low with the PCR-RFLP (since DNA fragments of same size but different sequence will band together); the DGGE protocol is known to distinguish between fragments with single nucleotide polymorphism (25).
The complex banding patterns generated by PCR-DGGE (13.2 ± 3.8 in cultivated and 20.4 ± 2.7 in metagenomic community subsets) represented the biological diversity of periodontal bacteria in the three states of oral health studied.
There was no significant difference between smokers and disease, suggesting similar bacterial community composition and diversity (p = 0.08, Student’s t-test). The diversity index (Table 2) was significantly lower in healthy groups (0.926 ± 0.07, p = 0.003) than in the other two groups. This suggests a more cohesive community which changes with gingival/sub-gingival inflammation resulting in increased proportion of inhabiting taxa. Indeed, Camelo-Castillo et al, 2015 demonstrated higher taxonomic diversity in dental plaques associated with no gingivitis compared to gingivitis [25] [26] . It is to be expected that the precise community profile in disease state will shift with the stage of illness and how chronic the condition has been. Subjects in this study had no chronic manifestation of the periodontal disease (no clinical examination was performed to evaluate their disease state) and may be at the inception phase where the homeostatic balance between the periodontopathic bacteria and host was disrupted. Cluster analysis, MDS and diversity indices clearly showed the smokers’ community in congruence with the disease group. Divergence among various studies could be explained by differences in patient populations, microbial sampling techniques, number of samples studied, detection methods of putative pathogens, species examined and also differences in evaluation and expression of data (counts versus proportions versus site or subject prevalence) [27] .
It is remarkable that bacterial diversity in healthy subjects was significantly high (p < 0.05) in metagenomic subset compared to that of cultivated subsets for all the samples (Figure 9). By contrast the diversity profile for smokers and disease subjects were similar for metagenomics and cultured communities. This strongly suggests that in the healthy oral state, there is a large variety of yet-to-be-cultured bacteria unlike the bacterial consortium of individuals with changing (declining) oral health. In fact, studies have shown that some bacteria associated with health are poorly known, and include members of TM7 phylum, a bacterial group which does not have a single member cultured in the laboratory but which appears to be widely present in the oral cavity [12] . These data underscore the need to develop culture techniques that improve recovery of bacteria of a healthy human mouth while indicating that disturbed or oral disease mouths are replete of cultivable members.
The PCR-RFLP technique spanned the nine hypervariable regions (V1-V9) of the 16S gene to improve it resolution of the bacterial consortia. It generated a set of stable and consistent profile in the three study groups as did the PCR-DGGE confirming the observed trends. Previous reports have found the existence of a so-called core bacteria in the mouth of smokers and non-smokers despite enormous variations between subjects. The prevalent genera of this core bacteria included Streptococcus, phylum Candidatus, Prevotella and Xanthomonas spp [28] [29] .
Distinctive bands were consistent for healthy and disturbed oral niche (Figure 7(a)) and may serve as bacterial predictors of oral health conditions. Sequencing these bands in future studies will shed further light on their roles.
The rigorous microbial diversity and cluster analysis of the three study groups, clearly shows that the bacteria consortium of smokers is the intersecting group between health and periodontal disease (Table 2; Figure 6 and Figure 7).
6. Conclusion
The experiment was successful in determining not only that the oral microbes can be indicative of health, but also indicative of the level of health of the host. This study has added a layer to the developing understanding of the role of microbes in health by proving that microbial communities can be indicative of the health status of its host.
Acknowledgements
This work has been supported in part by the FAU foundation grant to Microbial Biotech Lab and FAU graduate research grant. We also thank the Department of Biological Sciences at Florida Atlantic University.
Conflict of Interest
The authors declare no conflict of interest.
Cite this paper
Chakraborty, S. and Esiobu, N. (2017) Metagenomic rDNA Profiles of the Oral Microbiome in Healthy Subjects Are Distinctive Compared to That in Smokers and Disease Subjects Despite High Variations between Subjects. Advances in Microbiology, 7, 158-174. https://doi.org/10.4236/aim.2017.72013
References
- 1. Chen, T., Yu, W-Han, Izard, J., Baranova, O.V., Lakshmanan, A. and Dewhirst, F.E. (2010) The Human Oral Microbiome Database: A Web Accessible Resource for Investigating Oral Microbe Taxonomic and Genomic Information. Database, 2010, Article ID: baq013.
