American Journal of Analytical Chemistry
Vol.07 No.07(2016), Article ID:68654,12 pages
10.4236/ajac.2016.77051
A New and Enantioselective Chromatographic Method for Linagliptin on Amylose Coated Chiral Stationary Phase
Cholleti Vijay Kumar1,2*, Pasula Aparna2, Pavan Kumar Vasa3, Y. Ravindra Kumar1, Nitin Haridas Dhekale1
1Dr. Reddy’s Laboratories Ltd., Active Pharmaceutical Ingredients, IPDO, Hyderabad, India
2Department of Chemistry, J. N. T. University, Hyderabad, India
3Department of Pediatrics, SUNY Stony Brook Medical Center, New York, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 14 June 2016; accepted 17 July 2016; published 20 July 2016
ABSTRACT
A new and enantioselective liquid chromatographic method was developed for estimation of S-Li- nagliptin in Linagliptin (LINA) drug substances. The desired enantiomeric separation was achieved on Chiralpak AD-H (250*4.6 mm*5 µm) column with the mobile phase composition of ethanol, methanol and diethylamine in a ratio of 90:10:0.1 (v/v/v) with flow rate of 0.5 mL∙min−1 and column oven temperature 30˚C and the eluted compounds were monitored at 225 nm. In the proposed chiral method, USP resolutions between both the enantiomers were more than 5.0. Limit of detection and Limit of quantitation of S-LINA was found to be 0.03 µg∙mL−1 and 0.10 µg∙mL−1 respectively. Linearity study was conducted from LOQ to 150% and correlation coefficient found to be 0.9997. Accuracy was within the range of 98.6% to 101.5%. To prove selectivity power of the method specificity study was conducted by subjecting drug substance to acid, base, hydrolysis, oxidation and photolysis and ensured the peak purity of analyte in degraded samples. Moreover, the method has been fully validated as per ICH guidelines. The proposed method is precise, accurate, linear, rugged, robust and suitable for accurate quantification of S-LINA in LINA drug substance.
Keywords:
Enantiomers, Amylose Based Stationary Phase, Method Development, Method Validation, Specificity

1. Introduction
LINA is chemically (R) 8-(3-aminopiperidin-1-yl)-7-(but-2-yn-1-yl)-3-methyl-1-[(4-methylquinazolin-2-yl) methyl]-3, 7-dihydro-1H-purine-2, 6-dione with molecular formula C25H28N8O2. Linagliptin is a xanthine based DPP-4 (Dipeptidyl peptidase-4) inhibitor, which can be orally taken for treatment of type-II diabetes, which is approved by US Food and Drug Administration.
Type 2 diabetes mellitus is a progressive disease, and it occurs with increasing prevalence in the elderly and those with other comorbidities. Blood glucose control presents a challenge that is magnified by these co-existing problems. To achieve glycemic targets, many patients need more than one antidiabetic drug, and additional medications are often required as glucose control deteriorates [1] . Consequently, the development of new antidiabetic drugs that can help meet this challenge has been a field of intensive research.
The dipeptidyl peptidase-4 inhibitors are one of the recently developed therapeutic classes for treatment of hyperglycemia in Type 2 diabetes mellitus. The various agents in the class have differing chemical structures, but all acts by inhibiting the DPP-4 enzyme, thus prolonging the life of incretin hormones, which in turn raise insulin levels and suppress glucagon secretion in a glucose-dependent manner. As a class, DPP-4 inhibitors have been shown to provide significant improvements in glycosylated hemoglobin (HbA1c), and to have a good safety profile. In addition, their glucose-dependent mechanism of action is associated with a low rate of hypoglycemic events [2] .
High-throughput screening using an assay to detect inhibition of DPP-4 led to the discovery of linagliptin, a xanthine-based molecule with a high selectivity for DPP-4. The pharmacokinetics and pharmacodynamics of linagliptin have been reviewed in detail elsewhere [3] . Of note, unlike other DPP-4 inhibitors, which are predominantly excreted via the kidneys, linagliptin is mainly excreted unchanged via the enterohepatic system [4] .
