Journal of Sustainable Bioenergy Systems
Vol.04 No.03(2014), Article ID:50260,10 pages
10.4236/jsbs.2014.43017
Impact of Chain Length of Saturated Fatty Acids during Their Heterogeneously Catalyzed Deoxygenation
Shrikant Mohite1, Udo Armbruster2, Manfred Richter2, Andreas Martin2*
1ARMACO Chemical Process Systems, Mumbai, India
2Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Rostock, Germany
Email: *andreas.martin@catalysis.de
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 14 July 2014; revised 15 August 2014; accepted 5 September 2014
ABSTRACT
Fatty acids with different chain length were deoxygenated in the absence of hydrogen (caprylic acid (CA), lauric acid (LA) and stearic acid (SA)). The catalytic tests were carried over Pd-contain- ing catalysts out in a batch reactor under inert gas for 6 h at 250˚C to 350˚C and pressures from 18 to 75 bar in the absence of additionally fed hydrogen. Pd-containing catalysts were tested; the best performing catalyst was 10% Pd/C with 63% undecane yield at 327˚C. These catalysts were used for a comparative decarboxylation of CA, LA and SA. At equal reaction conditions (300˚C, 6 h), the chain length of the fatty acid had a strong impact on the conversion, which was steadily increasing, whereas the alkane selectivity ran through a maximum. This work demonstrated the usability of Pd-containing catalysts for the decarboxylation of various fatty acids in the absence of additionally fed hydrogen with respect to the manufacture of hydrocarbons that can be used as blending components for fuels.
Keywords:
Deoxygenation, Decarboxylation, Fatty Acids, Long-Chain Alkanes, Catalysts

1. Introduction
The increasing energy demand and depleting petroleum reserves stimulated the research towards finding alternatives for fossil fuels. At present, about 210 million metric tons of diesel fuel is consumed in EU27 every year [1] . During the last ten years, plant oils from different sources increasingly have served as sustainable feed stock to produce the so-called “Biodiesel” being manufactured by transesterification of triglycerides with methanol to fatty acid methyl esters (FAME) [2] . FAME was produced in EU27 with 11.2 million metric tons per annum in 2010 [1] (16.5 million metric tons worldwide [3] ). However, it makes only 4.5% of the EU27 diesel fuel consumption. FAME has a high cetane number and caloric values close to fossil-based diesel and, in general, it is blended to conventional diesel (B7 variant in Germany, i.e., 7% FAME admixture). However, the unsaturation in the chain and oxygen content lead to instability that affects the chemical properties and leads to corrosion of wetted materials like polymer gaskets. Thus, various attempts were carried out in the recent years on hydroprocessing and up-grade of biomass derived oils aiming at using such feedstock as blending component for fuels [4] [5] .
Usually, transesterifications carried out in the presence of homogeneous basic catalysts (e.g. sodium or potassium hydroxide or methanolate) [2] . Anyway, feedstock containing high free fatty acid content enhances saponification with such catalysts, and potassium or sodium soaps are hard to remove from aqueous glycerol phase [6] . Thus, such raw material is treated in the presence of acidic catalysts (e.g. H2SO4). In addition, various attempts are known to develop heterogeneous catalysts based on CaO [7] or CsF/CaO [8] due to their process advantages or to immobilize enzymes for their use as biocatalyst [9] , otherwise the reaction proceeds at a slower rate and the catalysts may suffer from deactivation.
To make “Biodiesel” equal to diesel fuel and to overcome the above mentioned drawbacks, oxygen and unsaturation have to be removed from the raw materials by cracking, hydrogen treatment and/or deoxygenation, for example.
There are several routes for deoxygenation of free fatty acids. Removal of oxygen as CO2 (giving alkane) and/or CO (giving alkene) is thermodynamically more favorable than removing oxygen as molecular O2 according to free enthalpy values [10] . Decarboxylation seems to be more favorable than decarbonylation, because Gibb’s free energy is lower, and in addition, required heat of reaction is much less [10] . Additionally, in the presence of hydrogen, fatty acids could be hydro-decarbonylated to get alkane, carbon monoxide and water or be hydrogenated to form straight chains with methyl group in end position and water [10] [11] .
In 1936, Bertram [12] presented a first short report on decarboxylation of stearic acid (SA, C18:0) by using grey selenium at 310˚C - 325˚C (50% conversion after 15 h). Some further reports on transition metal (Pd, Rh) complexes as homogeneous catalysts followed, e.g. for decarbonylation of stearic acid into a mixture of heptadecenes [13] . Maier et al. [14] carried out gas phase reactions on Pd/SiO2 catalyst in the presence of hydrogen and suggested that lower reaction temperature is preferable as it reduces the possibility of aromatization. These runs revealed 98% conversion of heptanoic acid and 97% conversion of octanoic acid. The authors also reported 20% conversion of heptanoic acid and 64% conversion of octanoic acid, respectively, when using Ni/Al2O3 catalyst. Very recently, Ford et al. reported on the use of supported Pd catalysts for the deoxygenation of various carboxylic acids in the C10 - C18 range at 300˚C under 5% H2 atmosphere [15] . Ping et al. recently described the decarboxylation of stearic acid in the absence of hydrogen (N2, 300˚C, 6 h, 85% - 90% conversion, 100% selectivity to n-heptadecane) over 5% Pd supported catalyst using an ultra-porous silica mesocellular foam [16] . The authors reported on a very uniform distribution of ~2 nm particles received from microscopic and chemisorption results. A review of Santillan-Jimenez and Crocker summarizes the progress on the deoxygenation/decarbony- lation of fatty acids of the last years from different points of view [17] .