- 2. Al-Hebshi, N.N., Nasher, A.T., Idris, A.M. and Chen, T. (2015). Robust Species Taxonomy Assignment Algorithm for 16S rRNA NGS Reads: Application to Oral Carcinoma Samples. Journal of Oral Microbiology, 7. https://doi.org/10.3402/jom.v7.28934
- 3. Horz, H.-P. and Conrads, G. (2007) Diagnosis and Anti-Infective Therapy of Periodontitis. Expert Review of Anti-Infective Therapy, 5, 11. https://doi.org/10.1586/14787210.5.4.703
- 4. Paster, B.J., Boches, S. K., Galvin, J.L., Ericson, R.E., Lau, C.N., Levanos, V.A., Sahasrabudhe, A. and Dewhirst, F.E. (2001) Bacterial Diversity in Human Subgingival plaque. Journal of Bacteriology, 183, 3770-3783. https://doi.org/10.1128/JB.183.12.3770-3783.2001
- 5. Beck, J.D. and Offenbacher, S. (2005) Systemic Effects of Periodontitis: Epidemiology of Periodontal Disease and Cardiovascular Disease. Journal of Periodontology, 76, 2089-2100.
https://doi.org/10.1902/jop.2005.76.11-S.2089 - 6. Genco, R.J., Grossi, S.G., Ho, A., Nishimura, F. and Murayama, Y. (2005) A Proposed Model Linking Inflammation to Obesity, Diabetes, and Periodontal Infections. Journal of Periodontology, 76, 2075-2084. https://doi.org/10.1902/jop.2005.76.11-S.2075
- 7. Gill, S.R., Pop, M., Deboy, R.T., Eckburg, P.B., Turnbaugh, P.J., Samuel, B.S., Gordon, J.I., Relman, D.A., Fraser-Liggett, C.M. and Nelson, K.E. (2006) Metagenomic Analysis of the Human Distal Gut Microbiome. Science, 312, 1355-1359. https://doi.org/10.1126/science.1124234
- 8. Meurman, J.H. (2010) Oral Microbiota and Cancer. Journal of Oral Microbiology, 2, 5195.
https://doi.org/10.3402/jom.v2i0.5195 - 9. Ahn, J., Chen, C.Y. and Hayes, R.B. (2012) Oral Microbiome and Oral and Gastrointestinal Cancer Risk. Cancer Causes & Control, 23, 399-404.
https://doi.org/10.1007/s10552-011-9892-7 - 10. Brook, I. and Gober, A.E. (2005) Recovery of Potential Pathogens and Interfering Bacteria in the Nasopharynx of Smokers and Nonsmokers. Chest, 127, 2072-2075.
https://doi.org/10.1378/chest.127.6.2072 - 11. Charlson, E.S. (2010) Disordered Microbial Communities in the Upper Respiratory Tract of Cigarette Smokers. PLoS ONE, 5, e15216. https://doi.org/10.1371/journal.pone.0015216
- 12. Atanasova, K.R. and Yilmaz, Ö. (2015) Prelude to Oral Microbes and Chronic Diseases: Past, Present and Future. Microbes and Infection, 17, 473-483.
https://doi.org/10.1016/j.micinf.2015.03.007
http://www.sciencedirect.com/science/article/pii/S1286457915000507 - 13. Franzosa, E.A., Huang, K., Meadow, J.F., Gevers, D., Lemon, K.P., Bohannan, B.J.M. and Huttenhower, C. (2015) Identifying Personal Microbiomes Using Metagenomic Codes. PNAS, 112, E2930-E2938. https://doi.org/10.1073/pnas.1423854112
- 14. Anne-Lise, B., Hovig, E. and Brøgger, A. (1988) Detection of Base Mutations in Genomic DNA Using Denaturing Gradient Gel Electrophoresis (DGGE) Followed by Transfer and Hybridization with Gene-Specific Probes. Mutation Research/ Fundamental and Molecular Mechanisms of Mutagenesis, 202, 77-83.