The separation of chiral isomers can be carried out by using HPLC through direct and indirect methods. Indirect methods are based on adding a chiral additive to the mobile phase. The optical isomers react with the chiral additives, and then the derivatives are separated on an achiral stationary phase. Direct methods separate the isomers on a chiral stationary phase. But in recent development into the chiral separation technology the enantiomers can be separated directly over a chiral stationary phase, they must form short-lived diastereomeric molecular complexes of non-identical stability by interacting rapidly and reversibly [5] . There are five different categories of HPLC chiral stationary phases which are commercially available: brush type, synthetic polymer, cyclo- dextrin bonded, ligand exchange, and protein.
The polysaccharides coated on silica are the most widely used synthetic polymer for preparation of stationary phase. The polysaccharide phase is comprised of derivatized cellulose or amylose coated on a silica support. Derivatization of the polysaccharide hydroxyl groups with various side chains gives different helical supramo- lecular structures. The curved groove of the helix is chiral, and can greatly favor the binding of one enantiomer over the other. The result is separation of the enantiomers. Interaction between analyte and synthetic polymer chiral stationary phases are based on both attractive interactions (H-bonding, pi-pi interaction, and/or dipole stacking) and inclusion complexes. Instead of a silica surface, inclusion complexes utilize cavities in which the analyte fits [6] . Synthetic polymer chiral stationary phases are most commonly used with non-polar mobile phases of alkanes (hexane or heptane) and a polar mobile phase modifier. An alcohol, such as ethanol or isopropyl alcohol, is generally the polar mobile phase modifier of choice. The structure of the alcohol plays an important role in the retention and resolution due to competition at or near the binding site.
LINA is a chiral molecule, and exists as S-LINA and R-LINA mentioned in (Figure 1(a) and Figure 1(b)). Both the enantiomers of drug substance may have different properties of toxicology, pharmacokinetics, and biological activities [7] . From past two decades, regulatory guidelines for chiral molecules became more stringent. According to ICH Q6A, it is required to control the unwanted enantiomers in drug substances, US FDA also recommends the study of each enantiomer activity of racemic drugs in the body [8] . Hence it is important to monitor the content of undesired isomer in chiral drug while developing process to synthesize the same. For estimation of undesired enantiomer, techniques like gas chromatography, high performance liquid chromatography, supercritical fluid chromatography, thin layer chromatography and capillary electrophoresis are available in literature [9] . As more versatile chiral stationary phases are available for HPLC, this technique is more prominent and user friendly to monitor the enantiomeric purity of chiral drug substance. Among available chiral stationary phases, most of chiral separations were reported by using derivatized polysaccharides [10] - [12] .
During through literature survey it is understood that there were few methods for single LINA estimation by spectrophotometry and HPLC [13] - [22] , whereas there is no single method available in literature for enantio-selec- tive separation of LINA. The purpose of the present research work was to develop a suitable, robust HPLC method for the determination of S-LINA in LINA. Hence, an attempt has been made to develop an accurate, rapid, specific
 (a)
(a) (b)
(b)
Figure 1. (a) R-Enantiomers of Linagliptin (R-LINA): (R)-8-(3-aminopiperidin-1-yl)-7-(but-2-yn-1-yl)-3-methyl-1-[(4-meth- ylquinazolin-2-yl) methyl]-3, 7-dihydro-1H-purine-2, 6-dione. (b) S-Enantiomers of Linagliptin (S-LINA): (S)-8-(3-aminopi- peridin-1-yl)-7-(but-2-yn-1-yl)-3-methyl-1-[(4-methylquinazolin-2-yl) methyl]-3, 7-dihydro-1H-purine-2, 6-dione.
and reproducible method for the quantification of S-LINA in LINA. Method was validation as per ICH guidelines. Specificity study also conducted to proven the selectivity power of the method.