Pure carboxylic acids were pyrolyzed to hydrocarbons on activated alumina at 450˚C and ambient pressure as reported by Leung et al. [18] These conditions favor decarbonylation more than decarboxylation; ketones were identified as intermediates [19] Moreover, in these studies the yield of alkene increased with acid chain length. Pestman et al. and Sugiyama et al. found ketones and aldehydes from carboxylic acids, too [20] - [22] . In hydrocracking batch experiments carried out by Gusmão et al. [23] , positional isomers of heptadecene and constitutional isomers of substituted aromatics (undecylbenzene) were detected.
Very systematic research work on decarboxylation of fatty acids was published during the last decade by the group of D. Yu. Murzin from Åbo Akademi University, Finland. Kubicková et al. reported on the decarboxylation of SA, ethyl stearate and tristearine over carbon supported metal catalysts leading to heptadecane [24] . There were no evidences for the formation of fatty alcohols and almost no octadecane was found, suggesting that the used catalysts were not able to promote hydrodecarboxylation [21] . Snåre et al. [10] reported on the decarboxylation of SA to heptadecane in a semi-batch reactor at 300˚C and ca. 6 bar using dodecane as solvent. Various noble metal- (Pd, Pt, Ir, Ru, Rh and Os) and nickel- or molybdenum-containing catalysts using carbon, magnesia, silica or alumina as carriers were tested. A complete conversion of SA with selectivity to heptadecane of ca. 99% was reached over a 5% Pd/C catalyst. Less palladium (1% Pd/C) gave less conversion and selectivity whereas higher Pd proportion (10% Pd/C) surprisingly led to poor performance. Apart from carbon-supported Pd catalysts, Ni exhibited good hydrocarbon selectivity on SiO2 and Cr2O3 supports. Other catalysts showed much less conversion and selectivity towards heptadecane; besides, some heptadecenes, cracking and heavier products were found. Simakova et al. also reported on the decarboxylation of different fatty acids (C17 - C20 and C22) in a semi-batch reactor over Pd supported on mesoporous carbon (Sibunit) [25] [26] The same group published further results of their work; they reported on linearly increasing conversion of stearic acid with temperature but inversely affected selectivity to alkanes over 5%Pd/C. The yield of aromatics increased by 9-fold with rising of temperature from 300˚C - 360˚C. [24] Moreover, high stirrer speed (1100 rpm) and low catalyst particle diameter established reaction without mass transfer limitation [27] . Rate of formation of n-heptadecane was doubled with decrease in SA concentration by one half. Impurities present in the feed such as palmitic acid (PA, C16:0), margaric acid (MA, C17:0) and arachidic acid (AA, C20:0) gave corresponding products. Nevertheless, selectivity of alkane was decreased with acid chain length. Reaction with behenic acid (BA, C22:0) gave less conversion as well as selectivity than with SA. All this research contributed to the periphery of Neste Oil’s (Porvoo, Finland) NExBTL process industrially converting triglycerides and/or fatty acids to hydrotreated vegetable oil (HVO) as diesel fuel blends [28] .
The conversion of biomass derived fatty acids and/or their esters towards hydrocarbons by decarboxylation may improve their properties for fuel applications. Although some research on decarboxylation is known, further improvements are required. In particular, suitable catalysts and the renouncement of additionally fed hydrogen might be future benefits. The presented concept using highly active and selective Pd-containing catalysts that work well in the absence of additional hydrogen seem to have future practical importance.
The primary objective of this work was a comparative study on the deoxygenation (decarboxylation) of model fatty acids (caprylic acid, CA (C8:0), lauric acid, LA (C12:0) and stearic acid, SA (C18:0)) to the corresponding alkanes containing one carbon atom less than the original molecule. CA and LA are found naturally in the milk of various mammals, in particular goats (CA), and they are a constituent of coconut oil and palm kernel oil (up to ca. 10%). These acids have niche applications only. SA is one of the most common saturated fatty acids found in most of the natural fats and oils as triglyceride and is mainly used for soaps, cosmetics and detergents. Pd-containing carbon supported catalysts were synthesized; batch tests were carried out in the absence of hydrogen to evaluate the potential of the solids in the decarboxylation reaction towards hydrocarbons and to determine activity and selectivity changes caused by different chain lengths.
2. Experimental
2.1. Catalyst Syntheses
Various Pd/C catalysts using a carbon support (650 m2/g, Merck) were prepared by a simple impregnation technique. The support material used for the catalyst syntheses was ground and dried for 15 h at 120˚C to remove adsorbed moisture. Required amount of Pd precursors was initially dissolved in diluted HCl; (e.g. 0.8331 g of PdCl2 (Acros) for 5% Pd/C) for preparation of catalysts with 1, 5 and 10 wt% loading. The solution was warmed for complete dissolution of PdCl2. The support material was added to the solution and stirred for 1 h. The solution was heated up to 100˚C for 5 - 7 h under constant stirring for water removal, followed by drying overnight (120˚C, 16 h). The as-synthesized catalyst precursors were calcined at 300˚C (N2, air, 4 h, 5 K/min). The subsequent reduction was carried out in-situ prior to reaction as described below.
2.2. Set-Up and Experimental Design
Figure 1 shows a sketch of the used set-up. A high-pressure compact 25 ml lab autoclave (Parr Instruments, Inc.) was used for the reactions. Dodecane (Acros) and tetradecane (Fluka) were chosen as solvents. For solvent addition, a cylinder with two inlets for N2 flushing was mounted at the top of the autoclave.
