International Journal of Clinical Medicine
Vol.4 No.7(2013), Article ID:33915,4 pages DOI:10.4236/ijcm.2013.47057
Sporadic Creutzfeldt-Jakob Disease Presenting with Visual Disturbance—A Case Report in China
![]()
1Department of Ophthalmology, Eye and ENT Hospital, Fudan University, Shanghai, China; 2Department of Neurology, Beijing Tongren Hospital, Capital Medical University, Beijing, China.
Email: *zxjsusan1@yahoo.com
Copyright © 2013 Guohong Tian et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 30th, 2013; revised May 20th, 2013; accepted May 27th, 2013
Keywords: Creutzfeldt-Jakob disease; visual disturbance; Heidenhain variant; diffusion weighted imaging
ABSTRACT
Creutzfeldt-Jakob disease (CJD) is a rare but fatal neurodegenerative prion disease. Classic CJD comprises a clinical triad of rapidly progressive dementia, myoclonus, and EEG abnormality. At initial presentation, this classic triad is present only in a minority of cases. Visual impairment is one of the predominant manifestations in the course of CJD, especially in Heidenhain variant phenotype. We reported a case presenting with progressive blurred vision, along with other neurological symptoms, who diagnosed as sporadic CJD with cortical blindness in China.
1. Case Report
A 56-year-old, male, Asian, retired porcelain worker was referred to neurological consultation because of bilateral blurred visual. His symptoms had been developed gradually over the past 2 months accompanied by dizziness, unsteadiness while walking, as well as the family noticed the changing of personality, decreased speech, and deterioration in memory. He did not report fever, headache, eye pain, skin rashes or weight loss. His past medical history is only significant for a heart attack 5 years ago. He did not undergo any recent surgeries or receive any transfusion of blood products in the past. He lived with his family in a small town after he was retired 6 years ago.
On physical examination, he was a well-nourished man. He had no thyomegaly, lymphadenopath, or jaundice. The cardiac, respiratory and abdominal examinations were normal. The Neuro-Ophthalmic examination revealed that he was poorly cooperating with the exam and disoriented to place and date, slurred speech and could only follow simple commands. Visual acuities were finger count at 10 ft OU. Pupils were equal in size and show normal response to light and near. Extraocular movements were normal, but optokinetic nystagmus could not be induced. Pursuit and saccadic function was difficult to evaluate. Funduscopy examination revealed normal optic nerves and maculae OU. Strength was normal, but he had increased muscle tone throughout. There was no myoclonic jerks, but tremor of the extremities was present at rest and worsened with movement. Dysmetria was present on finger-nose testing and rapid alternating movements were slow and clumsy. His gait was widebased and ataxic; he required assistance for standing. Reflexes were normal, and symmetric and plantar responses were flexor.
Routine Blood tests were normal (table 1) including complete blood cell, sugars, urea, creatinine, electrolytes and liver function. ESR and CRP were within normal limits. Serum levels of anti-thyroid peroxides (TPO) and anti-thyroglobulin antibodies (TGA) were slight elevated. The ELISA for HAV, HBV, HIV and VDRL test were all negative. Serum B12, folic acid levels were in the normal range. Testing for antinuclear antibodies (ANA), antineutrophil cytoplasmic antibodies (ANCA), and double-stranded DAN antibodies were negative. The panels for paraneoplastic were all negative. The lumber puncture was performed with opening pressure of 130 mm H2O. Cerebrospinal fluid (CSF) analysis reveals no cells, with normal glucose and protein levels, normal cytology. The bacterial cultures, smears for acid fast bacilli, Cryptococcus were negative. The CSF sample sent to Center for Disease Control and Prevention (CDC) in Beijing for western blot 14-3-3 protein which was positive.

Table 1. Blood tests and cerebrospinal fluids analysis.
A 12-lead electroencephalography showed diffused slow waves and periodic burst of 1 - 2 Hz sharp and slowwave complexs at centro-temporal and occipital cortex (Figure 1).
Magnetic resonance imaging (MRI) of the brain with gadolinium enhancement showed nonspecific atrophy of cerebral in T1, T2, and attenuated inversion recovery (FLAIR) sequences. The add-on diffusion weighted imaging (DWI) revealed hyperintensity throughout the cortical ribbon in bilateral occipital lobes and extented to the lateral-media temporal, which have corresponding hypointensity in the apparent diffusion coefficient (ADC) sequences (Figure 2).