2. Experimental
2.1. Chemicals
Individual enantiomers of Linagliptin were prepared and provided by Dr. Reddys Laboratories Limited, IPDO, Hyderabad, India. Ethanol, Methanol and Diethyl amine was purchased from Merck
2.2. Chromatographic Conditions and Equipment
All experiments were carried out on a Waters e2695 separation module consisting of 2998 photodiode array detector and Agilent 1260 series ternary pump with variable wavelength detector. Empower 2 software was used for signal monitoring and data processing. The method was developed by using Chiralpak AD-H (250*4.6 mm*5 µm) column which contains Amylose Tris (3, 5-Dimethylphenylcarbamate) as stationary phase. Mobile phase consists of ethanol, methanol and diethylamine in a ratio of 90:10:0.1 (v/v/v) with a flow rate of 0.5 mL∙min−1, 5 µL injection volume, 30˚C column oven temperature and chromatographic signal was monitored at 225 nm. Chromatograms were summarized in (Figures 2(a)-(c)).
2.3. Preparation of Sample Solutions
LINA samples are prepared in the methanol (1 mg∙mL−1). The system suitability was prepared by mixing equal portion of R-LINA and S-LINA at 0.2 mg∙mL−1 in methanol.
3. Results and Discussion
3.1. Method Development and Optimization
The main goal of method development was to achieve separation of S-LINA from R-LINA. As part of method development screened various chiral columns namely Chiralcel OD-H, Chiralcel OJ-H, Chiralpak-IA, Chiralpak-IB,
 (a)
(a) (b)
(b) (c)
(c)
Figure 2. (a) Typical system suitability chromatogram of LINA enantiomers; (b) Typical chromatogram of LINA sample; (c) Typical chromatogram of 0.15% spiked S-LINA in LINA sample.
Chiralpak-IC and Chiralpak-IE manufactured by Diacel
After careful screening of columns and mobile phase compositions it was observed that Chiralpak AD-H provides better resolution between S-LINA and R-LINA using n-Hexane, Ethanol and diethylamine in the ratio of (10%:90%:0.1% v/v/v) as mobile phase. To improve the chromatographic efficiency and resolution between enantiomers further mobile phase composition was altered and prepared the mobile phase in the composition of ethanol, methanol and diethylamine in the ratio of (90:10:0.1 v/v/v). In this optimized method the typical retention times of S-LINA and R-LINA were approximately about 12.3 min. and about 17.2 min. respectively. Resolutions between two enantiomers are found more than 5 and total run time of method was within 35 minutes. The complete screening of chiral columns and mobile phase compositions were summarized in Table 1 and (Figures 3(a)-(d)).

Table 1. Summary of method development experiments.
Note: n-Hex: n-Hexane; MeoH: Methanol; EtoH: Ethanol; DEA: Diethyl amine; IPA: Isopropyl alcohol; ACN: Acetonitrile; NA: Not applicable; No Res: No resolution.
3.2. Method Validation
3.2.1. Method Precision
The developed method was validated in terms of repeatability (Intra-day) and intermediate precision. Repeatability was established at four different levels (LOQ, 50%, 100%, and 150%) to the specification limit of 0.15% as per ICH guidelines. Performed the study by spiking the S-LINA in test sample at four different concentration as per above.
The same study has been repeated to generate intermediate precision for two consecutive days by using different instruments, different columns and different analyst. The intermediate precision and method repeatability are measured by calculating % relative standard deviation (% RSD) for area of S-LINA. The % RSD values for intra-day precision and inter-day precision were found less than 0.5%, these values indicate that developed method was precise.
3.2.2. Detection Limit and Quantification Limit
Limit of detection (LOD) and limit of quantitation (LOQ) were established based on signal to noise ratio. LOD is the lowest analyte concentration that can be detected and LOQ is the lowest analyte concentration that can be quantified. LOD and LOQ values are established by injecting a series of diluted solutions of S-LINA and calculated signal to noise ratio (S/N ratio). The LOD and LOQ values achieved for S-LINA were 0.03 µg∙mL−1 and 0.10 µg∙mL−1 respectively.
