Open Journal of Medical Microbiology
Vol.3 No.3(2013), Article ID:37052,6 pages DOI:10.4236/ojmm.2013.33026
A Simple LDT-PCR Method for Detection of Various Nucleic Acid Sequence Changes in a Small Region of Genes—Application for the Identification of Gene Mutations in MTB rpoB Gene Associated with Drug Resistance
Division of Biology, OSEL/CDRH/FDA
Email: *xing.tang@fda.hhs.gov
Copyright © 2013 Xing Tang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 10, 2013; revised May 10, 2013; accepted May 18, 2013
Keywords: Low Denaturing Temperature PCR (LDT-PCR); Mycobacterium Tuberculosis (MTB); Ribosome RNA Polymerase Subunit B (rpoB); Drug Resistance; Homoduplex; Heteroduplex; Microarray
ABSTRACT
A modified low denaturing temperature PCR (LDT-PCR) method combined with DNA microarray technique is developed in our lab for quick and effective identification of various mutations in an 81 base pair region of Mycobacterium Tuberculosis (MTB) ribosome RNA polymerase subunit B (rpoB) gene associated with rifampin resistance. By incurporation of wild type (wt) allele fragments that had been PCR amplified previously, the target PCR fragments coming from mutant clinical MTB samples were co-denaturized with incorporated wt type allele fragment at 94˚C and then let them randomly form matched structures (homoduplex) and allele mismatch-containing structures (heteroduplex), respectively, when the temperature cooled down to 70˚C. After the temperature was raised to 80˚C, the heteroduplex double stranded fragments were preferentially denatured and resulted in PCR amplification as well as fluorescence incurporation. Since the homoduplex fragments need a higher temperature to be denatured, they were kept in double-stranded status at that temperature and failed to be PCR amplified. By hybridization of LDT-PCR products with the probes spotted on microarray slides, the fluorescent signals representing the presence of gene mutations were detected. We have tested this method on 35 clinical MTB samples and obtained satisfied results.
1. Introduction
Tuberculosis is the second leading cause of adult mortality among infectious diseases and is responsible for about 2 million deaths per year worldwide. The World Health Organization has estimated that in the next two decades 150 million people will develop tuberculosis and 36 million individuals will die from tuberculosis if control of this disease is not improved [1]. Recent years, the success of tuberculosis control and treatment has been threatened by the emergence of drug-resistant tuberculosis strains [2]. This increasing prevalence of drug-resistance MTB infection has become a significant public health concern because of the difficulty and expense associated with clinical management. The diagnosis of MTB drug-resistance infections has been limited by the absence of rapid diagnostic techniques. Using sub-optimal procedures, two to four weeks are often required to identify drug-resistant microorganisms. Therefore, new molecular methods for the rapid detection of drug-resistant MTB are desperately needed.
Many of the mutations that cause drug resistance in M. tuberculosis are point mutations in known chromosomal genes [3]. For example, point mutations in the M. tuberculosis genes katG, which codes for catalase-peroxidase G, rpoB, which encodes the ribosomal RNA polymerase β subunit, and rpsL, which codes for ribosomal protein S12, are associated with resistance to isoniazid, rifampin and streptomycin, respectively. A number of molecular techniques are available for the detection of these point mutations in M. tuberculosis genes associated with drug resistance. Examples include direct sequencing of PCR products [4], PCR single-stranded conformational polymorphism analysis [4], molecular beacons [5], real-time PCR [6] and others [3]. However, many of these methods are either time-consuming or are not high-throughput. Microarray technology is widely being used for highthroughput, molecular studies on gene expression, genotyping, detection of microorganisms, detection of single nucleotide polymorphisms and detection of mutations. For example, microarrays combined with allele specific PCR have been used for genotyping of single nucleotide polymorphisms [7-9] and for mutation detection in cancer genes [10,11]. Oligonucleotide microarrays [12,13] have been used for detection of point mutations in M. tuberculosis rpoB genes associated with drug resistance. Our lab had developed a universal detection method combined DNA microarray and allele-specific techniques using tag-associated primers and successfully identified 10 different point mutations in MTB rpoB, katG, and rpsL genes [14].
