American Journal of Analytical Chemistry
Vol.06 No.01(2015), Article ID:53053,8 pages
10.4236/ajac.2015.61004
Effect of Hydrogen in Adsorption and Direct Dissociation of CO on Fe (100) Surface: A DFT Study
Sebastián Amaya-Roncancio1*, Daniel Humberto Linares1*, Hélio Anderson Duarte2, Germán Lener1, Karim Sapag1
1Department of Physics, Applied Physics Institute, National University of San Luis, San Luis, Argentina
2Department of Chemistry―ICEx, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
Email: *samaya@unsl.edu.ar, *dlinares@unsl.edu.ar
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 16 October 2014; revised 3 December 2014; accepted 17 December 2014
ABSTRACT
Density functional theory was employed to investigate the effect of the hydrogen in the adsorption and direct dissociation of CO on Fe (100) surface. The preadsorption of hydrogen with coverages of 0, 1/3 and 2/3 monolayer (ML) was used in the present investigation. In the case of 1/3 ML of hydrogen, two configurations of adsorption were studied. The presence of hydrogen shows a major transference of electronic density from Fe surface to CO adsorbed, increasing the adsorption energy of CO from 2.00 eV in clean surface, to 2.76 eV in 2/3 ML of hydrogen. Furthermore, the activation barrier for direct dissociation of CO was 1.13 eV and for the recombination energy 2.28 eV on clean Fe (100) surface. In the same way, the activation barrier for CO in the presence of coadsorbed hydrogen was slightly affected presenting values of 1.06 eV and 1.16 eV to 1/3 ML configurations and 0.98 eV for 2/3 ML of hydrogen. Finally, the recombination energy decreases to 1.63 eV and 1.49 eV for 1/3 ML configurations and to 1.23 eV for 2/3 ML of coadsorbed hydrogen. These results indicate that the CO adsorption and dissociation are favored in the presence of hydrogenated surfaces.
Keywords:
Adsorption, Dissociation, Carbon Monoxide, Density Functional Calculations, Iron

1. Introduction
The interaction between carbon monoxide and transition metal surfaces has been subject of many experimental and theoretical investigations and it is an important step in many industrial processes such as Fischer-Tropsch synthesis (FTS) [1] . FTS technology has become important because of the rapid hydrocarbon resource depletion, the unpredictable prices of crude oil, the increased fuel demands worldwide, and the environmental regulations. Among many transition metals having FTS activity, iron-based catalysts are preferred due to their low price, high activity, and low CH4 selectivity [2] [3] . Several experimental techniques have been used to investigate the adsorption of CO on Fe (100). These techniques include Temperature-Programmed Desorption (TPD) [4] [5] , Ultraviolet Photoelectron Spectroscopy (UPS) [5] , X-ray Photoelectron Spectroscopy (XPS) [6] , Electron- Energy Loss Spectroscopy (EELS) [7] , X-ray Photoelectron Diffraction (XPD) [8] , and Near-Edge X-ray Absorption fine structure (NEXAFS) [9] [10] . Experimentally, it is known that CO adsorbs molecularly on Fe (100) at low temperatures (at all coverages) giving rise to three distinctive TPD peaks, which are labeled as α1 (220 K), α2 (305 K), and α3 (440 K) corresponding to three molecular desorption states [4] [5] . The activation energies of desorption of α1, α2, and α3 states, calculated using the Redhead equation and a pre-exponential factor of 1013 are 12.9, 18.0, and 26.3 kcal∙mol−1 respectively. In addition, there is a fourth desorption state, labeled β, that appears at 820 K and is caused by the recombination of atomic carbon and oxygen on the surface. In 1981, Erley [7] studied CO adsorption on Fe (1 1 0) using Low Energy Electron Diffraction (LEED) and High Resolution Electron Energy Loss Spectroscopy (HREELS) at 120 K, displaying four high-symmetry sites on Fe (110), the long-bridge (LB), the quasi-threefold (TF), the