3.2.3. Linearity
Linearity is performed by injecting a series of diluted solutions of S-LINA ranging from LOQ to 150%. The linearity curve was plotted for peak area of S-LINA and concentration using least squares method. The correlation
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 3. Method development chromatograms. (a) Typical chromatogram of method development experiment. Chromatographic conditions; Column: Chiralpak AD-H (250*4.6 mm*5 µm), mobile phase composition: 30:70:0.1 (v/v/v) n-hexane: isopropyl alcohol: diethyl amine at a flow rate of 0.5 mL∙min−1 with column oven temperature 30˚C. No resolution between S-LINA and R-LINA. (b) Typical chromatogram of method development experiment. Chromatographic conditions; Column: Chiralpak AD-H (250*4.6 mm*5 µm), mobile phase composition: 10:90:0.1 (v/v/v) n-hexane: ethanol: diethyl amine at a flow rate of 0.5 mL∙min−1 with column oven temperature 30˚C. Resolution between S-LINA and R-LINA was 2.2. (c) Typical chromatogram of method development experiment. Chromatographic conditions; Column: Chiralpak AD-H (250*4.6 mm*5 µm), mobile phase composition: 100:0.1 (v/v) Ethanol: Diethyl amine at a flow rate of 0.5 mL∙min−1 with column oven temperature 30˚C. Resolution between S-LINA and R-LINA was 3.8. (d) Typical chromatogram of method development experiment. Chromatographic conditions; Column: Chiralpak AD-H (250*4.6 mm*5 µm), mobile phase composition: 90:10:0.1 (v/v/v) Ethanol: Methanol: Diethyl amine at a flow rate of 0.5 mL∙min−1 with column oven temperature 30˚C. Resolution between S-LINA and R-LINA was 5.5.
coefficient between concentration and peak area was 0.9997. Slope, y-intercept and % y-intercept at 100% level were calculated.
3.2.4. Accuracy
The recovery studies were performed at LOQ, 50%, 100% and 150% to the specification level. Each concentration level was prepared in triplicate, and the recovery was calculated based on mentioned in Equation (1).
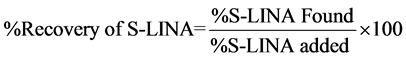 (1)
(1)
The accuracy values are in between 98.6% to 101.5%.
3.2.5. Solution and Mobile Phase Stability
Establishing solution and mobile phase stability by keeping the system suitability solution, Reference solution, test solution and spiked solution separately in tightly closed volumetric flasks at room temperature for 48 hr. during which they were analysed at 12 hrs intervals. Stability of mobile phase was demonstrated by analysis of freshly prepared sample solution at 12 hrs intervals for 48 hrs and comparing the results with those obtained from freshly prepared reference solution. Mobile phase was kept constant during the study period. From the data it can be concluded that mobile phase and sample solutions were found to be stable for 48 hrs.
3.2.6. Robustness
Robustness was studied by altering chromatographic conditions like flow rate and column oven temperature and found to be there is no significant change observed in resolution between S-LINA and R-LINA.
Validation parameters were summarized in Tables 2-4.
Table 2. Summarized data of method validation.
Table 3. S-LINA anova.
3.2.7. Specificity
Specificity is the ability of the method to measure the analyte in presence of degradation products. The specificity of developed method for LINA was performed in the presence of degradation products. Stress studies were performed at concentration of 1 mg∙mL−1 for LINA drug substance. Linagliptin was subjected to Thermal, photolytic, water, oxidation, Acid and Basic conditions were applied. Each degraded sample was injected as such and spiked with enantiomer. To evaluate the ability of proposed method to separate S-LINA from its degradation products, stressed samples were analyzed by using PDA detector and peak purities were calculated, and found that purity angle was within the purity threshold limit in all stressed samples which demonstrates the homogeneity of analyte peak. S-LINA content remains same in all stressed samples. Enantiomer was well separated from the obtained degradation products. Stress conditions for LINA and purity plot were summarized in Table 5 and Figure 4.