MTB rpoB is the only target of rifampin which is the most important anti-TB drug. A region containing 81 base pairs of nucleic acids in rpoB gene has been found to be a key position corresponding to rifampin treatment; any mutations happened in this region and changed amino acid sequences will cause the failure of rifampin treatment [3]. To date, more than 60 different mutations, including point mutations, deletions, and insertions, have been found in this region and related to the drug resistance of rifampin. None of the methods mention above, except DNA sequencing, is capable of identifying all the mutations in a single measurement.
Recent years, a LDT-PCR method has been developed for selective identification of low-level unknown mutations [15-17].
In order to efficiently screen out drug-resistance MTB strains, our lab developed a modified LDT-PCR method combined with DNA microarray technique with a simple measurement to identify various nucleic acid mutations in MTB rpoB gene and obtained satisfactory results.
2. Materials and Methods
2.1. DNA Isolation
Strain H37Rv (No. 27294/TMC 102; ATCC*, Manassas, VA) served as the source of wild-type (i.e., natural sequence) M. tuberculosis genomic DNA. Clinical isolates of M. tuberculosis and the clinical drug resistance-susceptibility profiles of these isolates were obtained from collaborators at the National Institutes of Health (Bethesda, MD) and the Korean Institute of Tuberculosis, Korean National Tuberculosis Association (Seoul, South Korea). M. tuberculosis genomic DNAs were isolated (in a biosafety level 3 lab) from cultured bacteria using a standard phenol:chloroform method. Cultured M. tuberculosis was transferred to a 1.5 ml Eppendorf tube, pelleted by centrifugation and suspended in 0.36 ml of Marmur’s solution (0.1 M NaCl, 0.01 M EDTA, pH 8.0). Forty µl of 5% SDS, 2 µl of 20 mg/ml proteinase K, and 2 µl of 20 mg/ml RNase A were added to the suspension. The M. tuberculosis lysate was incubated at 37˚C overnight. An equal volume of buffersaturated phenol was added into the digest and thoroughly mixed by vortexing for 10 min. After centrifugation the top phase was transferred into a clean tube, extracted with an equal volume of chloroform:isoamyl alcohol (24:1) by vortexing for 10 min, and then centrifuged again. The top phase was transferred to a clean tube, and 0.1 volume of 3 M sodium acetate (pH 4.8) plus 2.5 volume of absolute ethanol were added. After vortexing briefly, the mixture was kept at −20˚C for 2 h and centrifuged at full speed for 15 min. The pellet was washed with 0.5 ml of 70% ethanol, dried, and suspended in 200 µl distilled water. The presence of genomic DNA was confirmed by agarose gel electrophoresis.
2.2. Oligonucleotide Primers and Probes
The design of PCR primers and probes used for microarray analysis was based on the published genome sequence of M. tuberculosis [14]. The sequences of forward and reverse primers are 5’-GATCAAGGAGTTCTTCG-3 and 5’-GCTCACGTGACAGACC-3’, respectively. This pair of primers generated a 123 base pair PCR fragment containing the key position in MTB rpoB gene. A 60 base pair microarray probe sequenced as 5’- CGACAGTCGGCGCTTGTGGGTCAACCCCGACAGCGGGTTGTTCTGGACCATGAATTGGCTCAGCTGGCTGGT-3’ was also designed based on the published sequence data of MTB rpoB gene. Both primers and probe were prepared by the Department of Health and Human Services, Center for Biologics Evaluation and Research Facility for Biotechnology Resources (U.S. Food and Drug Administration, Rockville, MD) with a Model 394 automated DNA synthesizer (Applied Biosystems, Foster City, CA) using cyanoethyl phosphoramidite nucleosides. The DNA probe was amine-group linked for a tight attachment to the slide.
