American Journal of Analytical Chemistry
Vol. 3 No. 6 (2012) , Article ID: 20041 , 6 pages DOI:10.4236/ajac.2012.36056
Determination and Validation of Markers for Heat-Induced Damage in Wool Proteins
1Food and Bio-based Products, Lincoln Research Centre, AgResearch, Lincoln, New Zealand
2Department of Wine, Food and Molecular Biosciences, Lincoln University, Lincoln, New Zealand
3The Biomolecular Interaction Centre, University of Canterbury, Christchurch, New Zealand
4The Riddet Institute, Massey University, Palmerston North, New Zealand
Email: anita.grosvenor@agresearch.co.nz
Received April 2, 2012; revised May 9, 2012; accepted May 23, 2012
Keywords: Wool Proteins; Protein Damage; Molecular Markers; Mass Spectrometry; Hydrothermal; Heat
ABSTRACT
Protein-based animal fibres of commercial importance are frequently exposed to elevated temperatures during processing treatments. Hydrothermal processes cause protein deterioration, impacting negatively on the value or condition of these materials. This study was designed to investigate hydrothermal damage in wool proteins at the molecular level. The effect of hydrothermal damage on Type I and II intermediate filament proteins (keratins) extracted from wool was characterised using advanced quantitative techniques based on isobaric iTRAQ labelling and mass spectrometry. Many native peptides were observed to be degraded and modified. Amongst these, twenty keratin peptides were observed to consistently degrade during hydrothermal exposure. These peptides acted as molecular markers of damage – specific indicators of the extent of heat-induced protein damage. This technology will be of value in assessing the severity of damage imparted after high temperature exposure of protein-based animal fibres such as wool and cashmere during processes such as dyeing and carbonising, or even after high temperature human hair treatments. The identification of molecular damage markers identified within wool and other materials provides a new route to sensitive and specific evaluation of the effects of protein deterioration. It is anticipated that the utilisation of such markers will facilitate the development of targeted approaches to minimising processing damage to high-value fibres and protein-based biomaterials.
1. Introduction
Proteins are vulnerable to the degradative effects of oxidation, which results in lowered structural integrity and/or functionality due to chemical modifications to the protein molecular structure. In wool, oxidative modification results in lowered strength and abrasion resistance, along with undesirable colour changes [1,2]. Protein oxidation occurs primarily through the action of reactive oxygen species (ROS) and their derivatives, which, in combination, react with a large variety of functional groups [3]. Despite this general reactivity, protein oxidation is quite selective, with certain portions of these large molecules more susceptible to modification than others [4]. Consequently, sensitive amino acid residues such as asparagine, glutamine, the sulphur residues and aromatic residues are more or less susceptible to modification depending upon their location within the protein structure, meaning that the degradation of proteins is difficult to predict without experimental evidence.
The effects of protein oxidation have been primarily characterised at the level of changes in amino acid composition, without location-specific information. Recent mass spectrometry-based characterisations of damaged wool and skin proteins [5-9] allowed the identification of damaged residues within specific proteins, and new proteomic methods have also been developed to quantify and classify oxidative damage within peptides and proteins [8,10,11].
Thermal modification represents a specific kind of protein damage that, unlike photomodification, has not been well characterised at the molecular level. We here evaluate the effects of hydrothermal (wet heat) damage to wool intermediate filament proteins (IFPs). To identify and track markers of degradation to structural keratins through a damaging protocol, IFPs from wool were immersed in hot water (90˚C). Such temperatures are commonly employed during wool processing, even though high temperature is known to contribute to wool degradation [12,13]. Detailed information of the molecular effects of heat on wool is minimal. Following hydrothermal exposure, damaged peptides within the proteins were tracked using commercially available isobaric labelling reagents for quantitation (iTRAQ) and reverse phase liquid chromatography (RP-LC) coupled to matrixassisted laser desorption/ionisation tandem mass spectrometry (MALDI-MS/MS) for separation and analysis.