First, the autoclave was filled with 100 mg of the catalyst powder and sealed. The system was flushed 3 - 4 times with N2 to ensure that no air was present in the autoclave. After leak test, a constant gas flow of 10% H2/N2 (30 ml/min) was passed through the autoclave for pre-reduction of the catalyst and then, heating was started. The temperature was increased uniformly with a rate of 5 K/min up to 200˚C for carbon supported cata-
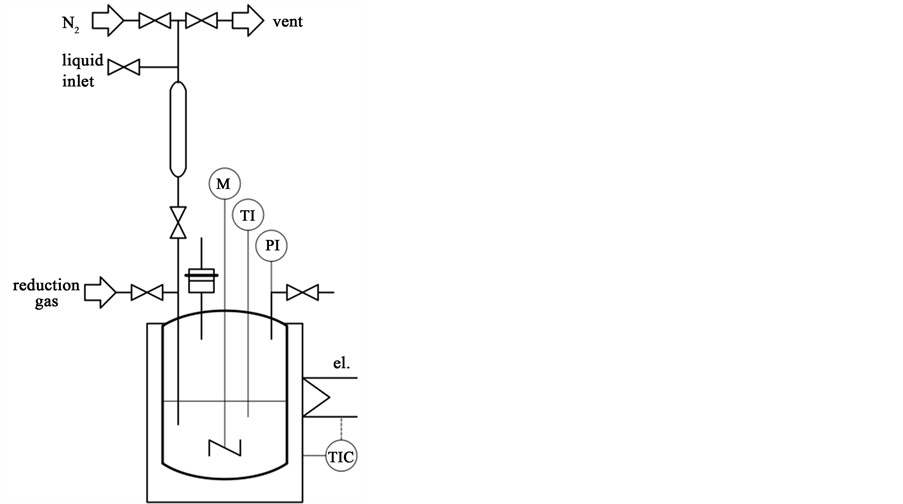
Figure 1. Scheme of the experimental set-up.
lysts according to literature data [10] . After reduction for 2 h, the autoclave was cooled down and the empty solvent cylinder was attached to the system. Then the solvent was introduced (10 ml each) into the cylinder that was flushed thoroughly with N2. Afterwards, the solvent was introduced into the autoclave under N2 atmosphere to avoid contact of the reduced catalyst with air. Finally, the desired quantity of the reactant (CA (Acros), LA (Acros) or SA (Merck)) was then added to the catalyst-solvent mixture via the mounted cylinder under N2 flow.
All the decarboxylation reactions were carried out at 250˚C - 350˚C and 18 - 75 bar reaction pressure (i.e., 10 - 35 bar initial pressure using N2) for 3 - 24 h, 500 rpm stirrer speed, 1.6 mmol/l (230 mg, 320 mg, 454 mg for CA, LA and SA, respectively) feed concentration and 100 mg of catalyst, in general.
After the completion of the reaction, the autoclave was cooled down, weighed and the content was removed; the solid residue (catalyst) was filtered off and used for further characterizations after washing with ethanol. 100 μl of diethylene glycol dibutyl ether (DEGDBE) were used as internal standard in 100 μl of each sample. Also 100 µl of trimethyl sulfonium hydroxide (TMSH) solution (0.1% in methanol) were used for methylation of acids to the corresponding methyl esters for an improved GC analysis. A sample dilution was done with 700 μl of methyl tertiary butyl ether so as to get smaller and well separated peaks. The as-prepared samples were analyzed with a HP 5890 Series II GC (FID) equipped with HP-5 column (cross-linked 5% phenyl methyl siloxane, 30 m × 0.32 mm × 0.25 μm), running the following temperature program: 50˚C - 10 K/min - 300˚C. 2 μl of sample were injected using a HP 7673 auto sampler. Helium was used as carrier gas. The accuracy of the analyses was within an error margin of ±5%. Gaseous samples were analyzed for carbon oxides with an off line gas chromatograph (Shimadzu GC 14B), however, mainly CO2 was present in the gas phase. The content of the gas collecting cylinder was flushed into a 6-port sampling valve and after injection the permanent gases were eluted with argon as carrier gas on a combination of two packed columns (PoraPak N, 3 m × 1/8”; Molsieve 13X, 3 m × 1/8”) and analyzed by thermal conductivity detector (TCD, temperature program: 40˚C – 10 K/min – 80˚C – 40 K/min – 120˚C).
The conversion (X) of the used fatty acid (R-COOH), the yield of the target alkane (Y) containing one carbon atom less than the parent acid (R-H) and its selectivity (S) were calculated by the following formulas:
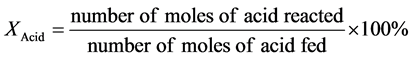


2.3. Solid Characterizations
Best performing Pd/C catalysts were characterized by N2-adsorption, elemental analysis (ICP-OES), X-ray diffraction (XRD) and transmission electron microscopy (TEM).
Catalyst surface area was determined by N2-adsorption according to the BET method (Gemini III 2375, Micromeritics) at −196˚C. Prior to the measurements a weighed amount of the catalyst was evacuated for 2 h at 150˚C to remove physically adsorbed water.
The elemental chemical composition of fresh and used catalyst samples were determined with ICP-OES (Optima 3000XL, Perkin-Elmer) using microwave pressure digestion (MDS 200; CEM) with hydrofluoric acid and aqua regia at 9 bar.
The XRD patterns were recorded using a STADI P (Stoe) set-up (Debye-Scherrer geometry, Ge-primary monochromator, CuKα) using Ni-filtered CuKα radiation (λ = 0.15418 nm). Data interpretation was carried out using the software X-POW (Stoe) and the database of the International Center for Diffraction Data (ICDD).