2.3. PCR and LDT-PCR
This experiment was composed of two steps of PCRs, or conventional PCR and LDT-PCR, both were performed in a GeneAmp® PCR Instrument System 9600 (Applied Biosystems, Foster City, CA).
For conventional PCR, the following protocol was used: Each 20 µl of PCR reagent contained 1× TaKaRa buffer, 200 µM each of dNTPs, 5 pM each of forward and reverses, 50 - 100 ng of M. tuberculosis DNA, and 1 units of TaKaRa Taq DNA polymerase. Cycling consisted of 94˚C for 3 min, followed by 35 cycles of 94˚C for 30 sec, 54˚C for 20 sec, and 72˚C for 45 sec. The final extension was 72˚C for 5 min. The size of the PCR products was analyzed by agarose gel electrophoresis. 5 µl of each PCR product was loaded in each lane.
For LDT-PCR, each 20 µl of PCR reagent contained 1× TaKaRa buffer, 5 pM each of forward and reverses, 1 µl of conventional PCR products from detected M. tuberculosis DNA sample and 1 µl of conventional PCR product from wild type H37Rv sample, 0.5 µM of Cy5 dye-labeled dCTP, and 1 units of TaKaRa Taq DNA polymerase. Cycling consisted of 94˚C for 2 min and then cooled down to 70˚C for 10 min, followed by 1 cycle of 80˚C for 30 sec, 54˚C for 20 sec, and 72˚C for 2 min. The LDT-PCR products were purified with ethanol precipitation method and finally eluted into 30 µl of distilled water.
2.4. DNA Sequencing
For verification of the wild type and mutant clinical isolates of M. tuberculosis strains, all the PCR products from the samples used in this study were sequenced by the Core Lab in the Department of Health and Human Services, Center for Biologics Evaluation and Research Facility for Biotechnology Resources (U.S. Food and Drug Administration, Rockville, MD).
2.5. Microarray Chip Preparation
DNA oligonucleotide probes were diluted in spotting solution (50 M NaOH, 0.01% SDS) at a concentration of 100 µM and spotted onto CSS-silylated glass microscope slides (Telechem International Inc., Fisher Scientific, Suwanee, GA) with an OmniGrid AccentTM robotic microarrayer (Genomic Solutions, Ann Arbor, MI). The average spot size was 400 µm. Total 8 sub-arrays, each contained double spots of same rpoB probe, were spotted on each slide.
2.6. Hybridization
The probe slides were pre-treated with 0.2% SDS at room temperature overnight and washed with distilled water 3 times for 5 minutes followed by rinsed in isopropanol for 1 min and than air dried. For hybridization of dye-labeled DNA targets and oligonucleotide probes, an equal volume of purified targets was mixed with hybridization solution (50% formamide, 10× SSC, 0.2% SDS, 0.2 mg/ml sperm DNA). Three µl of mixed target solution was added to one of subarray probe areas on the slide, covered with a cover slip and hybridized at room temperture in a moisturized chamber for 4 - 6 hours. This was followed with a series of washing solutions that descended from 4× SSC to 0.5× SSC, each for 5 min at room temperature. The results of hybridization on slides were visualized using a GenePix 4000B Array Scanner (Axon Instruments, Inc., Union City, CA) at 635 nm (Cy5). The data were analyzed with GenePix Pro 4.0 software (Axon Instruments, Inc.). For the data figures presented in this paper, each pattern of red spots was reproduced from the scanner computer screen diagram. Each red spot represents a successful hybridization between a specific Cy5 fluorescent dye-labeled target DNA and oligonucleotide probe.
3. Result and Discussion
3.1. Conventional PCR
Figure 1 shows the results of conventional PCR after 35 cycle amplification. A 123 bp DNA fragment was generated in all the samples including H37Rv wt control. The intensity of the fragment in each lane looked similar with clear background that represented the specificity of PCR reactions.