Quantitation using iTRAQ (and other isobaric approaches) relies upon the tagging of all peptides within a sample with a chemical label. Up to eight chemical labels of identical mass are available, for use on up to eight parallel samples, which can be combined after labelling for simultaneous RP-LC fractionation and MS/MS analysis —eliminating run-to-run variability. The fragmentation step during MS/MS causes these labels to cleave, and as the cleavage products are isotopically distinct, they appear as separate m/z peaks in the spectrum. The relative sizes of the iTRAQ reporter ion peaks relate directly to the relative abundance of each peptide in the samples, and the normal ions generated by the cleavage of the peptide itself (immonium, y-, band a-series ions) may still be used to determine the residue sequence of the peptide as usual [14].
The use of fractionation by RP-LC followed by sensitive tandem mass spectrometric analysis of all fractions and comparison to comprehensive in-house keratin databases permitted comprehensive protein and peptide identification. The use of iTRAQ reagents enabled quantitative comparisons between samples (thus providing a measurement of how individual peptides increased or decreased with time) by measuring the abundance of sample-specific tags after fragmentation in MS/MS.
2. Methods
2.1. Materials
Methyl methanethiosulfonate and 4-plex iTRAQ reagents were obtained from ABSciex (Foster City, CA); sodium tetrathionate from BDH (Poole, England). Reacti-Vials, HPLC-grade ethanol, LC/MS-grade water and trifluoroacetic acid were all obtained from Fisher Scientific (Loughborough, England). Triethylammonium bicarbonate and tris(2-carboxyethyl)phosphine were obtained from Sigma-Aldrich (St Louis, MO); LC-grade acetonitrile, sodium tetraborate and hydrochloric acid from J.T. Baker (Phillipsburg, NJ). Sequencing grade trypsin was obtained from Promega Corporation, (Madison WI). Acrylamide was obtained from BioRad Laboratories (Hercules, CA). α-Cyano-4-hydroxycinnamic acid and ammonium phosphate buffer were obtained from Bruker Daltonics (Bremen, Germany).
2.2. Wool Protein Extraction and Fractionation
Wool proteins were extracted from wool at pH 9.5, room temperature, and crudely fractionated using an adaptation of a protocol described previously [15]. Briefly, the extraction solution made use of sodium tetrathionate, a sulfitolysis reagent that reacts with cysteine residues to form sulfo-cysteine, which increases the solubility of highsulfur proteins [15]. The extracted keratins were precipitated as described in Thomas et al. (1986) and dialysed to form an enriched fraction of IFPs, which represent the major class of proteins within wool.
2.3. Hydrothermal Treatment
Wool IFPs were dissolved in 0.05 M sodium tetraborate, pH 8.5 (HCl) at 5.3 mg/ml and heated in sealed 1 ml ReactiVials at 90˚C for 0, 2, 4 and 8 hours. Samples were transferred immediately to –85˚C prior to LC-MALDIMS/MS analysis. This was performed in duplicate.
2.4. Digestion and Alkylation
Treated samples were diluted with triethylammonium bicarbonate (TEAB) to give ~ 50 µg protein in 20 µl 0.5 M TEAB, 0.025 M sodium tetraborate, pH 8.0. After reduction with 50 mM tris(2-carboxyethyl)phosphine at 56˚C for 45 min, alkylation was performed at room temperature with either 62 mM acrylamide or 40 mM methyl methanethiosulfonate for 30 min. Digestion was performed after the addition of acetonitrile to 7% with sequencing grade trypsin at 37˚C for 16 hours.
2.5. Mass Spectrometric Characterisation
Digested samples were labelled with 4-plex iTRAQ reagents in randomised order according to the manufacturer’s protocol. Briefly, the contents of iTRAQ reagent vials were diluted with 70 μl high-grade ethanol, and were transferred to randomly assigned sample digests in 0.5 M triethylammonium bicarbonate. Labelling was performed by incubation for one hour at ambient temperature, then equal volumes of each labelled sample were pooled to generate a combined sample.