short-bridge (SB), and the on top (OT) sites. In the same way, Erley [11] proposed an upright OT adsorption of CO at 0.25 ML and a tilted and displaced OT adsorption at 0.5 ML. Saiki et al. [8] reported a tilting angle of 55˚ ± 2˚ using XPD and Dwyer et al. [9] found an angle of 54.7˚ using NEXAFS. The α3 state has been proposed as a precursor for the dissociation of CO molecule on Fe(100), and the activation energy calculated from TPD has been suggested to be the apparent activation energy of dissociation of CO molecule instead of the activation energy of desorption of the CO molecules in the α3 state. In the same way, NEXAFS experiments revealed that CO molecule in α3 state is indeed tilted on the surface with an angle of 45˚ ± 10˚ between the CO molecular axis and the surface normal [10] . Despite of the interest in CO dissociation on iron catalysts for the chemical industry, and the several experimental works reported about the adsorption and dissociation of CO on iron surfaces [12] [13] , theoretical calculations are relatively limited. Most of the theoretical works reported were carried out using Hartree-Fock methods [14] [15] , and more recently, DFT calculations were performed on Fe surfaces and cluster models [16] . Sorescu and co-workers [17] -[19] have reported adsorption energies, structures, and CO stretching frequencies for several configurations of CO on Fe (100) at 0.25 ML. Thus, CO adsorption was found perpendicular to the surface at on top, bridge, and fourfold hollow site and tilted at four-fold hollow site. In addition, Sorescu et al. have studied several diffusion pathways and dissociation channels, reporting a CO stretching frequency of 1246 cm−1 for a CO molecule tilted on a fourfold hollow site with an angle between the CO molecular axis and the surface normal of 50˚. The effect of potassium promoters in adsorption and dissociation of CO on Fe (100), and the dissociative adsorption of H2 on the same metal surface was studied as well. In other way, Jiang et al. [20] have investigated the CO adsorption and dissociation in several coverage (0.25, 0.5, 1 ML) using different supper-cells p(1×2), p(2×2) and p(2×4), and have reported high dependence on coverage in direct dissociation, and on top preferential adsorption of CO in all studied coverages. The dissociation mechanism is followed by an endothermic diffusion of CO from top site to a long-bridge site with a barrier of 0.1 eV favoring the direct dissociation, where an activation energy of 1.52 eV was calculated for that mechanism. Besides of the direct dissociation of CO, the experimental results of Ojeda et al. [21] have provided evidence of two parallel CO activation pathways on CO-covered Fe catalysts, the direct and the H-assisted, both routes show a trend to form CH2 monomers. These two mechanisms differ in the oxygen rejection pathways, the unassisted CO activation removes O as CO2, while the H-assisted pathway forms H2O exclusively. In addition, to CO direct dissociation, H atom was found to assist CO dissociation through the formation of intermediate formyl (CHO) on several metals. In the same way, Shetty and coworkers [22] have reported that for compact faces, CHO route is favorable, whereas for corrugated facets, CO direct dissociation might predominate. In the present work we have carried out comprehensive calculations within spin-polarized density functional theory (DFT), to study the dissociative adsorption of H2 and the effect of the preadsorbed hydrogen in the CO adsorption on Fe(100) under (0, 1/3, 2/3) ML of hydrogen coverages. In addition, the mechanisms of direct dissociation of CO on clean and hydrogenated surfaces were studied. Our ultimate goal is to understand the hydrogen influence on the CO adsorption and dissociation, and provide an explanation of hydrogen influence in the initial steps of Fischer-Tropsch reactions on Fe (100).