3.2. LDT-PCR
The designation of LDT-PCR was based on following idea: In a short DNA fragment (<200 bp), if there is a nucleic acid mismatched structure, the fragment will be denatured at a lower denaturing temperature as compared with non-mismatched fragment. Since the binding of primers to their positions on template and the extension of PCR can only be processed in a complete denaturing status of the template. Thus, a suitable lower denaturing temperature that should enable double-strands of mismatched template to be separated but without completely denaturing homological template will result in mutant structure amplification by PCR.
In our experiment, after being heated at 94˚C for 2 min, both allele rpoB PCR fragments, which had been amplified by previous conventional PCR from an unknown sample and a wild type (H37Rv) sample respectively and co-existed in a same PCR tube, would be completely denatured. After the temperature cooled down to 70˚C and left for 10 min, the denatured fragments would randomly re-natured to form four different combinations in a consistent ratio as about one-fourth of each combination. If there were a mutation in unknown sample fragments, the half the mutant fragments would form a heteroallele structure with wild type fragments at 70˚C (Figure 2). In the next step, when the temperature wasraised to 80˚C, only fragments with heteroallele structure are denatured because one base mismatch in its structure would enable the primers to be seated at their specific positions; the fragments with homological structure would keep double strand status at this temperature andthe primers would failed to bind to their positions. Thus, after the temperature changed to 72˚C, only the fragments with heteroallele structure could be successfully