After transferal to autosampler vials, 5 μl of the combined sample was separated over 80 min at a flow rate of 300 nl/min with a 3% - 55% B (acetonitrile/0.1% trifluoroacetic acid) using a Proxeon Easy-nLC and a Proteineer fc fraction collection robot. Samples were loaded onto a 30 mm, 100 µm ID trap column (packed in-house with Microsorb C18 300-5 media, Varian, Palo, Alto, CA) and eluted over an 18 cm, 75 µm ID in-house packed C18 analytical column. Fractions were spotted post-column onto 600 µm AnchorChip plates (Bruker) along with saturated α-cyano-4-hydroxycinnamic acid in acetone mixed 1:10 with 6:3:1 ethanol/acetone/10 mM ammonium phosphate, 0.1% trifluoroacetic acid with a flow rate of 700 nl/min. External calibration was performed using a Peptide Calibration Standard (Bruker) containing ACTH clip (1-17) (2093.0862 Da), ACTH clip (18-39) (2465.1983 Da), angiotensin I (1296.6848 Da), angiotensin II (1046.5418 Da), bombesin (1619.8223 Da), somatostatin (3147.471 Da) and substance P (1347.7354 Da) diluted five-fold with matrix solution. An Ultraflex III MALDI-TOF/TOF mass spectrometer (Bruker) was used for automated sample characterisation. The settings for MS/MS and stable isotope labelling chemistry detection were adjusted for peak area recognition for iTRAQ reporter ions.
2.6. Data Analysis
To identify the proteins from which the sample peptides were derived, data was first processed through BioTools (Bruker), and searched against Ovis aries entries in an NCBInr database (NCBInr_20091103) augmented with in-house sheep sequences using Mascot v2.2.06 (Matrix Science, London, UK). Fixed modifications were specified as either propionamide or methylthio (C), and 4-plex iTRAQ (K and the N-terminus). Variable modifications were deamidation (Q and N), single oxidation (C, H, M, Y, W and F), double oxidation (C, Y, W and F), kynurenine (W), hydroxykynurenine (W), and dopa-quinone (Y). Enzyme specificity was set to semi-trypsin, with two allowable missed cleavages. Data was compiled and analysed using ProteinScape 2.1 (Bruker) with acceptance score thresholds for proteins and peptides set at 40 and 15, respectively.
3. Results
Wool-derived intermediate filament proteins (IFPs) were exposed to hydrothermal insult through immersion in water at 90˚C for 0, 2, 4 and 8 hours. Following protein reduction, alkylation and digestion, the resultant peptides were differentially labelled with isobaric iTRAQ reagents and analysed using RP-HPLC MALDI-MS/MS. Peptides were identified to specific proteins using automated database searching, and particular attention was paid to those peptides that decreased in abundance after hydrothermal exposure.
Mass spectrometric characterisation and quantitative evaluation using iTRAQ reporter ion abundance resulted in the identification of molecular damage markers for keratins. These peptides demonstrated measurable decreases in abundance when subjected to hydrothermal insult. In some cases, low abundance degradative products of these peptides were detected. Because of the complexity and dynamic range of biological extracts, detected and identified peptides susceptible to oxidative damage varied in their suitability for use as molecular markers of damage. The peptides described below represent those with characteristics suitable for use as reliable markers: sufficient abundance/ion signal to permit automated Mascot identification, consistent decreases in parent peptide abundance over the four time-points measured, and, in some cases, increases in a known degradation product.