Transmission Electron Microscopy (TEM) investigations were carried out at 200 kV on a CM-20 microscope (Philips/FEI) equipped with a Noran Six (Thermo Scientific) EDX spectrometer. Samples were prepared by depositing the catalyst on a carbon coated copper grid (300 mesh).
3. Results and Discussion
3.1. Characterization Results
The catalyst surface areas and pore sizes are summarized in Table 1. As expected, the highest surface area was found on 1% Pd/C catalyst (626 m2/g). With increasing of the Pd loading on activated carbon, the specific surface area decreased, probably due to pore filling or occlusion as indicated by the pore volume data. The catalyst with the highest Pd loading (10% Pd/C) exhibited 365 m2/g. The BET surface area measurement of the spent 5% Pd/C and 10% Pd/C showed that the catalysts undergo some alterations in the reaction. The surface area of these catalysts (5% Pd/C and 10% Pd/C) is found to decrease significantly. The fresh sample 10% Pd/C showed 365 m2/g but after 24 h reaction time, this catalyst revealed 180 m2/g only. This result might be due to a possible surface deposition of some oligomers formed from decarboxylation/decarbonylation intermediates. The pore volume decline of the spent samples points in the same direction. Santillan-Jimenez and Crocker also report in their review on the deactivation of noble metal catalysts used for such deoxygenations and their regeneration. [17] Therein, some papers are cited reporting on the loss of BET surface area, probably being a result of the occlusion of pores by organic deposits.
The results of the ICP studies for the Pd/C catalysts are shown in Table 2. For the catalysts with low Pd loading, it was found that Pd proportion was as high as desired. But for higher loading, less Pd was found in the catalyst than offered. The spent catalyst sample 10% Pd/C (from 24 h reaction using LA at 300˚C, 10 bar initial pressure (i.e., ca. 18 bar reaction pressure), 500 rpm) showed nearly the same Pd content than the fresh sample; only a decrease of ca. 1% was observed. Though this decrease is not much, it could be due to a slight Pd leaching in the course of the reaction [17] . Therefore, this low loss seems to be negligible in comparison to the loss of surface area.

Table 1. BET surface area, pore volume and pore diameter of supports and fresh catalysts.
Table 2. ICP analysis of Pd loading of catalysts.
adouble determination.
The XRD patterns (Figure 2) of calcined Pd/C catalysts reveal only in the case of 5% Pd/C and 10% Pd/C two reflections at 2θ = 40.42˚ assigned to Pd(111) and at 46.66° assigned to Pd(200) [29] . Solids with less Pd did not show these reflections due to the lower size of the particles.
TEM analyses revealed that the diameter of the Pd particles is not uniform; it varies from 5 to 30 nm for 10% Pd/C sample (Figure 3(a)). Besides some larger particles existing in the range of 20 - 30 nm many well-dis- persed Pd particles are also visible. The other two Pd-containing catalysts show a significantly smaller proportion of the big particles due to the lower loading; well-dispersed small particles are still present. TEM images of spent catalysts (e.g. 10% Pd/C, 24 h reaction, Figure 3(b)) reveal that the diameter of the bigger Pd particles remains unchanged but the proportion of the small particles in the range of 5 to 10 nm increased. This result points to a slight growing of smaller Pd particles, probably due to agglomeration of subnanometer particles during the catalytic reaction.
3.2. Catalytic Test Runs
The tests were addressed to the conversion of some model fatty acids (caprylic acid-CA, lauric acid-LA and stearic acid-SA) over Pd-containing catalysts under comparable reaction conditions mentioned above.
Previous blank tests with LA up to 320˚C in the absence of catalysts showed some thermal cracking up to a LA conversion of ca. 2% - 4% and low undecane yield of ca. 2% besides some other undesired products. The result of the screening of the Pd-containing catalysts using LA as reactant (see Table 3) showed that the 10% Pd/C revealed the highest yield of undecane (37%) at a conversion of 55%. 5%Pd/C and 1% Pd/C catalysts were less effective showing a conversion of ca. 32% and undecane yield between 14% - 17% only. Simultaneously with the formation of the desired undecane, we always observed the appearance of mainly CO2 as detected from gas phase analyses. This illustrates that the deoxygenation mainly follows the decarboxylation path.
The effect of reactant chain length on conversion and alkane selectivity using 10% Pd/C catalyst is depicted in Figure 4. The conversion of the three fatty acids clearly increases with acid chain length. In our experiments, the conversion for CA was 36%, LA conversion was 55% and that of SA 93%. This means that for the best- performing catalyst 10% Pd/C conversion is also increased by a factor of ca. 2.5 if fatty acid chain length grows from C8 to C18. But the selectivity towards alkanes showed a very different behavior. It was highest for LA (67%), and less for CA (by ~15%) and negligible in SA case (<13%). This could be explained to a large extent by the higher statistical chance of cracking of the SA reactant to smaller molecules. This finding is in good agreement with literature data. According to Leung et al. [15] , the production of organic liquids (pyrolysis products) increased slightly from 49 to 55% when acid chain length was increased from C4 to C12. (Note that their experiments were carried out on Pd/SiO2 at gas phase conditions). In addition, our results are supported by data of Murzin et al. [21] observed a decrease in selectivity to alkanes by 8% when they increased acid chain length from SA (C18) to behenic acid (C20). They did not investigate the effect of smaller acid chain length. In case of CA reactant a higher selectivity should be expected but the experiment revealed slightly less selectivity compared to the LA conversion. This might be explained by a superposition of cracking and adsorption effects leading to a blockage of surface sites, probably. Anyway, reactant and products might be cracked and thus influence conversion and selectivity. This has to be taken into account when discussing these results.