Because of the elevated serum levels of TPO and TGA antibodies, methylprednisolone 1 g was given for 3 days for concerning of Hashimoto’s encephalopathy, immuneencephalitis, primary central nervous system vasculitis, but without any improvement unfortunately. The patient became mute, incontinent and bedridden subsequently. His family insist on his coming back home and he passed away a month after discharged from hospital. The autopsy was not available because of ethical reasons.
2. Discussion
Sporadic Creutzfeldt-Jakob disease (sCJD) is a rare disease caused by prion proteins that undergo conformational changes, resulting in an invariably fatal transmissible spongiform encephalopathy. The worldwide annual incidence is estimated to be 1:1000,000 [1]. A recent report from Chinese Center for Disease Control and Prevention revealed that from 2006 to 2010, 261 patients were diagnosed as sCJD and 23 patients were diagnosed to have genetic human prion diseases in China, with most common presenting symptoms as progressive dementia, whereas the visual symptoms were not emphasized [2].
Our patient’s presenting symptoms were visual disturbance, progressive dementia and ataxia. Without the classic myoclonic jerks, he had other extrapyramidal signs as muscle rigid and tremor. The abnormal EEG, DWI hyperintensities of cortical in MRI, together with the positive 14-3-3 protein in CSF, resisted to corticosteroids therapeutic strongly support the diagnosis of probable sCJD even without the pathological confirmation [3].