Over 140 peptides (some native, others modified) were identified within the wool protein samples. The keratin peptides, ATAENEFVALK, ENAELESR and SNHEEE VNTLR, from the K31, K34 and K86 wool proteins, respectively, were observed using iTRAQ reporter ion ratios to degrade during hydrothermal exposure (see Figures 1 and 2), with concurrent formation of their deamidated products. These three peptides were identified using automated database searching with high scores of confidence (over 60). The peptide TAAENEFVALK and its deamidated product, from a cytoskeletal IFP, K75, were also observed to respectively degrade and form during exposure to heat. Consistent increases in the oxidative modification of the susceptible tyrosine and tryptophan residues (to form AEAES [Kyn] [dopa] R) accompanied the degradation of AEAESWYR (from Type II K86). Figure 3 illustrates the characteristic MS/MS fragmentation ions used for identification and quantitation of iTRAQ-labelled AEAESWYR in a pooled sample.
Additional peptides demonstrated consistent degradation patterns, but were not associated with consistent increases in degradation products. These are listed in the two lower sections of Figure 1. The confidence in the peptide identifications for these markers ranged more widely than the three most certain marker peptides (based on peptide ion scores, from 16 to 90). Parent peptide degradation, as determined by measuring iTRAQ reporter ion relative abundance, is represented graphically in the right-most column.
Many peptides that did not meet the criteria for selection as damage markers displayed modifi-cations after hydrothermal damage. These included a number of single or double deamidations (noted on IFP peptides DSLEN TLTETEAR, HQEKEQIK, IDPNIQRVR, LESEINTYR, LLEGEEQR, LNVEVDAAPTVDLNHVLNETR, LQA