Figure 5 and Figure 6 depict the conversion of the three fatty acids and the selectivity to the corresponding alkanes, respectively. These runs were carried out on two different Pd-containing catalysts (1% Pd/C and 10% Pd/C) while keeping the reaction parameters constant (300˚C, 18 bar, 6 h). It can be clearly seen that the conver-

Figure 2. XRD patterns of freshly calcined Pd/C catalysts.
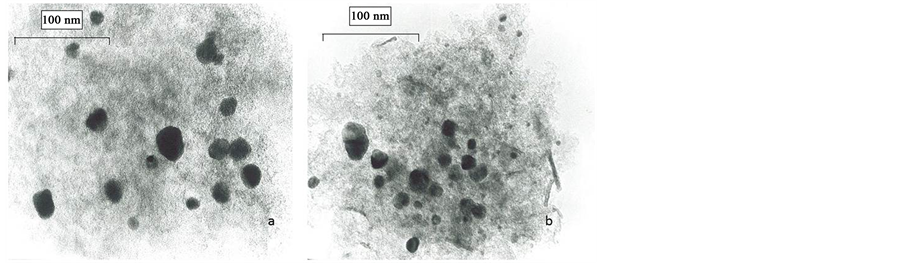
Figure 3. TEM images of parent (a) and spent ((b), after 24 h run) 10% PdC catalyst; magnification: 1:460,000.
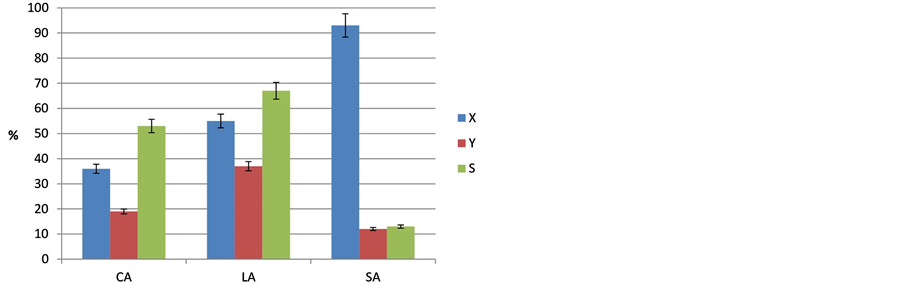
Figure 4. Impact of the acid chain length of the reactant (caprylic-CA, lauric-LA and stearic acid-SA) on the decarboxylation reaction at T = 300˚C (p = 18 bar, t = 6 h, stirrer speed = 500 rpm, catalyst = 10% Pd/C).
Table 3. Pd/C catalyst screening results for conversion of lauric acid (LA).
Conditions: 1.6 mmol of reactant, 100 mg of catalyst, 10 ml of solvent, 300˚C, 6 h, 500 rpm, initial N2 pressure 10 bar.
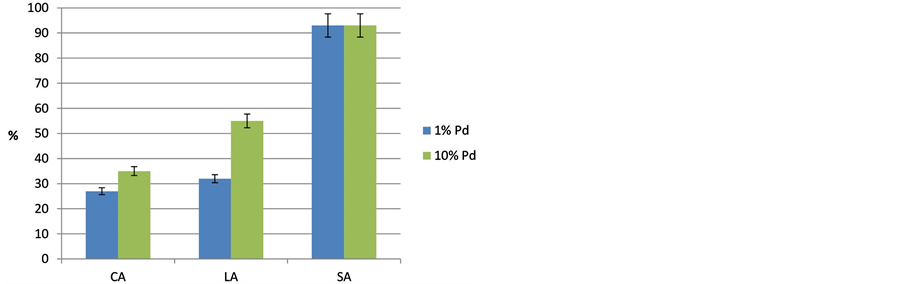
Figure 5. Influence of the Pd loading (1% Pd/C, 10% Pd/C) on the conversion of caprylic-CA, lauric-LA and stearic acid-SA at T = 300˚C (p = 18 bar, t = 6 h, stirrer speed = 500 rpm).
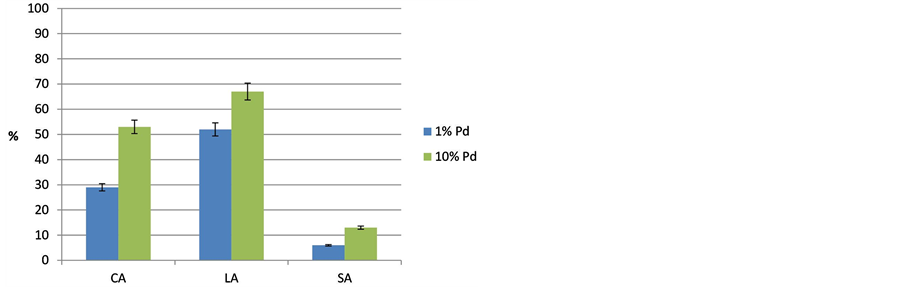
Figure 6. Influence of the Pd loading (1% Pd/C, 10% Pd/C) on the selectivity of the desired decarboxylation product from caprylic-CA, lauric-LA and stearic acid-SA at T = 300˚C (p = 18 bar, t = 6 h, stirrer speed = 500 rpm).
sion increases with chain length as discussed above. Furthermore, it is obvious that the Pd content of the two solids also shows an effect on the conversion; CA and LA conversions are significantly higher over 10%P d/C. This might be probably due to the ten-fold Pd-loading outperforming the effect of the different Pd particle size of both the Pd/C samples. Surprisingly, the conversion of SA on the 1% Pd/C sample doesn’t not fit to this picture, the tests with SA reveal almost the same result for both the catalysts (X = ca.93%). Probably, it is because of the much higher chances of SA cracking even though the Pd loading is rather low. LA was converted with 32% over 1% Pd/C, but for 10% loading the conversion further increased up to 55%. With LA, cracking is not as feasible as in the case of SA and therefore, it can be concluded that the increased number of Pd surface sites is responsible for such an increase. The same effect can be seen looking at CA; but conversion increased only from 27% to 35% because cracking is further hindered.