2. Computational Methods
We modeled the Fe (100) using a four-metal-layer slab model. Two layers were kept fixed with the bulk lattice parameter, and the other two layers free to relax. A p(2×3) supercell with a vacuum space of 8.6Ǻ was used to study the adsorption of CO and dissociative adsorption of H2 at 1/6 and 1/3 ML of coverage respectively. The same supercell was used to investigate the direct dissociation of CO coadsorbed with hydrogen. Bulk relaxation was achieved with a lattice parameter of 2.850 Å, while the experimental value is 2.8665 Å [23] . All calculations were carried out with the use of Quantum Espresso package [24] . Ultra-soft potentials with a scalar relativistic correction generated with Rappe-Rabe-Kaxiras-Joannopoulos method (RRKJUS) were used to describe the electron-ion interactions [25] [26] . The generalized gradient approximation (GGA) for the exchange/corre- lation density functional Perdew-Burke-Ernzerhof (PBE) was used [27] . Spin polarization calculations were used to ensure accurate results due to the magnetic nature of the Fe and Monkhorst-Pack meshes of 3 × 3 × 1 k-point sampling in the Brillouin zone were used [27] . For all calculations the chosen energy cutoff for the wave functions and for the charge density were equal to 544 eV and 2450 eV respectively, and the threshold for self-consistency used was 1 × 10−6 eV. The geometry relaxation was done using BFGS quasi-Newton algorithm until two conditions were simultaneously satisfied. First, the forces on each atom were less than 10−5 eV/Å and second, the energy difference of consecutive steps were less than 10−4 eV. The study of the minimum energy path for the different reaction was undertaken using the climbing image nudged elastic band method (CINEB) [28] [29] and the local minima were found through the conjugate gradient CG technique, employing DFT calculation with spin polarization. All the molecular and density plots were made with the XCrySDen package [30] .
3. Results
The adsorption of H2 and CO processes are the first steps for understanding the influence of the hydrogen in the mechanisms of adsorption and direct CO dissociation on iron surfaces. In order to investigate the crucial steps in the adsorption and dissociation processes, we analyzed the active sites and stable configurations of all the involved species as previously reported in the literature [31] -[33] . The adsorption energies and sites of involved species are shown in Table 1. The adsorption energy of a molecule in a surface was calculated using the following equation:
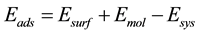 , (1)
, (1)
where Eads is the adsorption energy of the molecule, Esurf is the total energy of the clean slab or the slab with preadsorbed hydrogen, Emol is the total energy of the molecule at vacuum and Esys is the total energy of the molecule/slab [17] . On other hand, to analyze the electronic density generated by the interactions of the system, we calculated the pseudo charge density difference Δη according to Equation (2) and suggested elsewhere [34] .
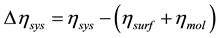 , (2)
, (2)
where ηsys is the pseudo charge density of the molecule/slab, ηsurf and ηmol are the pseudo charge density of the isolated slab (or preadsorbed hydrogen) and the gas phase CO molecule, respectively.

Table 1. Energetic and geometric parameters of CO adsorbed on Fe (100) with preadsorbed hydrogen.
*Reported value of CO adsorption on a p(2×2) supercell of clean Surface of Fe (100) by Sorescu et al. [17] .
3.1. Dissociative Adsorption of H2
Firstly the adsorption of H2 molecule on Fe (100) surface was analyzed in parallel orientation to the surface and being adsorbed at on top site. The split hydrogen adsorbed on bridge or hollow sites were also investigated as shown at Figure 2. These calculations have been done as follows: a molecule of H2 was fixed in z-axis, parallel to the surface at 4.3 Å and the atomic positions of Fe and H2 were allowed to relax (Figure 1(a)). It was found that close to the surface, the molecule dissociation takes place. Once the final adsorbed configurations have been determined; we refined the calculations using NEB. The first site of the surface that weakly interacts with the molecule is the top site, with an interaction distance of 1.69 Å above the surface (Figure 1(b)). This site makes H2to dissociate readily, promoting each H atom to bridge sites (Figure 2(a)), and finally spreading the H atoms to the hollow sites as shown Figure 2(b), Figure 2(c). Even though dissociated H2 is present in the bridge sites, the geometric relaxations and NEB calculations shows that hollow sites are the most favored places for adsorbing H2 [18] . The dissociative adsorption of H2 has a small activation barrier of 0.05 eV, but from the interaction of H2 with the Fe on top, Figure 1(b), the adsorption is mainly a non-activated process with an exothermic energy of 0.55 eV for atomic hydrogen adsorbed on the bridge sites (Figure 2(a)) and 0.83 eV on hollow sites Figure 2(b), Figure 2(c). No appreciable differences among energies in the final configurations of dissociative adsorption on hollow sites were found. This activation barrier values and adsorption geometries are in accordance with the values reported by Sorescu [18] . We need to emphasize, however, that the weakly bounded molecular state described above (Figure 1(b)) should be considered carefully. The exchange/correlation functional used does not have the right physics to account for van der Waals interactions and, consequently, might not provide an accurate description of such weakly bound states [18] [19] . The hollow site (Figure 2(b), Figure 2(c)), were used for the calculations of CO adsorption with preadsorbed hydrogen.