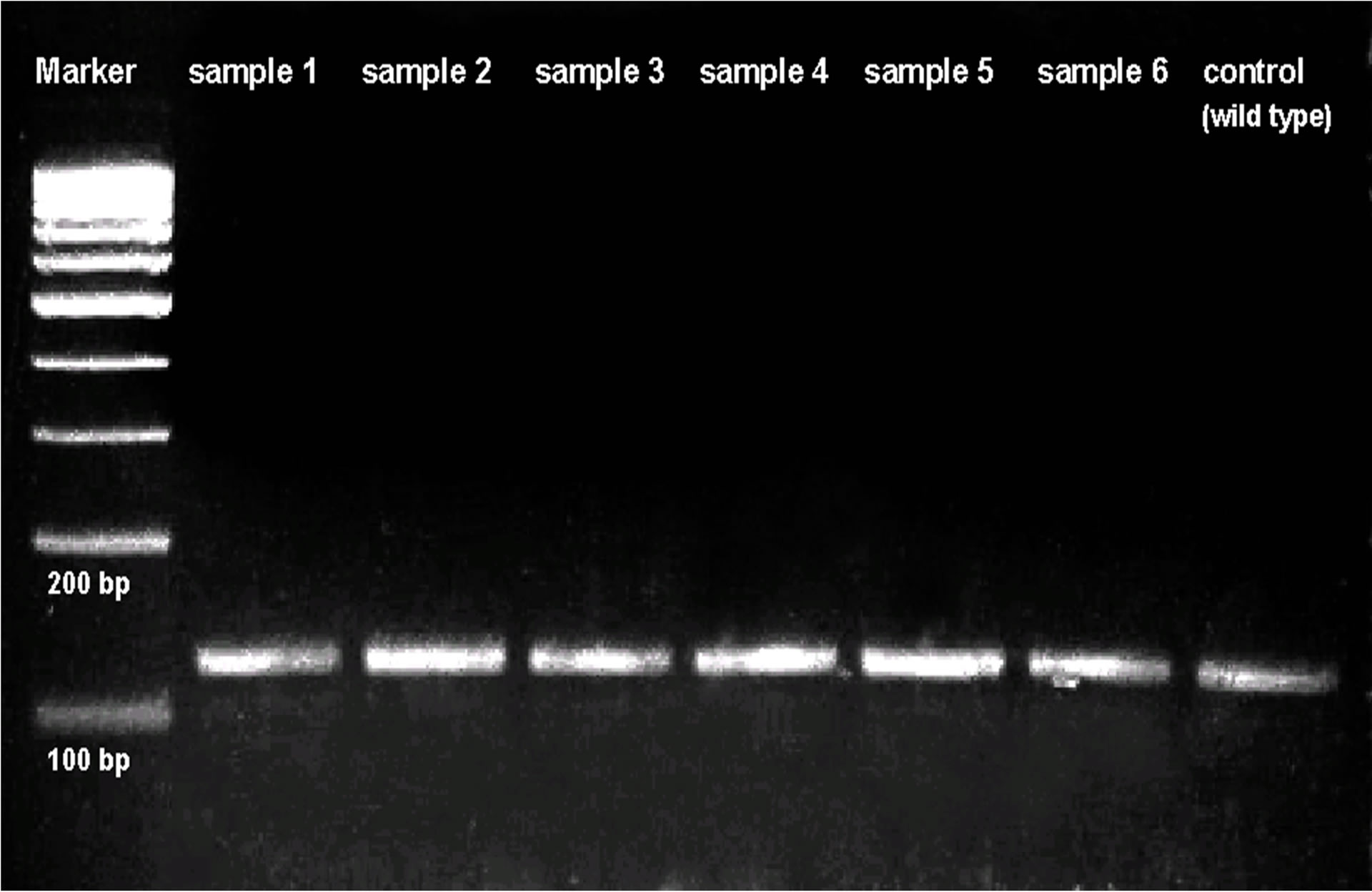
Figure 1. After 35 cycle amplification of conventional PCR, a 123 bp DNA fragment was generated in each of the samples including H37Rv wt control.
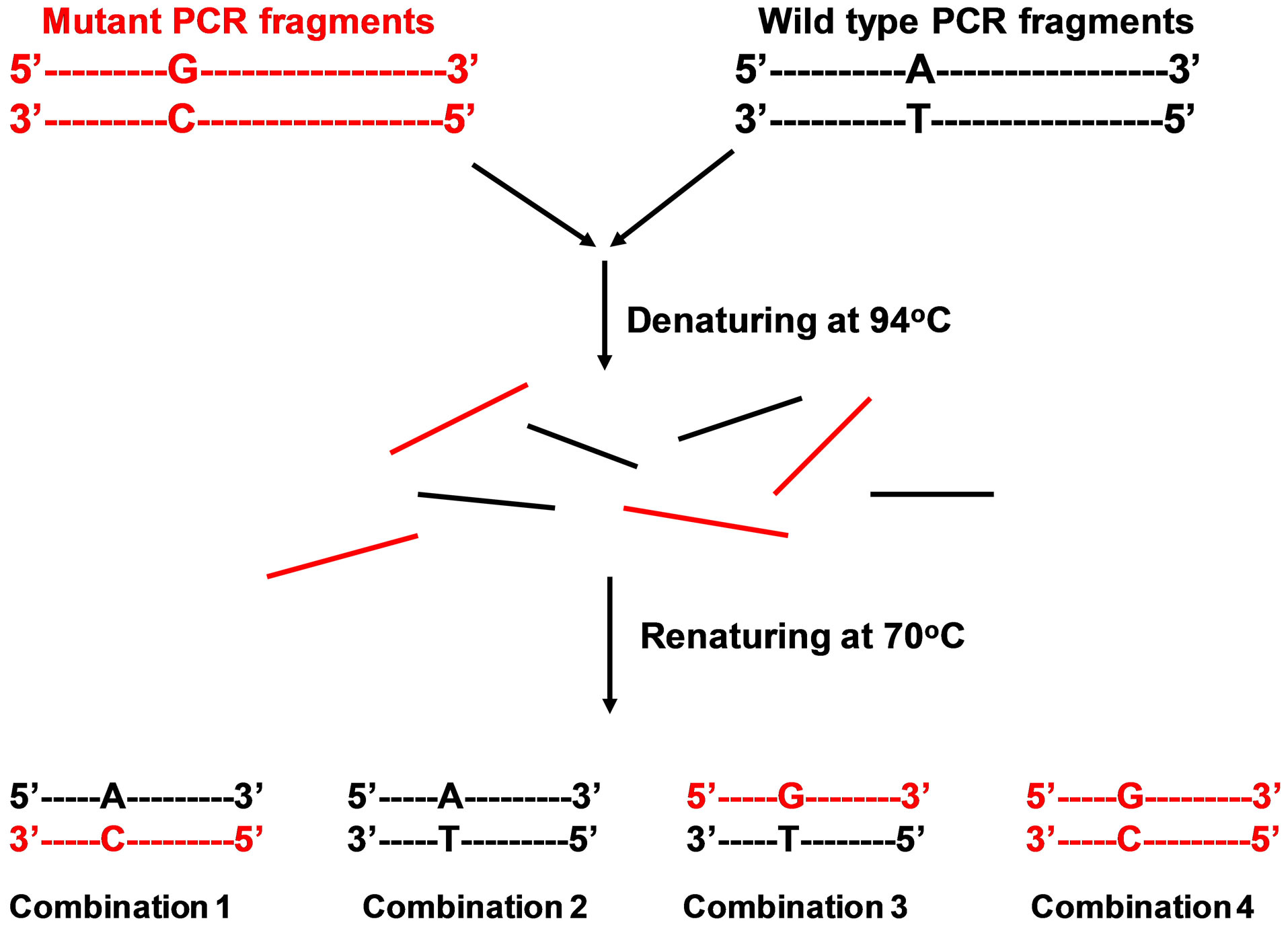
Figure 2. This diagram shows 4 hybridization combinations of wild and mutant PCR fragments formed after denature and re-nature procedure representing about one-fourth of each combination. Half of the mutant fragments form hetero-allele structures with wild type fragments.
copied by PCR extension and resulted in fluorescence incorporation (Figure 3). If the detected sample had no mutation (wild type), all the combinations would be homological structures, none of them would be completely denaturized at the 80˚C and thus no PCR extension and fluorescence incorporation would happen in the reaction. After LDT-PCR, each sample was purified by ethanol precipitation and then hybridized to the probes spotted on microarray chips for verify detection effecttiveness of LDT-PCR.
3.3. Microarray Hybridization
Figure 4 showed hybridization results of 6 subarrays hybridized with LDT-PCR products from samples of wild type, mutation 516, mutation 526, and mutation 531, respectively. The mutation 516 is A to T mutation and has a least binding force changes (2 hydrogen bonds), thus it showed a weaker signal (see right middle subarray