Table 4. S-LINA linearity.
Table 5. Summarized data of specificity study.
4. Conclusions
A simple, precise, accurate and cost effective chiral method was developed and validated as per ICH guidelines.
Enantiomers were well separated from each other and the method validation data showed satisfactory results for all tested method parameters. All the degradation products formed during stress conditions were well separated from the S-LINA and purity angle was within the purity threshold limit in all stressed samples indicating that the developed liquid chromatographic method was specific. This simple HPLC method was precise, accurate, linear, robust, sensitive and rugged. Developed method can be used for routine testing in quality control laboratories for estimation of S-LINA in LINA drug substance.

Figure 4. Linagliptin specificity chromatograms.
Acknowledgements
The authors wish to thank the management of Dr. Reddy’s Laboratories Ltd. for supporting this work. Co-opera- tion from colleagues of Research & Development and Analytical Research & Development of Dr. Reddy’s Laboratories Ltd. is appreciated.
Cite this paper
Cholleti Vijay Kumar,Pasula Aparna,Pavan Kumar Vasa,Y. Ravindra Kumar,Nitin Haridas Dhekale, (2016) A New and Enantioselective Chromatographic Method for Linagliptin on Amylose Coated Chiral Stationary Phase. American Journal of Analytical Chemistry,07,556-567. doi: 10.4236/ajac.2016.77051
References
- 1. Nathan, D.M., Buse, J.B., Davidson, M.B., Ferrannini, E., Holman, R.R. and Sherwin, R. (2009) Medical Management of Hyperglycemia in Type 2 Diabetes: A Consensus Algorithm for the Initiation and Adjustment of Therapy: A Consensus Statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care, 32, 193-203.
http://dx.doi.org/10.2337/dc08-9025 - 2. Richter, B., Bandeira-Echtler, E., Bergerhoff, K. and Lerch, C. (2008) Emerging Role of Dipeptidyl Peptidase-4 Inhibitors in the Management of Type 2 Diabetes. Vascular Health and Risk Management, 4, 753-768.
- 3. Scheen, A.J. (2011) Linagliptin for the Treatment of Type 2 Diabetes (Pharmacokinetic Evaluation). Expert Opinion on Drug Metabolism & Toxicology, 7, 1561-1576.
http://dx.doi.org/10.1517/17425255.2011.628986 - 4. Blech, S., Ludwig-Schwellinger, E., Graefe-Mody, E.U., Withopf, B. and Wagner, K. (2010) The Metabolism and Disposition of the Oral Dipeptidyl Peptidase-4 Inhibitor, Linagliptin, in Humans. Drug Metabolism and Disposition, 38, 667-678.
http://dx.doi.org/10.1124/dmd.109.031476 - 5. Perrin, S.P. and Pirkle, W.H. (1991) Commercially Available Brush-Type Chiral Selectors for Direct Resolution of Enantiomers. ACS Symposium Series, 471, 43-66.
http://dx.doi.org/10.1021/bk-1991-0471.ch003 - 6. Wainer, I.W., Alembik, M.C. and Smith, E. (1987) Resolution of Enantiomeric Amides on a Cellulose Tribenzoate Chiral Stationary Phase: Mobile Phase Modifier Effects on Retention and Stero-Selectivity. Journal of Chromatography A, 388, 65-74.
http://dx.doi.org/10.1016/S0021-9673(01)94466-8 - 7. Nguyen, L.A., He, H. and Pham-Huy, C. (2006) Chiral Drugs, an Overview. International Journal of Biomedical Science, 2, 85-100.
- 8. Vijaya Sri, K., Anusha, M. and Ravindra Reddy, S. (2015) A Rapid RP-HPLC Method Development and Validation for the Analysis of Linagliptinin Bulk and Pharmaceutical Dosage Form. Asian Journal of Pharmaceutical Analysis, 5, 16-20.
http://dx.doi.org/10.5958/2231-5675.2015.00003.4 - 9. Shekar Reddy, B.R.C., Vijaya Bhaskar Rao, N. and Saraswathi, K. (2014) A Validated Stability Indicating HPLC Assay Method for Linagliptin. Der Pharmacia Sinica, 5, 131-137.