3.2. Adsorption of CO with in Presence of Hydrogen
The effect of the hydrogen on the CO adsorption energy were investigated for 0, 1/3 and 2/3 ML hydrogen coverages. Initially the CO was adsorbed on the clean surface (0 ML of hydrogen adsorption) at hollow site. Afterwards, with the CO adsorbed in the hollow site, hydrogen atoms were added for 1/3 and 2/3 ML of hydrogen.
 (a) (b)
(a) (b)
Figure 1. Dissociative adsorption of H2 on Fe (100). (a) H2 parallel to iron surface; (b) First contact of H2 with iron on Top site. Fe in yellow, H in cyan.
 (a) (b) (c)
(a) (b) (c)
Figure 2. Dissociative adsorption of H2 on Fe (100). (a) H2 Adsorbed on Bridges sites; (b) H2 Adsorbed on adjacent Hollow sites; (c) H2 adsorbed in asymmetric Hollow sites.
Then, the whole system was relaxed including the hydrogen atoms and the CO molecule. Figure 3, shows the optimized CO/H2 co-adsorbed surfaces. The hollow sites are the preferred adsorption sites for CO and hydrogen as previously reported [14] -[19] [31] -[33] . The adsorption energies of CO were calculated using Equation (1) and are shown in Table 1. For 0 ML of adsorbed hydrogen, the adsorption energy of CO was 2.00 eV (see Table 1). The relaxed geometry presents OC-Fe bond distance of 1.91 Å and a tilt of 46.6˚ with respect to the iron surface in the hollow site (Figure 3(a)). These results are in good agreement with the previously reported calculations [17] . Table 1 shows the energetic and geometric parameters for the CO adsorption on Fe (100) surfaces with coasorbed hydrogen in different configurations. In the case of 1/3 ML two cases were found, the asymmetric and symmetric configurations of coadsorbed H around the CO (see Figure 3). For 1/3-asymmetric ML of H (Figure 3(b)), the orientation of CO remains as in 0 ML case, but the tilt angle slightly increase to 46.9˚. The adsorption energy of CO was 2.26 eV and the distance of CO to the surface was estimated to be 1.97 Å. For 1/3-symmetric ML of H (Figure 3(c)), the adsorbed CO moves to a transversal orientation with respect to H coadsorbed, and the CO tilt increase to 47.3˚. The adsorption energy of CO calculated with Equation (1) increases to 2.70 eV, and the CO distance to Fe surface was calculated to be 1.95 Å (Table 1). For 2/3 ML of hydrogen adsorbed on Fe (100) surface (Figure 3(d)) the adsorption energy was highly increased to 2.76 eV and the CO distance to the surface was increased to 1.98 Å with a tilt angle of 45.41˚. In general, the Fe C-O bond distance remain similar in all cases with a slight decrease of 0.03 Å from 0 ML to 2/3 ML of hydrogen coadsorbed (Table 1). The case of 2/3 ML of adsorbed hydrogen presents the largest adsorption energy (see Table 1). It is worth to emphasize that the proportion of adsorbates presented in 2/3 ML of H, corresponds to the proportion of reactants used experimentally in FTS [35] . The ΔE shown at the Table 1 is the relative adsorption energy with respect to the CO adsorbed in the clean surface. The negative sign of ΔE indicates that hydrogen favors the adsorption of CO on the surface [36] . The preadsorbed hydrogen leads to a remarkable stabilization of CO on the surface. This stabilization is estimated to be about 0.70 eV for 1/3-symmetric and 2/3 ML of preadsorbed hydrogen on the Fe (100) surface. Figure 3, shows Δη of the system calculated with Equation (2). This pseudo- differential indicates the generated electronic density from the interaction between CO molecule and clean surface or hydrogenated surface. Figure 3(a) shows how the Fe atoms transfer electronic density to the carbon of the adsorbed CO at 0 ML of preadsorbed hydrogen. For 1/3-asymmetric ML of H (Figure 3(b)), there is a major transference of electronic density from Fe to the C of the adsorbed CO molecule, favoring CO binding to the surface. For 1/3-symmetric ML, the CO molecule is reoriented favoring the interaction with the hydrogenated surface. The Δη indicates an increment of the accumulation lobules in the CO-surface interaction, and a major trans- ference of electronic density to C (see Figure 3(c)). From all adsorptions showed in Figure 3, it is possible to see a depletion of electronic density around the C-O bond, which indicates that the transfer of electron density from surface to C favors the CO dissociation on Fe, and the covalent interaction between Fe and C. The 2/3 ML of H does not show major differences with respect to pseudo difference electron density of 1/3-symmetric distribution.
