International Journal of Clinical Medicine
Vol.2 No.4(2011), Article ID:7543,10 pages DOI:10.4236/ijcm.2011.24064
Angiotensinogen Expression Is Enhanced in the Progression of Glomerular Disease
![]()
Department of Physiology, and Hypertension and Renal Center of Excellence Tulane University Health Sciences Center, New Orleans, USA.
Email: murushih@tulane.edu
Received April 11th, 2011; reised May 13rd, 2011; accepted July 17th 2011.
Keywords: Renin-Angiotensin System, Angiotensinogen, Glomerulonephritis, Glomerulosclerosis
ABSTRACT
Intrarenal renin-angiotensin system (RAS) activation plays a critical role in the development and progression of renal injury. In the kidney, all of the RAS components are present and intrarenal angiotensin II (Ang II) is formed by multiple independent mechanisms. Angiotensinogen (AGT) is the only known substrate for renin that is a rate-limiting enzyme of the RAS. Recently, enhanced intrarenal AGT levels have been shown to reflect the intrarenal RAS status in hypertension, chronic glomerular disease and diabetic nephropathy. In this review, we focus on AGT expression of the diseased glomeruli in the progression of glomerular disease. An anti-glomerular basement membrane nephritis rat model developed progressive proteinuria and glomerular crescent formation, accompanied by increased macrophage infiltration and glomerular expression of AGT and Ang II. The addition of Ang II type 1 receptor blocker to CC-chemokine recaptor 2 antagonist markedly attenuated the induction of macrophage infiltration, AGT and Ang II, and reduced glomerular crescent formation. Next, the levels of glomerular AGT expression and marker of reactive oxygen species in Zucker diabetic fatty (ZDF) obese rats were higher than those in ZDF lean rats. Hydrogen peroxide (H2O2) induced an increase in the AGT expression in primary rat mesangial cells. Furthermore, the H2O2-induced upregulation of AGT was inhibited by a mitogen-activated protein kinase kinase and a c-Jun N-terminal kinase inhibitor. These data suggest the potential contribution of enhanced AGT expression in glomeruli to the intrarenal RAS activation for the development of glomerular disease.
1. Introduction
The critical role of the renin-angiotensin system (RAS) in arterial pressure and sodium homeostasis has been widely recognized [1,2]. Angiotensin II (Ang II) is the most powerful biologically active product of the RAS [3]. Ang II directly constricts vascular smooth muscle cells, enhances myocardial contractility, stimulates aldosterone production, stimulates release of catecholamines from the adrenal medulla and sympathetic nerve endings, increases sympathetic nervous system activity, and stimulates thirst and salt appetite [3]. Recently, the focus of interest in the RAS has shifted toward the role of the local/tissue RAS in specific tissues [4]. Locally produced Ang II induces inflammation, cell growth, mitogenesis, apoptosis, migration, and differentiation; regulates the gene expression of bioactive substances; and activates multiple intracellular signaling pathways, all of which might contribute to tissue injury [3]. Intrarenal RAS activity has several pathophysiological functions for not only in blood pressure regulation but also in renal cell growth and production of glomerulosclerosis, which contributes to the development of renal fibrosis [5,6]. Indeed, previous studies have shown that angiotensin converting enzyme inhibitor (ACEi) and/or Ang II type 1 (AT1) receptor blocker (ARB) have beneficial effects in rats and humans with various renal diseases, and these effects are often considerably more significant than their suppressive effects on blood pressure [7,8].
Many diseases affect kidney function by attacking the glomeruli—the tiny units within the kidney in which blood is filtered [9]. Glomerular diseases include many conditions with a variety of genetic and environmental causes, but they fall into 2 major categories: glomerulonephritis and glomerulosclerosis. Although glomerulonephritis and glomerulosclerosis have different causes, they can both lead to kidney failure [9]. Activation of the Ang II-AT1 receptor pathway results in production of proinflammatory mediators, cell proliferation, and extracellular matrix synthesis that facilitate kidney damage and advance chronic kidney disease [10-13] (Figure 1). This review explores recent findings concerning the expression of angiotensinogen (AGT) in the intrarenal RAS activation in glomerular disease.