Figure 3. At a lower denaturing temperature (80˚C), only the hetero-allele fragments are denatured because of the one base mismatch in their structures, thus are enabling enables the primer binding and PCR extension as well as fluorescence (*Cy5-dCTP) incorporation. Fragments with homological structure in contrast, would keep double strand status at this temperature and the primers would fail to bind to their positions resulting no PCR extension.
in Figure 4); mutations at the positions 526 and 531 are both C to T mutations and have stronger binding force changes (3 hydrogen bonds), they have a more complete denaturizing status at 80˚C and result in a better PCR results, thus express stronger hybridization signals (see left 3 subarrays and right bottom subarray in Figure 4). Wild type sample has no mutation in its sequence and shows a very weak signal (see right top subarray in Figure 4) or no signal.
Based on the sequencing results, 6 of the 35 samples we tested have mutations at position 531; 2 of them have mutations at position 526, and one has mutation at position 516. Rests of the samples are wild type with no mutation in the detecting area. With the method we developed, all 9 mutant samples showed positive signals as displayed in Figure 4. Twenty-four of the wild type samples showed no signal or very weak signals. Only 2 of the wild type samples showed signals for unidentified reason, maybe a result of sample contamination or coinfection of both wild type and mutant MTB in a same patient. All the samples were tested in duplicate and obtained similar results.