 (a) (b) (c) (d)
(a) (b) (c) (d)
Figure 3. Geometries and pseudo-differential electronic density of adsorption for: (a) CO on clean surface; (b) CO on Fe with 1/3-asymmetric ML of H; (c) CO on Fe with 1/3-symmetric ML of H; (d) CO on Fe with 2/3 ML of H. Grey atoms: C, yellows atoms: Fe, red atoms: O, cyan atoms: H. Red lobules: accumulation of electronic density, blue lobules: depletion of electronic density. Value of isosurface: 0.005 eV/A3.
3.3. Direct Dissociation of CO in Presence of Hydrogen
The direct dissociation of CO on Fe (100) was calculated after finding the initial state (IS) of CO for 0, 1/3 and 2/3 ML of H as shown in Figure 3. The transition state (TS) of the reaction was calculated using NEB, and consist of a carbon atom at a hollow site and the oxygen atom close to a bridge site, see Figure 4(b). The final state (FS) of CO dissociation was found by geometrical optimization where the C and O atoms adsorb in adjacent hollow sites. Similar TS and FS geometries were found for all coverages of H (Figure 4(b), Figure 4(c)). The activation energy (EForward) of CO dissociation on clean Fe (100) was 1.13 eV (Table 2) and the recombination energy (EBack) was 2.28 eV showing the exothermic nature of the dissociation process (Table 2). The IS, TS and FS at 0 ML coverage of H are in good agreement with the other reported calculations of Wang et al. [31] , Tracy et al. [32] , Xun-hua et al. [33] , and Elahifard et al. [37] . The direct dissociation of CO in the presence of coadsorbed H present slight variations of forward barrier to all coverages studied, where 1.06 eV, 1.16 eV and 0.90 eV were found for 1/3-asymmetric, 1/3-symmetric and 2/3 ML of H, respectively. The recombination barrier decreases from 2.28 eV in 0 ML of H to 1.14 eV in 2/3 ML of H (see Table 2). In all cases, the CO dissociation remains exothermic. The calculations of Δη in FS for direct dissociation on clean surface and in the presence of 2/3 ML of H are shown in Figure 5. To the cases of 1/3-asymmetric and 1/3-symmetric ML of H no major difference for Δη was observed in comparison with 2/3 ML of H. Figure 5(b) shows more accumulation of electronic density on the surface in presence of H compared with the clean surface (Figure 5(a)). In the same way, the transference of electronic density to the dissociated C atom decreases in the presence of H. The presence of H in the direct dissociation could cause a decreasing of EBack by the destabilization of the surface in FS. In presence of H, the atoms adsorbed in FS (H, C and O) could be more reactive promoting the formation of CHn chains and elimination of O by H2O formation.
 (a) (b) (c)
(a) (b) (c)
Figure 4. Direct dissociation in presence of 1/3 ML of hydrogen. (a) Initial state; (b) Transition state; (c) Final state.