Visual disturbances are common, which reported in 20% of Heidenhain variant phenotype of sCJD, even in the early stages of the disease [4,5]. The Heidenhain variant predominately affects the occipital lobe, producing visual disturbances that may manifest as decreased visual acuity, metamorphopsia, dyschromatopsia, visual agnosia, homonymous hemianopia, palinopsia, or micropsia, and eventually cortical blindness. Oculomotor disorders, such as supranuclear gaze palsies, infranuclear gaze palsies, and apraxia of eyelid opening also have been reported [6].
The clinical diagnosis of sCJD often is difficult to make in the early stages of the disease. A recent study showed that diagnosis of sCJD is quite delayed with a mean time of 7.9 months from onset which was an average of two-thirds of the way through their disease course [7]. The most common misdiagnoses by physician were viral encephalitis, paraneoplastic disorder, depression, vertigo and Alzheimer disease, stroke, central nervous system vasculitis, and Hashimoto encephalopathy. A new evidence-based guideline from the American Academy of Neurology recommends clinicians order cerebrospinal fluid testing for protein 14-3-3 when sCJD is strongly suspected in a patient with rapidly progressive dementia
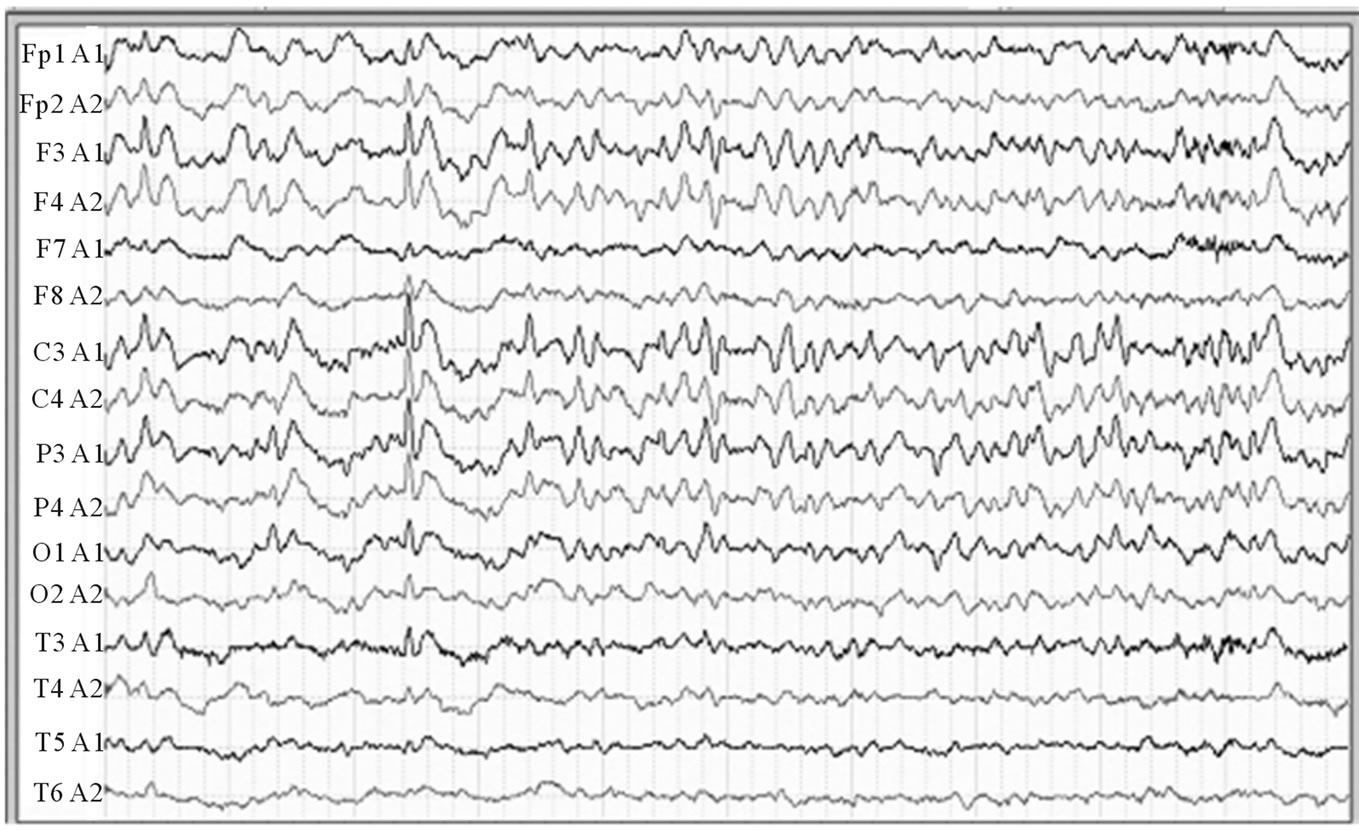
Figure 1. EEG reveals diffused absence of alpha wave and reduced normal fast rhythm. Burst of 1 - 2 Hz sharp and slow-wave complexs at centro-temporal and occipital cortex.
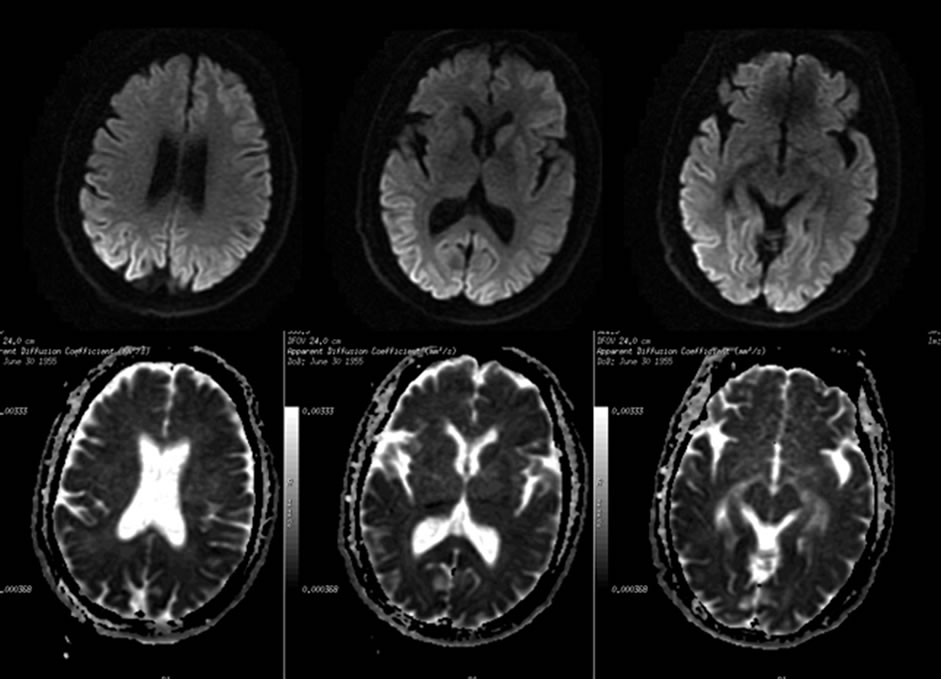
Figure 2. The axial DWI showed hyperintensities (arrows) throughout the cortical ribbon in bilateral occipital lobes and extent to lateral-temporal, which have corresponding hypointensity in the ADC sequences. The changes were not seen on the T1 and T2 sequence.