QHNLR, LSSELNSLQEVLEG, LTAEVENAK, LYNL GGTKR, LYQEEIR, NHEEEANSLR, QLERENAELESR, QNHEQEVNTLR, QNQEYQVLLDVR, QTEELNK, RLYEEEIR, SDLEANSEALIQEIDFLR, TKYETELGLR, TVNALEVELQAQHNLR and YEEEVALR). Additional oxidative modifications to aromatic residues that were noted included tyrosine derivatives (dopa in DAAYLNK, ERQNQEYRVLLDVR, QDMASLVKQY and VVTSSE QLQSYQSDIIDLR, dopa-quinone on LQEVLEGYK, SKYETELGLR and YQSYFR, topa-quinone on QSYQ SDIIDLR and SLYNLG); tryptophan derivatives (kynurenine in DVEEWYIR and WQLYQNQR, hydroxytryptophan in AEAESWYR, dihydroxytrypto-phan/ N-formylkynurenine in AEAESWYR and DVEEWYIR, hydroxykynurenine in FYQNQR); and a histidine derivative (hydroxyhistidine in LLKPLNLEIDHNAQR).
4. Discussion
Isobaric quantitation of hydrothermally damaged wool
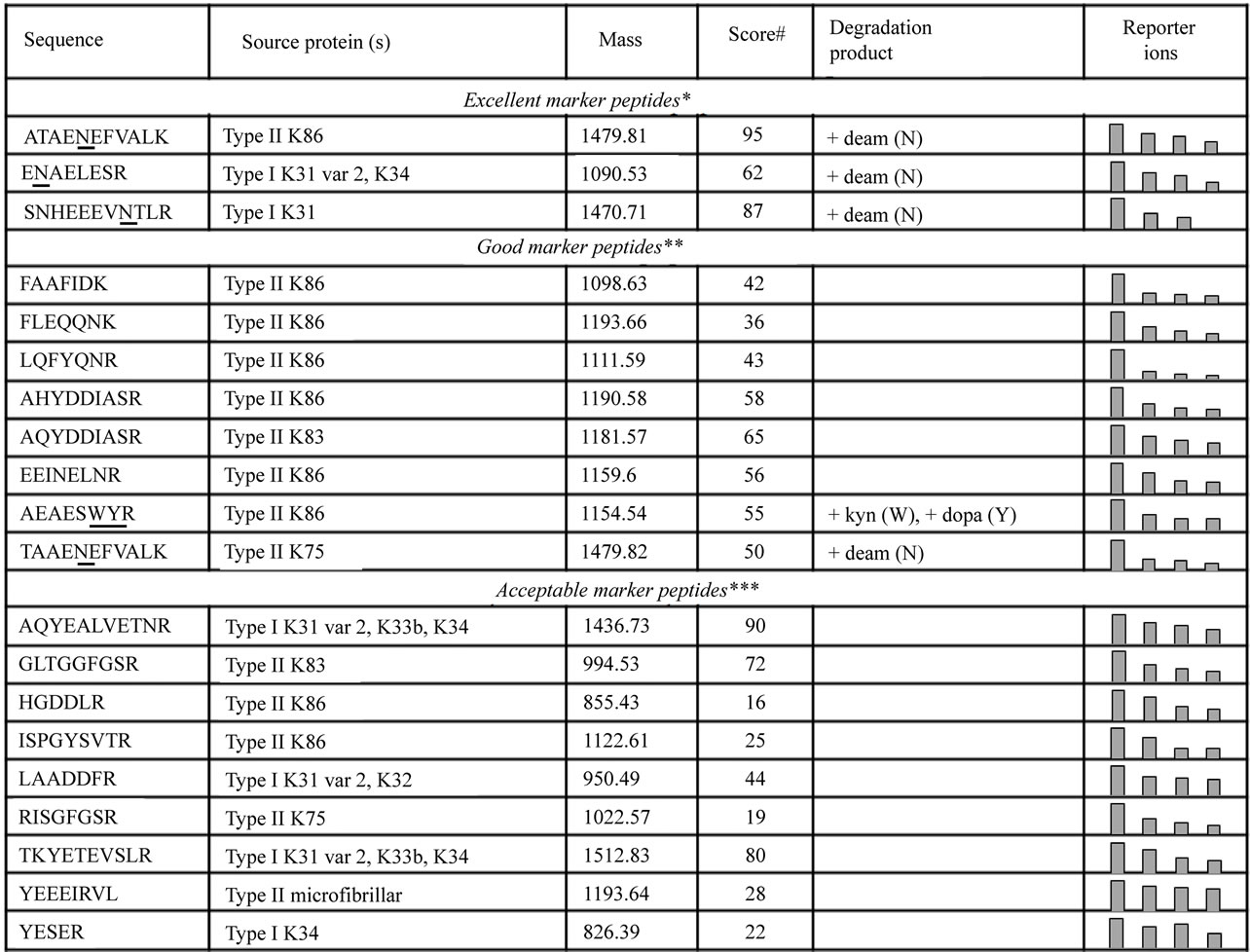
Figure 1. Marker peptides for molecular-level hydrothermal damage observed by MS/MS in keratin proteins. Relative abundance of parent peptide as determined by the measurement of iTRAQ reporter ions (peak area) represented graphically in the right-most column. # Highest score observed; * Degradation of parent peptide observed in both samples, degradation product observed to increase; ** Degradation of parent peptide observed in both samples, or in one sample accompanied by an increase in a degradation product; *** Degradation of parent peptides observed in one sample.
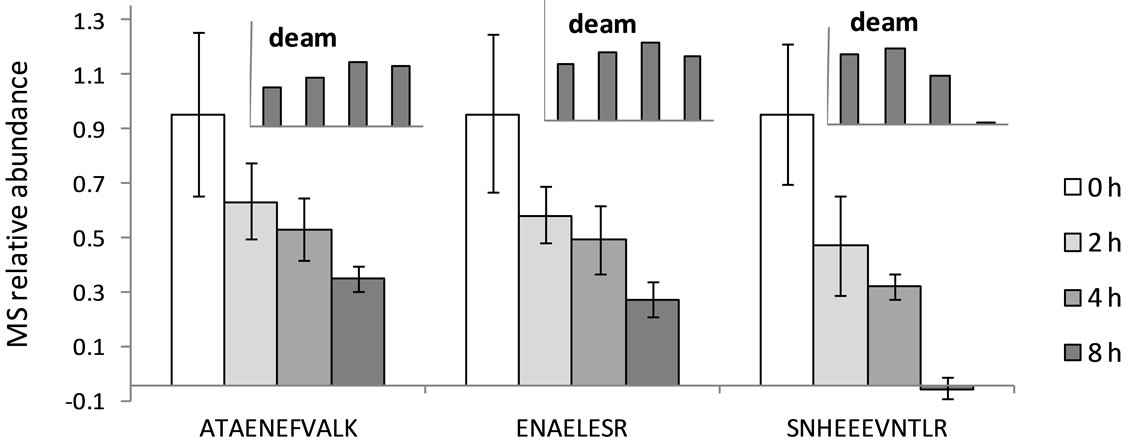
Figure 2. Decreasing relative abundance of marker peptides in wool keratins after exposure to hydrothermal insult at 90˚C, as determined by iTRAQ reporter ion abundance in MALDI-MS/MS. Error bars represent ± standard deviation (SD). Initial relative abundance defined arbitrarily as 1.0. Insets: Increasing relative abundance of deamidated products of ATAENEFVALK, ENAELESR, and SNHEEEVNTLR.