 (a) (b)
(a) (b)
Figure 5. Final State Geometries of dissociation of CO and pseudo-differential electronic density (Δη) in presence of. (a) 0 ML of H; (b) 2/3 ML of H. Value of isosurface: 0.005 eV/A3.
Table 2. Activation barriers of CO dissociation for clean surface and in presence of hydrogen.
*Reported values of direct dissociation of CO on p(2×2) supercell of Fe (100) by Elahifard et al. [37] .
4. Conclusion
Fe (100) slab model was used to investigate the dissociative adsorption of H2 and the effect of preadsorbed hydrogen on the CO adsorption and its direct dissociation. The CO adsorption energy increases from 2.00 eV for the clean surface to 2.76 eV for 2/3 ML of preadsorbed hydrogen on Fe (100) surface. Transference of electron density from the surface to the carbon has been observed leading to the increase of the stabilization of the CO. The depletion of the electron density around the C-O bond increases its reactivity towards the dissociation. However, the direct dissociation in the presence of coadsorbed hydrogen slightly affects the direct dissociation barrier about 1.13 - 0.90 eV. However, a remarkable decrease of the recombination barrier has been observed from 2.28 eV for the clean surface to 1.14 eV for 2/3 ML of hydrogen. The decrease of the electron density donated to the adsorbed carbon atom due to the presence of the preadsorbed hydrogen is probably the explanation for this result. In conclusion, the presence of the preadsorbed hydrogen leads to the increase of the CO adsorption energy and the decrease of the dissociation energy barrier. However, the reaction becomes less favored due to the increase of the dissociation energy (less negative) and the decrease of the recombination energy barrier, which affects the global reaction of the CO on the Fe (100) surface. In order to understand the complete reaction, it is necessary to accomplish the other steps of the reaction such as the formation of the H2O and CH2 on the surface.
Acknowledgements
The authors thanks to Universidad Nacional de San Luis, AMCYT, CAPES and SPU for financial support and to INCT-ACQUA for the computational facilities.
References
- Anderson, R.B. (1984) The Fischer-Tropsch Synthesis. Academic Press, Orlando.
- Nassir, M.H., Frahberger, B. and Dwyer, D.J. (1994) Coverage Dependence of CO Dissociation on Clean and Hydrogen Presaturated Fe(100) Surface. Surface Science, 312, 115-123. http://dx.doi.org/10.1016/0039-6028(94)90808-7
- Lu, J.-P., Albert, M.R. and Bernasek, S.L. (1990) Effects of Postdosed Species on Preadsorbed Carbon Monoxide on Iron (100): Adsorption Site Conversion Caused by Site Competition. The Journal of Physical Chemistry, 94, 6028- 6033. http://dx.doi.org/10.1021/j100378a074
- Cameron, S.D. and Dwyer, D.J. (1988) A Study of .pi.-Bonded Carbon Monoxide on Iron (100). Langmuir, 4, 282-288. http://dx.doi.org/10.1021/la00080a007
- Moon, D.W., Bernasek, S.L., Lu, J.-P., Gland, J.L. and Dwyer, D.J. (1987) Activation of Carbon Monoxide on Clean and Sulfur Modified Fe (100). Surface Science, 184, 90-108. http://dx.doi.org/10.1016/S0039-6028(87)80274-1
- Moon, D.W., Dwyer, D.J. and Bernasek, S.L. (1985) Adsorption of CO on the Clean and Sulfur Modified Fe (100) Surface. Surface Science, 163, 215-219. http://dx.doi.org/10.1016/0039-6028(85)90859-3
- Moon, D.W., Bernasek, S.L., Dwyer, D.J. and Gland, J.L. (1985) Observation of an Unusually Low Carbon Monoxide Stretching Frequency on Iron (100). Journal of the American Chemical Society, 107, 4363-4364. http://dx.doi.org/10.1021/ja00300a064