The selectivity data show a quite different behavior (Figure 6); it seems that it passes through a maximum for medium-chain acid (LA) as reactant. The selectivity of the expected alkane slightly increased from CA to LA but it dramatically dropped using SA due to further cracking and other side reactions as already evaluated above.
The effect of the variation of the reaction pressure from 18 to 75 bar was investigated using LA as reactant at 300˚C over 6 h (Figure 7). It was observed that the highest conversion of LA was 55% at 18 bar. Increase in reaction pressure from 18 to 45 bar (by increasing the initial pressure from 10 to 20 bar) decreased conversion of LA from 55% to 42% and further to 40% at 75 bar (35 bar initial pressure). Interestingly, the yield of undecane received was on the same level, i.e., 34% - 37%. Thus, the selectivity to undecane increases linearly up to 90% with increase in pressure. The reason might be that an elevated pressure suppresses parallel and/or consecutive side reactions, in particular those leading to an increased number of moles.
The temperature influence from 250˚C to 325˚C keeping all the other variables constant is plotted in Figure 8.
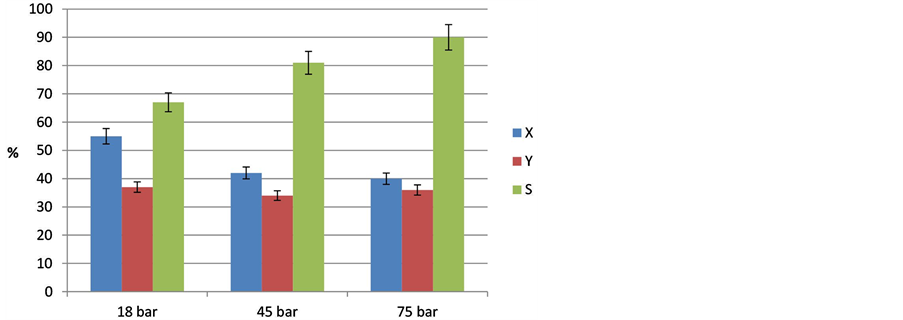
Figure 7. Influence of the reaction pressure of 18, 45 and 75 bar on the decarboxylation of lauric acid at T = 300˚C (t = 6 h, stirrer speed = 500 rpm, catalyst 10% Pd/C).
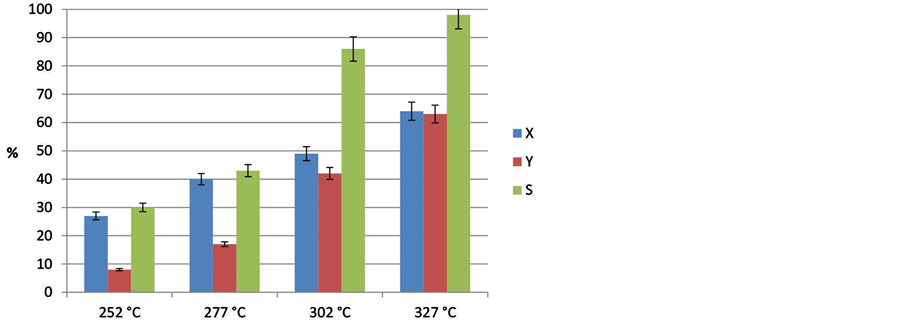
Figure 8. Effect of the reaction temperature on the decarboxylation of lauric acid (p = 18 bar, t = 6 h, stirrer speed = 500 rpm, feed concentration = 1.6 mmol/l, catalyst = 10% Pd/C).
As expected, temperature had a significant effect on conversion of LA. It is well known that these deoxygenations carried out in the absence of hydrogen are endothermic reactions [10] and higher temperatures favor not only conversion but also accelerate the desired alkane formation by decarboxylation. Thus, the formation of desired undecane increased with reaction temperature, i.e., decarboxylation gets dominant over cracking at high temperatures using such medium-chain reactant (in contrast to SA). In our case and using LA, the best results were received at 327˚C with a conversion of LA of 64% and selectivity to undecane of 98%. The increase in the temperature favors the decarboxylation reaction using LA, but at higher temperature and other fatty acids some other reactions may become significant. This could be cracking of higher chain-length hydrocarbons to lower chain-length hydrocarbons, aromatization or oligomerization to give dimers, as already suggested by Murzin et al. This is also supported by recent studies on the pyrolysis of carboxylic acids [30] .
In addition, some experiments were carried out with respect to the effect of the reaction time using LA (Figure 9). It can be easily seen that the reaction was very selective to undecane after 3 h reaction time. The conversion of LA increased with reaction time as expected, but the selectivity dropped from 98% after 3 h to 80% and 68% for 6 h and 12 h, respectively. It seems that undecane was converted consecutively probably to dimers and higher molecular compounds but also other side-products might be formed by cracking, however, both of these possible consecutive products were not analyzed by the used GC set up.
Both the results for the temperature and time variation point at an optimum window with regard to the optimum reaction conditions in the conversion of LA. On the other side, the chain length also affects the optimum decarboxylation conditions.