2. Intrarenal RAS
The biologically active peptides that are formed from AGT include Ang II and angiotensin-(1-7). The balance between the vasoconstricting actions of Ang II, mediated by the AT1 receptor, are countered by the vasodilating actions of Ang II, mediated by the AT2 receptor [14], and the action of angiotensin-(1-7) on the G protein-coupled receptor Mas [15].
Local/tissue RAS in specific tissues has become the focus of much recent interest [4]. Emerging evidence has demonstrated the importance of tissue-specific RAS in the brain [16], heart [17], adrenal glands [18], and vasclulature [19] as well as the kidneys [20]. In particular, renal RAS is unique because all of the components necessary to generate intrarenal Ang II are present along the nephron in both interstitial and intratubular compartments [3,21]. The presence of AGT gene transcripation in the proximal tubules has been shown using in situ hybridization [22]. AGT mRNA is expressed primarily in the proximal convoluted tubules and proximal straight tubules, with small amounts in glomeruli, vasa recta, and renal vasculature [23]. Renal AGT protein is abundant in the proximal convoluted tubules [24,25]. Strong positive immunostaining for AGT protein has been reported in proximal convoluted tubules and proximal straight tubules, and weak positive staining in glomeruli and vasa recta; however, distal tubules and collecting ducts were negative [26]. The AGT synthesized in the kidney is secreted into the lumen leading to Ang I generation. Low but measureable renin concentrations have been detected in proximal tubule fluid in rats [27].
Renin mRNA and renin-like activity have been demonstrated in cultured proximal tubular cells [28-30]. The brush border membrane of proximal human kidney tubules expresses abundant levels of ACE mRNA [31] and protein [32,33]. ACE has also been measured in proximal and distal tubular fluid but is greater in proximal tubule fluid [34]. Therefore, all of the major components required to generate Ang II are expressed within the kidney [3,20].
3. AGT in Intrarenal RAS Activation
AGT is the only known substrate for renin that is a rate-limiting enzyme of the RAS. Because the level of AGT is close to the Michaelis-Menten constant for renin, not only renin levels but also AGT levels can control
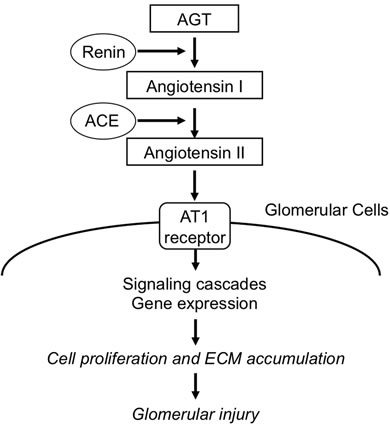
Figure 1. Working scheme of the renin-angiotensin system in glomerular disease. AGT: amgiotensinogen, ACE: angiotensin converting enzyme, AT1 receptor: angiotensin II type 1 receptor, ECM: extracellular matrix.
RAS activity, and AGT upregulation may lead to elevated angiotensin peptide levels and increased blood pressure [35,36]. Recent studies using experimental animal models and transgenic mice have documented AGT involvement in the activation of the RAS and development of hypertension [37-45]. Genetic manipulations that lead to AGT overexpression have consistently been shown to cause hypertension [46,47]. In human genetic studies, a linkage has been established between the AGT gene and hypertension [48-51]. Enhanced intrarenal AGT mRNA and/or protein levels have also been observed in multiple experimental models of hypertension including Ang II-dependent hypertensive rats [26,52-56], Dahl salt-sensitive hypertensive rats [57,58], and spontaneously hypertensive rats [59], as well as in kidney diseases including diabetic nephropathy [60-65], IgA nephropathy [66,67], and radiation nephropathy [68]. In addition, a direct quantitative method to measure urinary AGT using human AGT enzyme-linked immunosorbent assays (ELISA) was developed [69], which revealed significantly increased urinary AGT levels in patients with hypertension [70,71], chronic kidney disease [72-75] and diabetes [76, 77]. Thus, AGT plays an important role in the development and progression of hypertension and kidney disease [3,20].