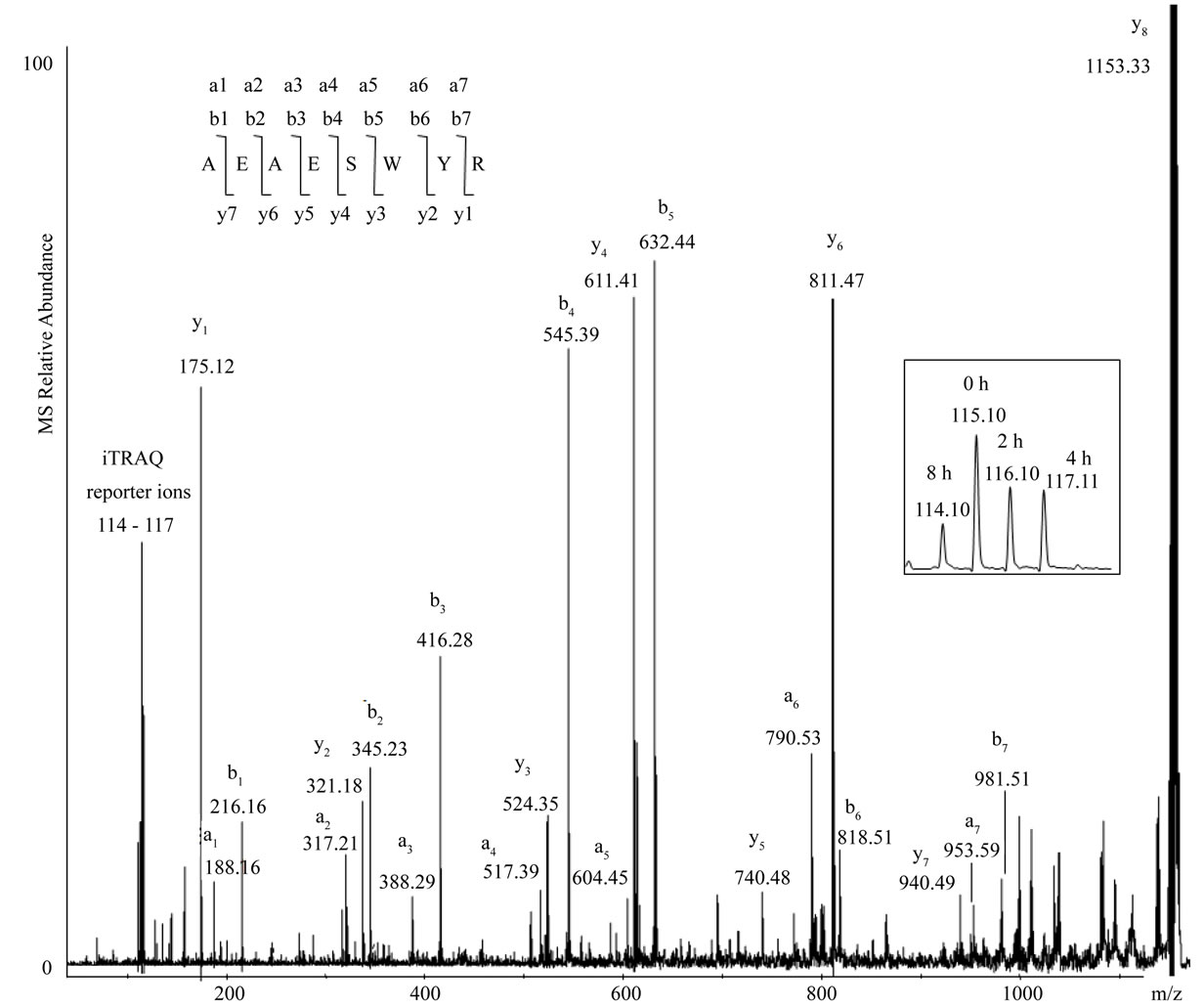
Figure 3. MS/MS scan of a damage marker peptide from wool IFP Type II K86, AEAESWYR, demonstrating the y-, aand b-ion series used for sequencing. Insert: expanded view of the iTRAQ reporter ion region of the scan. Note: the peak of the parent ion (labelled as y8) has been truncated due to its large size relative to the fragmentation peaks.
fractions allowed the observation of consistent degradation in a number of peptides. This resulted in the identification of peptides that served as markers of hydrothermal damage. These peptides were all present in the samples in sufficient abundance to allow detection and automated identification. Each damage marker proved sensitive to hydrothermal exposure, consistently decreasing in abundance over the treatment period. While degradation products were observed for many of the damage markers, those reported here demonstrated expected increases in abundance along with treatment. These modified peptides may themselves be utilised as (inverse) markers of damage. The peptides identified and selected here as damage markers demonstrate particular sensitivity to hydrothermal damage.
The identification and relative abundance profiling of peptide hydrothermal damage within keratins has provided a new set of tools to assess and track the extent of damage incurred in a fibrous protein system. This can be applied to validating the effectiveness of a protective treatment, or the detrimental side-effects of damaging exposure conditions. Three IFP peptide sequences in particular, ATAEN EFVALK, ENAELESR, and SNHEEEVNTLR) were selected as markers of oxidative damage and these three have a high level of