[8]. The guideline also notes that magnetic resonance imaging DWI and FLAIR sequences are more useful than electroencephalography [9].
The Heidenhain variant of sporadic Creutfeld-Jakob disease should be suspected in patients suffering from early visual disturbances, unremarkable ophthalmic examination, and subsequent rapid decline of their cognitive function [10].
As conclusion, although very rare, sCJD should be in the differential diagnosis lists when vision disturbance accompany progressive dementia, ataxia, myoclonous, and routine MRI is atypical for stroke or encephalitis. The further work-up for CSF 14-3-3 together with the DWI or Flair sequences of MRI are very helpful in diagnosis.
3. Acknowledgements
The authors thank Dr. Behzad Mansouri (Fellow of NeuroOphthalmology, Massachusetts Eye and Ear Infirmary) for editing English.
REFERENCES
- A. Ladogana, M. Puopolo, E. A. Croes, H. Budka, et al., “Mortality from Creutzfeldt-Jakob Disease and Related Disorders in Europe, Australia, and Canada,” Neurology, Vol. 64, No. 9, 2005, pp. 1586-1591. doi:10.1212/01.WNL.0000160117.56690.B2
- C. Gao, Q. Shi, C. Tian, C. Chen, J. Han, W. Zhou, B. Y. Zhang, H. Y. Jiang, J. Zhang and X. P. Dong, “The Epidemiological, Clinical, and Laboratory Features of Sporadic Creutzfeldt-Jakob Disease Patients in China: Surveillance Data from 2006 to 2010,” PLoS One, Vol. 6, No. 8, 2011, Article ID: e24231. doi:10.1371/journal.pone.0024231
- E. Eggenberger, “Prion Disease,” Neurologic Clinics, Vol. 25, No. 3, 2007, pp. 833-842. doi:10.1016/j.ncl.2007.03.006
- S. Kropp, W. J. Schulz-Schaeffer, M. Finkenstaedt, et al., “The Heidenhain Variant of Creutzfeldt-Jakob Disease,” Archives of Neurology, Vol. 56, No. 1, 1999, pp. 55-61. doi:10.1001/archneur.56.1.55
- S. A. Cooper, K. L. Murray, C. A. Heath, R. G. Will and R. S. Knight, “Isolated Visual Symptoms at Onset in Sporadic Creutzfeldt-Jakob Disease: The Clinical Phenotype of the ‘Heidenhain Variant’,” British Journal of Ophthalmology, Vol. 89, No. 10, 2005, pp. 1341-1342. doi:10.1136/bjo.2005.074856
- A. A. Proulx, M. J. Strong and D. A. Nicolle, “Creutzfeldt-Jakob Disease Presenting with Visual Manifestations,” Canadian Journal of Ophthalmology, Vol. 43, No. 5, 2008, pp. 591-595. doi:10.3129/I08-123
- R. W. Paterson, C. C. Torres-Chae, A. L. Kuo, T. Ando, E. A. Nguyen, K. Wong, S. J. Dearmond, A. Haman, P. Garcia, D. Y. Johnson, B. L. Miller and M. D. Geschwind, “Differential Diagnosis of Jakob-Creutzfeldt Disease,” Archives of Neurology, Vol. 24, 2012, pp. 1-5.
- T. Muayqil, G. Gronseth and R. Camicioli, “EvidenceBased Guideline: Diagnostic Accuracy of CSF 14-3-3 Protein in Sporadic Creutzfeldt-Jakob Disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology,” Neurology, Vol. 79, No. 14, 2012, pp. 1499-1506. doi:10.1212/WNL.0b013e31826d5fc3
- P. Vitali, E. Maccagnano, E. Caverzasi, R. G. Henry, A. Haman, C. Torres-Chae, D. Y. Johnson, B. L. Miller and M. D. Geschwind, “Diffusion-Weighted MRI Hyperintensity Patterns Differentiate CJD from Other Rapid Dementias,” Neurology, Vol. 76, No. 20, 2011, pp. 1711- 1719. doi:10.1212/WNL.0b013e31821a4439
- S. Prasad, E. B. Lee, J. H. Woo, A. Alavi and S. L. Galetta, “Photo Essay. MRI and Positron Emission Tomography Findings in Heidenhain Variant CreutzfeldtJakob Disease,” Journal of Neuro-Ophthalmology, Vol. 30, No. 3, 2010, pp. 260-262. doi:10.1097/WNO.0b013e3181e2aef7
NOTES
*Corresponding author.