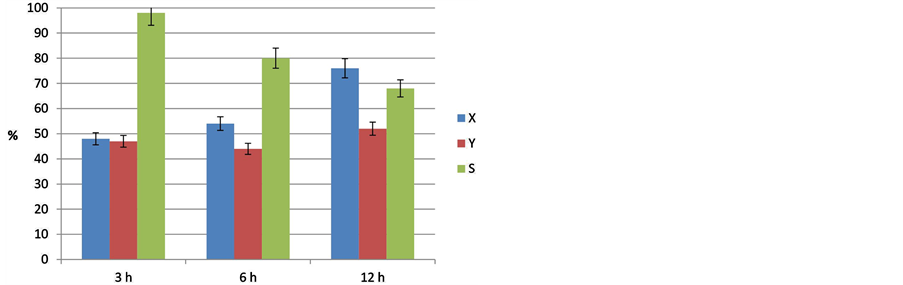
Figure 9. Influence of reaction time on decarboxylation of lauric acid at T = 300˚C (18 bar, stirrer speed = 500 rpm, catalyst 10% Pd/C).
4. Conclusions
The hydrogen-free decarboxylation of saturated C8, C12 and C18 fatty acids was carried out over Pd/C catalysts in batch mode. It was found that the 10% Pd/C catalyst revealed the best effect on the decarboxylation reaction of lauric acid. Best results were received at 327˚C with a conversion of 64% and an undecane yield of 63%. For comparison, higher chain-length acid (C18:0) gave an improved conversion but very low alkane selectivity; lower chain-length acid (C8:0) showed less conversion as expected but also alkane selectivity hung behind the selectivity reached using the C12:0 acid.
The effect of the reaction pressure and temperature, the fatty acid feed chain-length and the reaction time was studied intensively in this work. The increase in the reaction pressure was found to slightly decrease the conversion but increase the selectivity, keeping the yield for alkane constant. The reaction temperature demonstrated a promotional effect on the reaction as expected. The studies on the variation of the reaction time suggested that it is advantageous to stop reaction after ca. 3 h due to an increasing cracking or other consecutive reactions with prolonged reaction time. The increase of both the temperature and reaction time lead to a rising level of the fatty acid conversion and dropping alkane selectivity; this might clearly point to consecutive reactions forming undesired by-products. Regarding reaction thermodynamics, higher temperature might enhance cracking, whereas prolonged reaction time might lead to oligomers, which are hard to identify and quantify.
It looks that future work is needed, probably, for the involvement of bi-metallic catalysts (e.g. NiPd) on suitable carriers to extend the excellent performance of nanoscale Pd catalysts for the deoxygenation of medium- chain fatty acids in the absence of additional hydrogen to long-chain compounds such as stearic acid and to reduce costs for precious Pd.
Acknowledgements
The authors like to thank Drs. M. Schneider and M.-M. Pohl for XRD and TEM measurements, respectively, and Mrs. A. Simmula for ICP analyses. S. Mohite thanks LIKAT for financial support.
References
- Raps - die „Leit(d)”-Kultur. http://www.ufop.de/medien/downloads/biodiesel-and-co/allgemein/
- Leung, D.Y.C., Wu, X. and Leung, M.K.H. (2010) A Review on Biodiesel Production using Catalyzed Transesterification. Applied Energy, 87, 1083-1095. http://dx.doi.org/10.1016/j.apenergy.2009.10.006
- Internationale Biodiesel-Märkte. http://www.ufop.de/medien/downloads/biodiesel-and-co/sonstige/
- Al-Sabawi, M., Chen, J. and Ng, S. (2012) Fluid Catalytic Cracking of Biomass-Derived Oils and Their Blends with Petroleum Feedstocks: A Review. Energy & Fuels, 26, 5355-5372. http://dx.doi.org/10.1021/ef3006417
- Al-Sabawi, M. and Chen, J. (2012) Hydroprocessing of Biomass-Derived Oils and Their Blends with Petroleum Feedstocks: A Review. Energy Fuels, 26, 5373-5399. http://dx.doi.org/10.1021/ef3006405
- Fukuda, H., Kondo, A. and Noda, H. (2001) Biodiesel Fuel Production by Transesterification of Oils. Journal of Bioscience and Bioengineering, 92, 405-416. http://dx.doi.org/10.1016/S1389-1723(01)80288-7
- Liu, X., He, H., Wang, Y., Zhu, S. and Piao, X. (2008) Transesterification of Soybean Oil to Biodiesel using CaO as a Solid Base Catalyst. Fuel, 87, 216-221. http://dx.doi.org/10.1016/j.fuel.2007.04.013
- Liu, C.-C., Lu, W.-C. and Liu, T.-J. (2012) Transesterification of Soybean Oil Using CsF/CaO Catalysts. Energy Fuels, 26, 5400-5407. http://dx.doi.org/10.1021/ef300941w
- Xie, W. and Ma, N. (2009) Immobilized Lipase on Fe3O4 Nanoparticles as Biocatalyst for Biodiesel Production. Energy Fuels, 23, 1347-1353. http://dx.doi.org/10.1021/ef800648y
- Snåre, M., Kubicková, I., Paivi, M., Eränen, K. and Murzin, D.Yu. (2006) Heterogeneous Catalytic Deoxygenation of Stearic Acid for Production of Biodiesel. Industrial & Engineering Chemistry Research, 45, 5708-5715. http://dx.doi.org/10.1021/ie060334i
- Şenol, O., Viljava, T.R. and Krause, A. (2005) Hydrodeoxygenation of Aliphatic Esters on Sulphided NiMo/γ-Al2O3 and CoMo/γ-Al2O3 Catalyst: The Effect of Water. Catalysis Today, 106, 186-189. http://dx.doi.org/10.1016/j.cattod.2005.07.129
- Bertram, S. (1936) Action of Selenium on Stearic Acid. Chemisch Weekblad, 33, 457-459.