4. AGT in Anti-Glomerular Basement Membrane (GBM) Disease
Chronic glomerulonephritis that results in substantial renal damage is frequently characterized by relentless progression to end-stage renal disease. Renal Ang II, the production of which is enhanced in chronic glomerulonephritis, can elevate the intraglomerular pressure, increase glomerular cell hypertrophy, and augmented extracellular matrix accumulation [78,79]. ACEi and/or ARB markedly decelerate, and can even prevent, renal deterioration in renal disease [1,78,80,81]. This may reflect the relatively short-term nature and small size of these studies, but may also be an indication that factors other than Ang II play an important role in the progresssion of renal disease.
Anti-GBM disease or Goodpasture’s syndrome is a crescentic glomerulonephritis that is characterized by the formation and deposition of antibodies on the basement membranes of glomeruli and alveoli [82]. Patients present with renal failure, dyspnea, hemoptysis, a sudden decrease in the hemoglobin level, pallor, and circulatory disturbances. Most patients with advanced disease do not respond to plasmapheresis or immunosuppression therapy [83]. While kidney transplantation is an option, a patient should wait for 6 months or after the disappearance of serum anti-GBM antibodies before undergoing kidney transplantation because of the risk of recurrence [82]. Therefore, a novel therapeutic strategy is needed. Studies based on anti-GBM antibody have focused on elucidating the molecular and cellular mechanisms involved in the pathogenesis of this disease. Understanding the mechanisms of proinflammatory responses help facilitate the identification of therapeutic targets that arrest the progression of anti-GBM disease.
Glomerular crescents are defined as the presence of 2 or more layers of cells in the Bowman’s space. Monocyte/macrophages and parietal epithelial cells are the principal mediators of crescent formation [84]. The presence of crescents in glomeruli is a marker of severe injury [90]. After a single injection of anti-GBM antibodies, marked crescent formations were observed in almost all glomeruli as a result of severe glomerular damage. CCR2 antagonist (CA) or ARB alone moderately normalized the crescent formation [91]. The dose of CA or ARB was determined by previous reports [92,93] and could be adequate to preclude the effects of the MCP-1/CCR2 signal pathway and RAS. Their combination significantly blocked the development of crescent formation, preventing the infiltration of macrophages [91]. Consistently, the combination therapy markedly reduced proteinuria.
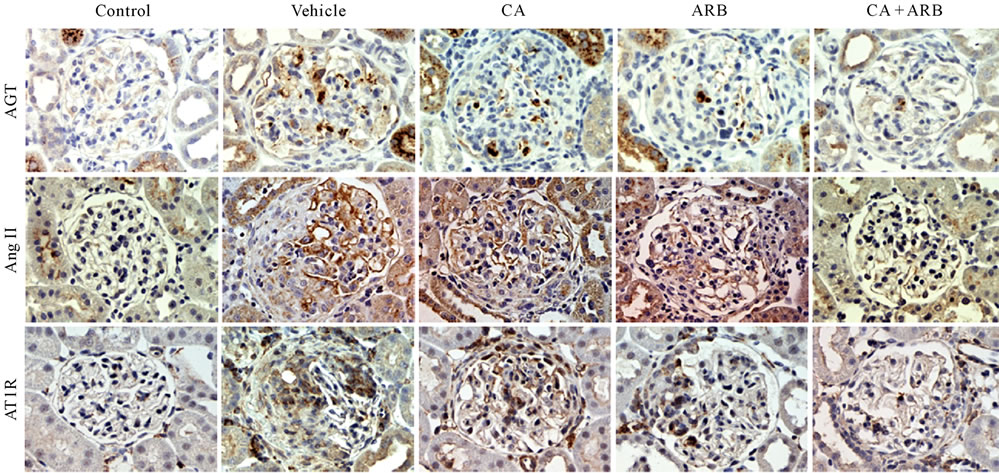
It is well known that intrarenal RAS activation is a major mediator of progressive renal injury in glomerulonephritis [78-81]. In this anti-GBM disease model, the glomerular expression levels of AGT, Ang II and AT1 receptors were increased compared with control rats [91]. While CA or ARB treatment moderately reduced the increase of these components in glomeruli, CA plus ARB treatment further prevented these increases (Figures 2(a)-(d)). Urinary AGT levels were paralleled with the expression levels of RAS components in glomeruli (Figure 2(e)). The disturbance in the expression of these components likely plays an important role in the pathogenesis of the crescentic formation in glomerulonephritis. Furthermore, Ang II reportedly upregulated AGT and Ang II receptor expressions and ARB prevented the increase of AGT, suggesting positive Ang II feedback in the kidney [94]. Interestingly, ARB treatment prevented increases in kidney and renal interstitial fluid Ang II concentration in the Ang II-infused rat [95]. Thus, from these findings, combination therapy suppressed these expressions more effectively than did CA or ARB alone, cutting intrarenal RAS activation. In previous studies, the RAS activation was shown to be involved in the formation of glomerular crescents [96,97]. Together, these data clearly indicate that blocking the RAS is a key target and that AGT expression reflects intrarenal RAS activation in treatment of anti-GBM disease.