- 10. Madhusudhan, P., Radhakrishna Reddy, M. and Devanna, N. (2015) RPHPLC Method Development and Validation for Simultaneous Determination of Linagliptin and Empagliflozine in Tablet Dosage Form. International Advanced Research Journal in Science, Engineering and Technology, 2, 95-99.
- 11. Padmaja, N. and Veerabhadram, G. (2015) Development and Validation of Analytical Method for Simultaneous Estimation of Empagliflozin and Linagliptin in Bulk Drugs and Combined Dosage Forms Using UV-Visible Spectroscopy. Der Pharmacia Lettre, 7, 306-312.
- 12. Mallikarjuna Rao, N. and Gowri Sankar, D. (2015) Rp-Hplc Method For Simultaneous Estimation and Stability Indicating Study of Metformin and Linagliptin in Pure and Pharmaceutical Dosage Forms. International Journal of Pharmacy and Pharmaceutical Sciences, 7, 191-197.
- 13. Vemula, P., Dodda, D., Balekari, U., Panga, S. and Ciddi, V. (2015) Simultaneous Determination of Linagliptin and Metformin by Reverse Phase-High Performance Liquid Chromatography Method: An Application in Quantitative Analysis of Pharmaceutical Dosage Forms. Journal of Advanced Pharmaceutical Technology and Research, 6, 25-28.
http://dx.doi.org/10.4103/2231-4040.150368 - 14. Sujatha, K. and Seshagiri Rao, J.V.L.N. (2013) New RP-HPLC Method for the Estimation of Linagliptin in Tablet Dosage Forms. Indo American Journal of Pharmaceutical Research, 3, 8376-8381.
- 15. Kavitha. K.Y., Geetha, H., Kaviarasu, M. and Venkatnarayanan, R. (2013) Development and Validation of Stability Indicating RP-HPLC Method for the Simultaneous Estimation of Linagliptin and Metformin in Pure and Pharmaceutical Dosage Form. Journal of Chemical and Pharmaceutical Research, 5, 230-235.
- 16. Janardhan Swamy, A. and Harinadha Baba, K. (2013) Analytical Method Development and Method Validation for the Simultaneous Estimation of Metformin HCL and Linagliptin in Bulk and Tablet Dosage Form by RP-HPLC Method. International Journal of Pharmaceutics, 3, 594-600.
- 17. Lakshmi, B. and Reddy, T.V. (2012) A Novel Rp-Hplc Method for the Quantification of Linagliptin in Formulations. Journal of Atoms and Molecules, an International Online Journal, Jamonline, 2, 155-164.
- 18. Yamamoto, C., Inagaki, S. and Okamoto, Y. (2006) Enantioseparation Using Alkoxyphenyl Carbamates of Cellulose and Amylose as Chiral Stationary Phase for High-Performance Liquid Chromatography. Journal of Separation Science, 29, 915-923.
http://dx.doi.org/10.1002/jssc.200500514 - 19. Ye, Y.K., Lord, B.S., Yin, L. and Stringham, R.W. (2002) Enantioseparation of Amino Acids on a Polysaccharide-Based Chiral Stationary Phase. Journal of Chromatography A, 945, 147-159.
http://dx.doi.org/10.1016/S0021-9673(01)01490-X - 20. Lammerhofer, M. and Lindner, W. Handbook of Analytical Separations, Recent Developments in Liquid Chromatographic Enantioseparation. Separation Methods in Drug Synthesis and Purification, 1, 337-437.
- 21. Gubitz, G. and Schmid, M.G. Methods in Molecular Biology. Chiral Separations Methods and Protocols, 243.
- 22. ICH (1999) Topic Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products. Chemical Substances.
NOTES

*Corresponding author.