4. Summary
A simple and reliable method for identification of various type nucleic acid mutations in a small area of DNA sequence has been developed. The method can be used to quickly screen out point mutation associated drug-resistant MTB strains from clinical samples. We believe thatthis detection method can be employed to identify various

Figure 4. The detection results of 6 DNA subarrays at the same slide hybridized with LDT-PCR products from samples of wild type, mutation 516, mutation 526, and mutation 531, respectively. The mutation 516 (right middle subarray) shows a weaker signal because of it is an A to T mutation and has the least binding force changes (2 hydrogen bonds). Mutations at the positions 526 and 531 are both C to T mutations and have bigger binding force changes (3 hydrogen bonds), thus they show better PCR results and stronger signals (see left 3 subarrays and right bottom subarray). Wild type sample has no mutation in its sequence and shows a very weak signal (see right top subarray) or no signal.
genomic mutations in other applications. A suitable lower denaturing temperature for a specific target is critical in this method. The determination of this suitable temperature depends on the size and nucleic acid compositions of amplified PCR fragment.
REFERENCES
- M. A. Espinal, A. Laszlo, L. Simonsen, F. Boulahbal, S. J. Kim, A. Reniero, S. Hoffner, H. L. Rieder, N. Binkin, C. Dye, R. Williams and M. C. Ravigilione, “Global Trends in Resistance to Antituberculosis Drugs. World Health Organization—International Union against Tuberculosis and Lung Disease Working Group on Anti-Tuberculosis Drug Resistance Surveillance,” The New England Journal of Medicine, Vol. 344, 2001, pp. 1294-1303. doi:10.1056/NEJM200104263441706
- C. Dye, M. A. Espinal, C. J. Watt, C. Mbiaga and B. G. Williams, “Worldwide Incidence of Multidrug-Resistant Tuberculosis,” Journal of Infectious Diseases, Vol. 185, No. 8, 2002, pp. 1197-1202. doi:10.1086/339818
- S. Ramaswamy and J. M. Musser, “Molecular Genetic Basis of Antimicrobial Agent Resistance in Mycobacterium tuberculosis: 1998 Update,” The International Journal of Tuberculosis and Lung Disease, Vol. 79, No. 1, 1998, pp. 3-29. doi:10.1054/tuld.1998.0002
- D. A. Rouse, Z. Li, G. H. Bai and S. L. Morris, “Characterization of the katG and inhA Genes of Isoniazid-Resistant Clinical Isolates of Mycobacterium tuberculosis,” Antimicrobial Agents and Chemotherapy, Vol. 39, No. 11, 1995, pp. 2472-2477. doi:10.1128/AAC.39.11.2472
- A. S. Piatek, A. Telenti, M. R. Murray, H. El-Haij, W. R. Jacobs, F. R. Kramer and D. Alland, “Genotypic Analysis of Mycobacterium tuberculosis in Two Distinct Populations Using Molecular Beacons: Implications for Rapid Susceptibility Testing,” Antimicrobial Agents and Chemotherapy, Vol. 44, No. 1, 2000, pp. 103-110. doi:10.1128/AAC.44.1.103-110.2000
- H. R. Van Doorn, E. C. J. Class, K. E. Templeton, A. G. M. van der Zanden, , A. te Koppele Vije, M. D. de Jong, J. Dankert and E. J. Kuijper, “Detection of a Point Mutation Associated with High-Level Isoniazid Resistance in Mycobacterium tuberculosis by Using Real-Time PCR Technology with 3’-Minor Groove Binder-DNA Probes,” Journal of Clinical Microbiology, Vol. 41, No. 10, 2003, pp. 4630-4635. doi:10.1128/JCM.41.10.4630-4635.2003
- A. J. Flavell, V. N. Bolshakov, A. Booth, R. Jing, J. Russell, T. H. Ellis and P. Isaac, “A Microarray-Based High Throughput Molecular Marker Genotyping Method: The Tagged Microarray Marker (TAM) Approach,” Nucleic Acids Research, Vol. 31, No. 19, 2003, p. e115. doi:10.1093/nar/gng113
- D. O’Meara, A. Ahmadian, J. Odeberg and J. Lundeberg, “SNP Typing by Apyrase-Mediated Allele-Specific Primer Extension on DNA Microarrays,” Nucleic Acids Research, Vol. 30, No. 15, 2002, p. e75. doi:10.1093/nar/gnf074
- C. Su, C. Hott, B. H. Brownstein and L. D. Sibley, “Typing Single-Nucleotide Polymorphisms in Toxoplasma Gondii by Allele-Specific Primer Extension and Microarray Detection,” Methods in Molecular Biology, Vol. 270, 2004, pp. 249-262.