5. AGT in Diabetic Nephropathy
Glomerulosclerosis refers to scarring of the glomeruli. In several sclerotic conditions, a systemic disease such as diabetes is responsible. Glomerulosclerosis is caused by the activation of glomerular cells to produce scar material. This may be stimulated by molecules called growth factors, which may be made by glomerular cells themselves or may be brought to the glomerulus by circulating blood that enters the glomerular filter.
Diabetic nephropathy is the most common etiology of end-stage renal failure in patients starting dialysis [98]. A pathological feature of diabetic nephropathy is the thickening of the GBM and expansion of the mesangium due to accumulation of extracellular matrix [99]. The detailed mechanisms responsible for the development and progression of diabetic nephropathy have yet to be fully elucidated. However, the renoprotective effects of drugs that block or interfere with the RAS indicate that inappropriate activation of the RAS contributes to diabetic nephropathy [100,101]. ACEi reduces proteinuria in patients with diabetes [100]. In a large-scale clinical study, captopril, an ACEi, provided protection against deterioration of renal function in diabetic nephropathy patients with type 1 diabetes [101]. Similarly, losartan, an ARB, conferred significant renal benefits in diabetic nephropathy patients with type 2 diabetes [102]. More recently, olmesartan, another ARB, has been shown to suppress the incidence of microalbuminuria in patients with type 2 diabetes [103,104]. Thus, the renoprotective effects of ACEi and ARBs have been established in various studies. Combination therapy with ACEi and ARBs provides greater renoprotection than with ACEi alone in diabetic renal disease [105,106] suggesting that ACE-independent
 (a)
(a)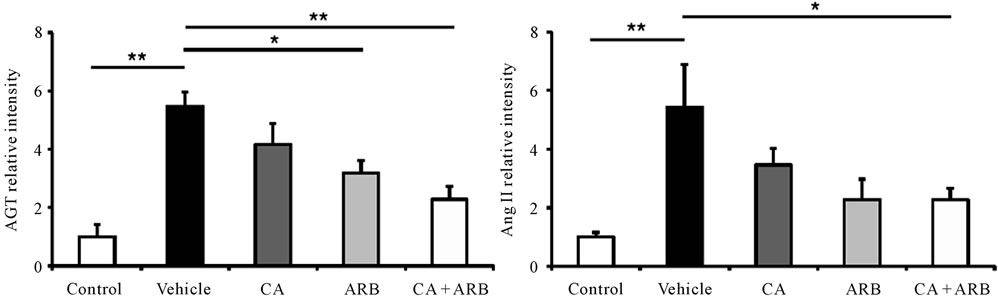 (b)
(b)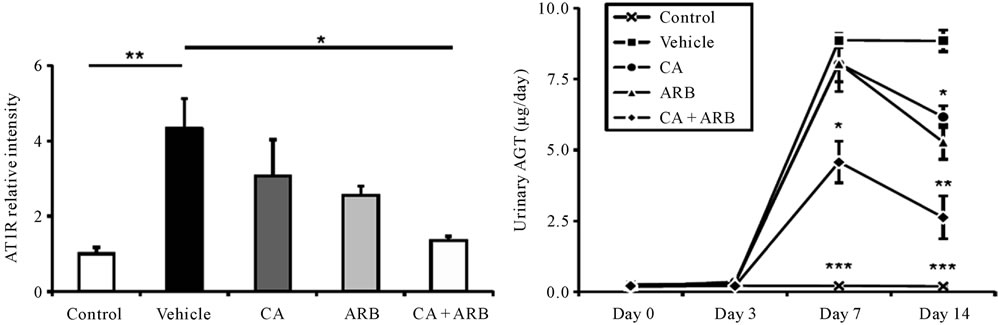 (c)
(c)
Figure 2. Effects of the treatments on intrarenal AGT, Ang II, and AT1 receptor (AT1R) expression and urinary AGT expression in an anti-GBM disease rat model. (a) AGT (top), Ang II (middle), and AT1R (bottom) immnostained kidney sections. Densitometric analyses of AGT (b), Ang II (c) or AT1R (d) expressions determined by immunostaining. Data are mean +/– SEM. *P < 0.05 (vs. vehicle); **P < 0.01 (vs. v vehicle); ***P < 0.001 (vs. vehicle).