homology with peptides derived from human hair and skin proteins. These three marker peptides demonstrated measureable and consistent sensitivity to hydrothermal exposure, along with hydrothermally-induced formation of deamidated products. A second level of potential marker peptides were also identified, but were less consistent in their behaviour in this study.
Although these peptides have been selected for their use in evaluating protein damage in wool, similar markers may be utilised to evaluate damage in related substrates such as cashmere, angora, or even human skin or hair and keratin-based biomaterials. This study demonstrated that quantitative mass spectrometry using isobaric labels permits the acquisition of data sensitive enough to detect the treatment-induced changes in the abundance of specific peptides. We anticipate that the development of targeted protective treatments for protein-based fibres and textiles, or of gentler high temperature treatment steps for processing and product care, will be assisted by the advanced damage tracking protocols presented here.
5. Acknowledgements
This study was kindly supported through a Tertiary Education Commission Bright Futures Enterprise Scholarship, a Vernon-Wiley Trust Fellowship, and a Wool Research Organisation of New Zealand Inc. and New Zealand Wool Industry Charitable Trust Post-Graduate Scholarship. Additional support was provided by the New Zealand Foundation for Research Science and Technology (FRST C10X0710).
REFERENCES
- F. G. France and I. L. Weatherall, “The Effect of Light on Wool,” Proceedings of the 9th International Wool Textile Research Conference, Biella, Italy, 28 June 1995, Vol. 2, pp. 100-108.