https://doi.org/10.1016/0027-5107(88)90166-2 - 15. Jagathrakshakan, S.N., Sethumadhava, R.J., Mehta, D.T. and Ramanathan, A. (2015) 16S rRNA Gene-Based Metagenomic Analysis Identifies a Novel Bacterial Co-Prevalence Pattern in Dental Caries. European Journal of Dentistry, 9, 127-132. https://doi.org/10.4103/1305-7456.149661
- 16. Caporaso, J.G. (1998) QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nature Methods, 7, 335-420.
- 17. Martin-Laurent, F., Philippot, L., Hallet, S., Chaussod, R., Germon, J.C., Soulas, G. and Catroux, G. (2001) DNA Extraction from Soils: Old Bias for New Microbial Diversity Analysis Methods. Applied and Environmental Microbiology, 67, 2354-2359.
https://doi.org/10.1128/AEM.67.5.2354-2359.2001 - 18. Li, Y., Ku, C.Y.S., Xu, J., Saxena, D. and Caufield, P.W. (2005) Survey of Oral Microbial Diversity Using PCR-Based Denaturing Gradient Gel Electrophoresis. Journal of Dental Research, 84, 559-564. https://doi.org/10.1177/154405910508400614
- 19. Chen, Z., Trivedi, H.M., Chhun, N., et al. (2011) Using DGGE and 16S rRNA Gene Sequence Analysis to Evaluate Changes in Oral Bacterial Composition. The Chinese Journal of Dental Research, 14, 95-103.
- 20. Vauterin, L. and Vauterin, P. (1992) Computer-Aided Comparison of Electrophoretic Patterns for Grouping and Identification of Microorganisms. Environmental Microbiology, 1, 37-41.
- 21. Sneath, P.H.A. and Sokal, R.R. (1973) Numerical Taxonomy: The Principles and Practice of Numerical Classification. W. H. Freeman, San Francisco.
- 22. Smalla, K., Wieland, G., Buchner, A., Zock, A., Parzy, J., Kaiser, S., Roskot, N., Heuer, H. and Berg, G. (2001) Bulk and Rhizosphere Soil Bacterial Communities Studied by Denaturing Gradient Gel Electrophoresis: Plant-Dependent Enrichment and Seasonal Shifts Revealed. Applied and Environmental Microbiology, 67, 4742-4751.
https://doi.org/10.1128/AEM.67.10.4742-4751.2001 - 23. Shannon, C.E. and Weaver, W. (1949) The Mathematical Theory of Communication. University of Illinois Press, Urbana.
- 24. Pielou, E.C. (1969) An Introduction to Mathematical Ecology. Wiley, New York.
- 25. Camelo-Castillo, A.J., Mira, A., Pico, A., Nibali, L., Henderson, B., Donos, N. and Tomás, I. (2015) Subgingival Microbiota in Health Compared to Periodontitis and the Influence of Smoking. Frontiers in Microbiology, 6, 119. https://doi.org/10.3389/fmicb.2015.00119
- 26. Gafan, G.P. (2005) Statistical Analyses of Complex Denaturing Gradient Gel Electrophoresis Profiles. Journal of Clinical Microbiology, 43, 3971-3978.
https://doi.org/10.1128/JCM.43.8.3971-3978.2005 - 27. Socransky, S.S. and Haffajee, A.D. (1992) The Bacterial Etiology of Destructive Periodontal Disease: Current Concepts. Journal of Periodontology, 63, 322-331.
- 28. Dewhirst, F.E. (2010) The Human oral Microbiome. Journal of bacteriology 192, 5002-5017. https://doi.org/10.1128/JB.00542-10
- 29. Chakraborty, S., Persaud, V., Vanegas, S., Gautier, G. and Esiobu, N. (2014) Analysis of the Human Oral Microbiome of Smokers and Non-Smokers Using PCR-RFLP and Ribotyping. Advances in Microbiology, 4, 681-691. https://doi.org/10.4236/aim.2014.410073