pathways for Ang II formation may be of significance.
The Zucker diabetic fatty (ZDF) obese rat is considered to be an excellent animal model of type 2 diabetes because it presents a physiological and metabolic profile similar to that seen in humans [107,108]. The ZDF obese rat is characterized by hyperglycemia, hyperinsulinemia, hyperlipidemia, moderate hypertension, moderate obesity and progressive renal injury [109,110]. The ZDF obese rat exhibits progressive diabetic nephropathy at approximately 20 weeks of age [107]. Mizuno et al. [108] demonstrated that pharmacological blockade of the RAS with an ARB reduced proteinuria and delayed the progression of renal disease in diabetic nephropathy in ZDF obese rats. This suggests that the activated intrarenal RAS plays an important role in the development of diabetic nephropathy in ZDF obese rats. Meanwhile, previous studies revealed that the reactive oxygen species (ROS)-related increase in AGT plays an important role in the development of renal injury in genetic salt-sensitive hypertension [57,58]. Moreover, it has been recently reported that the activated intrarenal ROS-AGT axis plays a role in the development of IgA nephropathy in patients at an early stage [66].
Therefore, ZDF obese rats were examined in terms of enhanced ROS-associated augmentation of intrarenal AGT was involved in the development of nephropathy. As a result, the levels of glomerular immunoreactivity for 4-HNE and urinary excretion of 8-isoprostane, a marker of ROS-in ZDF obese rats were higher than those in ZDF lean rats [64,111]. The levels of glomerular AGT immunoreactivity and in ZDF obese rats were higher than those in ZDF lean rats. Double staining with AGT and Thy1.1 antibodies showed that the majority of AGT in glomeruli was seen in mesangial cells [111]. Relative ratios of intrarenal Ang II immunoreactivity were significantly increased in ZDF obese rat glomeruli compared with controls [64]. These data suggest that the sequential activation of the ROS-AGT-RAS axis plays an important role in the development of diabetic nephropathy in ZDF obese rats.
The signal transduction pathways involved in AGT expression are currently being investigated. Recent studies showed that in immortalized human renal proximal tubular epithelial cells, Ang II acts synergistically with interleukin-6 to increased AGT expression through activation of nuclear factor-κB and the signal transducer and activator of trascription-3 [112]. It has also been reported that in immortalized rat proximal tubular cells, a high glucose concentration stimulates AGT expression through ROS generation and subsequent p38 mitogen-activated protein kinase (MAPK) expression [113]. However, the signal transduction pathway that induces AGT expression has not yet been completely elucidated yet. To clarify the signal transduction pathway for glomerular AGT expression, primary rat mesangial cells were treated with hydrogen peroxide (H2O2) [111]. H2O2 induced an increase in AGT expression in a doseand time-dependent manner, and the H2O2-induced upregulation of AGT was suppressed by catalase. Furthermore, the H2O2-induced upregulation of AGT was inhibited by a MAPK kinase (MEK) inhibitor and a c-Jun N-terminal kinase (JNK) inhibitor, but not inhibited by a p-38 MAPK inhibitor. These data suggest that the majority of AGT was induced in mesangial cells in glomeruli under pathological conditions such as diabetic nephropathy, and that AGT expression in mesangial cells was mediated by H2O2 and the subsequent activation of the extracellular-regulated kinase (ERK)/JNK pathway.