- O. Ericsson, A. Sivertsson, J. Lundeberg and A. Ahmadian, “Microarray-Based Resequencing by Apyrase-Mediated Allele-Specific Extension,” Electrophoresis, Vol. 24, No. 19-20, 2003, pp. 3330-3338. doi:10.1002/elps.200305583
- N. P. Gerry, N. E. Witowski, J. Day, R. P. Hammer, G. Barany and F. Barany, “Universal DNA Microarray Method for Multiplex Detection of Low Abundance Point Mutations,” Journal of Molecular Biology, Vol. 292, No. 2, 1999, pp. 251-262. doi:10.1006/jmbi.1999.3063
- A. Troesch, H. Nguyen, C. G. Miyada, S. Desvarenne, T. R. Gingeras, P. M. Kaplan, P. Cros and C. Mabilat, “Mycobacterium Species Identification and Rifampin Resistance Testing with High-Density DNA Probe Arrays,” Journal of Clinical Microbiology, Vol. 37, No. 1, 1999, pp. 49-55.
- J. Yue, W. Shi, J. Xie, Y. Li, E. Zeng, L. Liang and H. Wang, “Detection of Rifampin-Resistant Mycobacterium tuberculosis Strains by Using a Specialized Oligonucleotide Microarray,” Diagnostic Microbiology and Infectious Disease, Vol. 48, No. 1, 2004, pp. 47-54. doi:10.1016/j.diagmicrobio.2003.08.005
- X. Tang, L. M. Sheldon, J. L. Langone and L. E. Bockstahler, “Microarray and Allele Specific PCR Detection of Point Mutations in Mycobacterium Tuberculosis Genes Associated with Drug Resistance,” Journal of Microbiological Methods, Vol. 63, No. 3, 2006, pp. 318-330. doi:10.1016/j.mimet.2005.04.026
- C. A. Milbury, J. Li and G. M. Makrigiorgos, “COLDPCR-Enhanced High-Resolution Melting Enables Rapid and Selective Identification of Low-Level Unknown Mutations,” Clinical Chemistry, Vol. 55, No. 12, 2009, pp. 2130-2143. doi:10.1373/clinchem.2009.131029
- J. Li, C. A. Milbury, C. Li and G. M. Makrigiorgos, “Two-Round Coamplification at Lower Denaturation Temperature-PCR (COLD-PCR)-Based Sanger Sequencing Identifies a Novel Spectrum of Low-Level Mutations in Lung Adenocarcinoma,” Human Mutation, Vol. 30, No. 11, 2009, pp. 1583-1590. doi:10.1002/humu.21112
- I. Mancini, C. Santucci, R. Sestini, L. Simi, N. Pratesi, F. Cianchi, R. Valanzano, P. Pinzani and C. Orlando, “The Use of COLD-PCR and High-Resolution Melting Analysis Improves the Limit of Detection of KRAS and BRAF Mutations in Colorectal Cancer,” The Journal of Molecular Diagnostics, Vol. 12, No. 5, 2010, pp. 705-711. doi:10.2353/jmoldx.2010.100018
NOTES
*Corresponding author.