- Saiki, R.S., Herman, G.S., Yamada, M., Osterwalder, J. and Fadley, C.S. (1989) Structure of an Unusual Tilted State of CO on Fe (001) from X-Ray Photoelectron Diffraction. Physical Review Letters, 63, 283-286. http://dx.doi.org/10.1103/PhysRevLett.63.283
- Dwyer, D.J., Rausenberger, B., Lu, J.-P., Bernasek, S.L., Fischer, D.A., Cameron, S.D., Parker, D.H. and Gland, J.L. (1989) Fluorescence Yield near Edge Spectroscopy of π-Bonded CO on Fe (100). Surface Science, 224, 375-385. http://dx.doi.org/10.1016/0039-6028(89)90921-7
- Moon, D.W., Cameron, S.D., Zaera, F., Eberhardt, W., Carr, R., Bernasek, S.L., Gland, J.L. and Dwyer, D.J. (1987) A Tilted Precursor for CO Dissociation on the Fe (100) Surface. Surface Science, 180, L123-L128. http://dx.doi.org/10.1016/0039-6028(87)90033-1
- Erley, W. (1981) Vibrational Spectra of CO Chemisorbed on Fe (110). Journal of Vacuum Science & Technology, 18, 472-475. http://dx.doi.org/10.1116/1.570768
- Whitman, L.J., Richter, L.J., Gurney, B.A., Villarrubia, J.S. and Ho, W. (1989) CO Adsorption Site Occupations on Fe (111) vs Coverage and Temperature: The Kinetics of Adsorption and Reaction. The Journal of Chemical Physics, 90, 2050-2062. http://dx.doi.org/10.1063/1.455996
- Bartosch, C.E., Whitman, L.J. and Ho, W. (1986) The Adsorption, Interconversion, and Dissociation of CO on Fe (111). The Journal of Chemical Physics, 85, 1052-1060. http://dx.doi.org/10.1063/1.451298
- Mehandru, S.P. and Anderson, A.B. (1988) Binding and Orientations of CO on Fe (110), (100), and (111): A Surface Structure Effect from Molecular Orbital Theory. Surface Science, 201, 345-360. http://dx.doi.org/10.1016/0039-6028(88)90617-6
- Blyholder, G. and Lawless, M. (1993) A Theoretical Study of the Site of CO Dissociation on Fe (100). Surface Science, 290, 155-162. http://dx.doi.org/10.1016/0039-6028(93)90597-D
- Meehan, T.E. and Head, J.D. (1991) A Theoretical Comparison of CO Bonding on the Fe (100) Surface. Surface Science, 243, L55-L62. http://dx.doi.org/10.1016/0039-6028(91)90334-O
- Sorescu, D.C., Thompson, D.L., Hurley, M.M. and Chabalowski, C.F. (2002) First-Principles Calculations of the Adsorption, Diffusion, and Dissociation of a CO Molecule on the Fe (100) Surface. Physical Review B, 66, Article ID: 035416. http://dx.doi.org/10.1103/PhysRevB.66.035416
- Sorescu, D.C. (2005) First Principles Calculations of the Adsorption and Diffusion of Hydrogen on Fe (100) Surface and in the Bulk. Catalysis Today, 105, 44-65. http://dx.doi.org/10.1016/j.cattod.2005.04.010
- Sorescu, D.C. (2011) Adsorption and Activation of CO Coadsorbed with K on Fe(100) Surface: A Plane-Wave DFT Study. Surface Science, 605, 401-414. http://dx.doi.org/10.1016/j.susc.2010.11.009
- Jiang, D.E. and Carter, E.A. (2004) Adsorption and Dissociation of CO on Fe (110) from First Principles. Surface Science, 570, 167-177. http://dx.doi.org/10.1016/j.susc.2004.07.035
- Ojeda, M., Nabar, R., Nilekar, A.U., Ishikawa, A., Mavrikakis, M. and Iglesia, E. (2010) CO Activation Pathways and the Mechanism of Fischer-Tropsch Synthesis. Journal of Catalysis, 272, 287-297. http://dx.doi.org/10.1016/j.jcat.2010.04.012
- Shetty, S.G., Jansen, A.P.J. and Van Santen, R.A. (2009) Direct Versus Hydrogen-Assisted CO Dissociation. Journal of the American Chemical Society, 131, 12874-12875. http://dx.doi.org/10.1021/ja9044482
- Kohlhaas, R., Dunner P. and Schmitz-Pranghe N. (1967) Uber die temperaturabhangigkeit der gitterparameter von eisen, kobalt und nickel im bereich hoher temperaturen. Zeitschrift fur Angewandte Physik, 23, 245-249. http://www.crystallography.net
- Giannozzi, P., et al. (2009) QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. Journal of Physics-Condensed Matter, 21, 1-19.