- Foglia, T.A. and Barr, P.A. (1975) Decarbonylation Dehydration of Fatty Acids to Alkenes in the Presence of Transition Metal Complexes. Journal of the American Oil Chemists Society, 53, 737-741.
- Maier, W.F., Roth, W., Thies, I. and von RaguéSchleyer, P. (1982) Hydrogenolysis, IV. Gas Phase Decarboxylation of Carboxylic Acids. Chemische Berichte, 115, 808-812. http://dx.doi.org/10.1002/cber.19821150245
- Ford, J.P., Immer, J.G. and Lamb, H.H. (2012) Palladium Catalysts for Fatty Acid Deoxygenation: Influence of the Support and Fatty Acid Chain Length on Decarboxylation Kinetics. Topics in Catalysis, 55, 175-184. http://dx.doi.org/10.1007/s11244-012-9786-2
- Ping, E., Wallace, R., Pierson, J., Fuller, T.F. and Jones, C.W. (2010) Highly Dispersed Palladium Nanoparticles on Ultra-Porous Silica Mesocellular Foam for the Catalytic Decarboxylation of Stearic Acid. Microporous and Mesoporous Materials, 132, 174-180. http://dx.doi.org/10.1016/j.micromeso.2010.02.017
- Santillan-Jimenez, E. and Crocker, M. (2012) Catalytic Deoxygenation of Fatty Acids and Their Derivatives to Hydro- carbon Fuels via Decarboxylation/Decarbonylation. Journal of Chemical Technology and Biotechnology, 87, 1041- 1050. http://dx.doi.org/10.1002/jctb.3775
- Leung, A., Boocock, D.G.B. and Konar, S.K. (1995) Pathway for the Catalytic Conversion of Carboxylic Acids to Hydrocarbons over Activated Alumina. Energy Fuels, 9, 913-920. http://dx.doi.org/10.1021/ef00053a026
- Hites, R.A. and Biemann, K. (1972) Mechanism of Ketonic Decarboxylation. Pyrolysis of Calcium Decanoate. Journal of the American Chemical Society, 94, 5772-5777. http://dx.doi.org/10.1021/ja00771a039
- Pestman, R., van Duijne, A., Pieterse, J.A.Z. and Ponec, V. (1995) The Formation of Ketones and Aldehydes from Car- boxylic Acids, Structure-Activity Relationship for Two Competitive Reactions. Journal of Molecular Catalysis A: Chemical, 103, 175-180. http://dx.doi.org/10.1016/1381-1169(95)00138-7
- Pestman, R., Koster, R.M., van Duijne, A., Pieterse, J.A.Z. and Ponec, V. (1997) Reactions of Carboxylic Acids on Oxides: 2. Bimolecular Reaction of Aliphatic Acids to Ketones. Journal of Catalysis, 168, 265-272. http://dx.doi.org/10.1006/jcat.1997.1624
- Sugiyama, S., Sato, K., Yamasaki, S., Kawashiro, K. and Hayashi, H. (1992) Ketones from Carboxylic Acids over Su- pported Magnesium Oxide and Related Catalysts. Catalysis Letters, 14, 127-135. http://dx.doi.org/10.1007/BF00764227
- Gusmão, J., Brodzki, D., Djéga-Mariadassou, G. and Frety, R. (1989) Utilization of Vegetable Oils as an Alternative Source for Diesel-Type Fuel: Hydrocracking on Reduced Ni/SiO2 and Sulphided Ni-Mo/γ-Al2O3. Catalysis Today, 5, 533-544. http://dx.doi.org/10.1016/0920-5861(89)80017-3
- Kubicková, I., Snåre, M., Eränen, K., Mäki-Arvela, P. and Murzin, D.Y. (2005) Hydrocarbons for Diesel Fuel via Decarboxylation of Vegetable Oils. Catalysis Today, 106, 197-200. http://dx.doi.org/10.1016/j.cattod.2005.07.188
- Simakova, I., Simakova, O., Mäki-Arvela, P. and Murzin, D.Y. (2010) Decarboxylation of Fatty Acids over Pd Supported on Mesoporous Carbon. Catalysis Today, 150, 28-31. http://dx.doi.org/10.1016/j.cattod.2009.07.064
- Simakova, I., Simakova, O., Mäki-Arvela, P., Simakov, A., Estrada, M. and Murzin, D.Y. (2009) Deoxygenation of Palmitic and Stearic Acid over Supported Pd Catalysts: Effect of Metal Dispersion. Applied Catalysis A: General, 355, 100-108. http://dx.doi.org/10.1016/j.apcata.2008.12.001
- Mäki-Arvela, P., Kubicková, I., Snåre, M., Eränen, K. and Murzin, D.Y. (2007) Catalytic Deoxygenation of Fatty Acids and Their Derivatives. Energy Fuels, 21, 30-41. http://dx.doi.org/10.1021/ef060455v
- http://www.nesteoil.com/default.asp?path=1,41,11991,22708,22709,22710
- Kaszkur, Z. (2000) Nanopowder Diffraction Analysis beyond the Bragg Law Applied to Palladium. Journal of Applied Crystallography, 33, 87-94. http://dx.doi.org/10.1107/S002188989901290X
- Maher, K.D., Kirkwood, K.M., Gray, M.R. and Bressler, D.C. (2008) Pyrolytic Decarboxylation and Cracking of Stearic Acid. Industrial & Engineering Chemistry Research, 47, 5328-5336. http://dx.doi.org/10.1021/ie0714551
NOTES

*Corresponding author.