- K. R. Millington, “Photoyellowing of Wool. Part 2: Photoyellowing Mechanisms and Methods of Prevention,” Coloration Technology, Vol. 122, No. 6, 2006, pp. 301- 316. doi:10.1111/j.1478-4408.2006.00045.x
- M. J. Davies, “The Oxidative Environment and Protein Damage,” Biochimica et Biophysica Acta—Proteins & Proteomics, Vol. 1703, No. 2, 2005, pp. 93-109. doi:10.1016/j.bbapap.2004.08.007
- S. Guedes, R. Vitorino, R. Domingues, F. Amado and P. Domingues, “Oxidation of Bovine Serum Albumin: Identification of Oxidation Products and Structural Modifications,” Rapid Communications in Mass Spectrometry, Vol. 23, No. 15, 2009, pp. 2307-2315. doi:10.1002/rcm.4149
- J. M. Dyer, S. D. Bringans and W. G. Bryson, “Characterisation of Photo-Oxidation Products within Photoyellowed Wool Proteins: Tryptophan and Tyrosine Derived Chromophores,” Photochemical and Photobiological Science, Vol. 5, No. 7, 2006, pp. 698-706. doi:10.1039/b603030k
- J. M. Dyer, S. D. Bringans and W. G. Bryson, “Determination of Photo-Oxidation Products within Photoyellowed Bleached Wool Proteins,” Photochemistry and Photobiology, Vol. 82, No. 2, 2006, pp. 551-557. doi:10.1562/2005-08-29-RA-663
- J. M. Dyer, S. Clerens, C. D. Cornellison, C. J. Murphy, G. Maurdev and K. R. Millington, “Photoproducts Formed in the Photoyellowing of Collagen in the Presence of a Fluorescent Whitening Agent,” Photochemistry and Photobiology, Vol. 85, No. 6, 2009, pp. 1314-1321. doi:10.1111/j.1751-1097.2009.00614.x
- J. M. Dyer, J. E. Plowman, G. Krsinic, S. Deb-Choudhury, H. Koehn, K. R. Millington and S. Clerens, “Proteomic Evaluation and Location of UVB-Induced Photomodification in Wool,” Photochemistry and Photobiology: B Biology, Vol. 98, 2010, pp. 118-127.
- A. J. Grosvenor, J. D. Morton and J. M. Dyer, “Proteomic Characterisation of Hydrothermal Redox Damage,” Journal of the Science of Food and Agriculture, Vol. 91, No. 15, 2010, pp. 2806-2813. doi:10.1002/jsfa.4525
- A. J. Grosvenor, J. D. Morton and J. M. Dyer, “Profiling of Residue-Level Photo-Oxidative Damage in Peptides,” Amino Acids, Vol. 39, No. 1, 2009, pp. 285-296. doi:10.1007/s00726-009-0440-7
- A. J. Grosvenor, J. D. Morton and J. M. Dyer, “Isobaric Labelling Approach to the Tracking and Relative Quantitation of Peptide Damage at the Primary Structural Level,” Journal of Agricultural and Food Chemistry, Vol. 58, No. 24, 2010, pp. 12672-12677. doi:10.1021/jf103309b
- M. Bide, “Wool Dyeing: What’s New?” Process Improvement, Vol. 24, 1992, pp. 17-22.
- W. S. Simpson, “Prevention of Dyebath Yellowing,” In: W. S. Simpson and G. H. Crawshaw, Eds., Wool: Science and Technology, Woodhead Publishing Limited, Cambridge, 2002, pp. 216-217.
- P. L. Ross, Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski, S. Pillai, S. Dey, S. Daniels, S. Purkayastha, P. Juhasz, S. Martin, M. BartletJones, F. He, A. Jacobson and D. J. Pappin, “Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Reagents,” Molecular & Cellular Proteomics, Vol. 3, 2004, pp. 1154-1169. doi:10.1074/mcp.M400129-MCP200
- H. Thomas, A. Conrads, K. H. Phan, M. van de Löcht and H. Zahn, “In Vitro Reconstitution of Wool Intermediate Filaments,” International Journal of Biological Macromolecules, Vol. 8, No. 5, 1986, pp. 258-264. doi:10.1016/0141-8130(86)90038-3