- Fe.pbe-nd-rrkjus.UPF. http://www.quantum.espresso.org
- H.pbe-nd-rrkjus.UPF, O.pbe-nd-rrkjus.UPF and C.pbe-nd-rrkjus.UPF. http://www.quantum.espresso.org
- Payne, M.C., Teter, M.P., Allan, C., Arias, T.A. and Joannopoulos, J.D. (1992) Iterative Minimization Techniques for ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Reviews of Modern Physics, 64, 1045-1097. http://dx.doi.org/10.1103/RevModPhys.64.1045
- Henkelman, G., Uberuaga, B.P. and Jonsson, H. (2000) A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. Journal of Chemical Physics, 113, 9901-9904. http://dx.doi.org/10.1063/1.1329672
- Borthwick, D., Fiorin, V., Jenkins, S.J. and King, D.A. (2008) Facile Dissociation of CO on Fe {211}: Evidence from Microcalorimetry and First-Principles Theory. Surface Science, 602, 2325-2332. http://dx.doi.org/10.1016/j.susc.2008.05.014
- Kokalj, A. (2003) Computer Graphics and Graphical User Interfaces as Tools in Simulations of Matter at the Atomic Scale. Computational Materials Science, 28, 155-168. http://dx.doi.org/10.1016/S0927-0256(03)00104-6
- Wang, T., Tian, X.X., Li, Y.W., Wang, J.G., Beller, M. and Jiao, H.J. (2014) Coverage Dependent CO Adsorption and Dissociation Mechanisms on Iron Surfaces from DFT Computations. Journal of Physical Chemistry C, 118, 1095-1101. http://dx.doi.org/10.1021/jp4103367
- Bromfield, T.C., Ferro, D.C. and Niemantsverdriet, J.W. (2005) A DFT Study of the Adsorption and Dissociation of CO on Fe (100): Influence of Surface Coverage on the Nature of Accessible Adsorption States. ChemPhysChem, 6, 254-260. http://dx.doi.org/10.1002/cphc.200400452
- Zhao, X.H., Li, Y.W., Wang, J.G. and Huo, C.F. (2011) CO Adsorption, CO Dissociation, and C-C Coupling on Cu Monolayer-Covered Fe (100). Journal of Fuel Chemistry and Technology, 39, 956-960.
- Corso, A.D., de Gironcoli, S. and Baron, S. (2002) The Interaction of Ethylene with Perfect and Defective Ag (001) Surfaces. Journal of Physical Chemistry B, 106, 9839-9846.
- Blanco, A.A.G., Amaya, M.G., Roca Jalil, M.E., Nazzarro, M., Oliva, M.I. and Sapag, K. (2011) Effect of the Synthesis Method on Co-Catalysts Based on MCM-41 for the Fischer-Tropsch Reaction. 54, Topics in Catalysis, 190-200.
- Liu, H.Y., Yan, R.X., Zhang, R.G., Wang, B.J. and Xie, K.C. (2011) A DFT Theoretical Study of CH4 Dissociation on Gold-Alloyed Ni (111) Surface. Journal of Natural Gas Chemistry, 20, 611-617. http://dx.doi.org/10.1016/S1003-9953(10)60252-6
- Elahifard, M.R., Pérez Jigato, M. and Niemantsverdriet, J.W. (2012) Direct versus Hydrogen-Assisted CO Dissociation on the Fe (100) Surface: A DFT Study. ChemPhysChem, 13, 89-91. http://dx.doi.org/10.1002/cphc.201100759
NOTES

*Corresponding authors.