6. Conclusions
This review reveals that AGT expression is enhanced in the glomerular injury associated with RAS activation. Additional studies are needed to clarify the mechanism for AGT overexpression by intrarenal RAS activation in glomerular disease. Intrarenal RAS activation clearly plays a pivotal role in the development of glomerular disease. Accordingly, the assessment of glomerular AGT expression may be of substantial importance. We believe that the investigation of AGT expression in the glomeruli could provide a novel pharmacological strategy for the treatment of glomerular disease.
7. Acknowledgements
We acknowledge the critical discussion and/or excellent technical assistance from L. Gabriel Navar, Naro Ohashi, Kayoko Miyata, Ryousuke Satou, Toshie Saito, Masumi Kamiyama, Akemi Katsurada, M. Patrick Sweeny, G. Michael Upchurch, Nina A. Perrault, Jessica L. Mucci, and Salem I. Elkhayat.
8. Sources of Funding
These studies were supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK072408) and the National Center for Research Resources (P20RR017659).
REFERENCES
- S. Anderson, H. G. Rennke and B. M. Brenner, “Therapeutic Advantage of Converting Enzyme Inhibitors in Arresting Progressive Renal Disease Associated with Systemic Hypertension in the Rat,” Journal of Clinical Investigation, Vol. 77, No. 6, 1986, pp. 1993-2000. doi:10.1172/JCI112528
- L. G. Navar, L. M. Harrison-Bernard, J. D. Imig, et al., “Intrarenal Angiotensin II Generation and Renal Effects of AT1 Receptor Blockade,” Journal of the American Society of Nephrology, Vol. 10 No. S12, 1999, pp. S266- S272.
- H. Kobori, M. Nangaku, L. G. Navar and A. Nishiyama, “The Intrarenal Renin-Angiotensin System: From Physiology to the Pathobiology of Hypertension and Kidney Disease,” Pharmacological Reviews, Vol. 59, No. 3, 2007, pp. 251-287. doi:10.1124/pr.59.3.3
- V. J. Dzau and R. Re, “Tissue Angiotensin System in Cardiovascular Medicine. A Paradigm Shift?” Circulation, Vol. 89, No. 1, 1994, pp. 493-498.
- S. Kagami, W. A. Border, D. E. Miller and N. A. Noble, “Angiotensin II Stimulates Extracellular Matrix Protein Synthesis through Induction of Transforming Growth Factor-Beta Expression in Rat Glomerular Mesangial Cells,” Journal of Clinical Investigation, Vol. 93, No. 6, 1994, pp. 2431-2437. doi:10.1172/JCI117251
- M. Ruiz-Ortega and J. Egido, “Angiotensin II Modulates Cell Growth-Related Events and Synthesis of Matrix Proteins in Renal Interstitial Fibroblasts,” Kidney International, Vol. 52, No. 6, 1997, pp. 1497-1510. doi:10.1038/ki.1997.480
- Y. Horita, M. Tadokoro, K. Taura, et al., “Low-Dose Combination Therapy with Temocapril and Losartan Reduces Proteinuria in Normotensive Patients with Immunoglobulin a Nephropathy,” Hypertension Research, Vol. 27, No. 12, 2004, pp. 963-970. doi:10.1291/hypres.27.963
- M. Ravid, D. Brosh, Z. Levi, et al., “Use of Enalapril to Attenuate Decline in Renal Function in Normotensive, Normoalbuminuric Patients with Type 2 Diabetes Mellitus. A Randomized, Controlled Trial,” Annals of Internal Medicine, Vol. 128, No. 12, 1998, pp. 982-988.
- NIDDK, “Glomerular Diseases,” NIH Publication, Bethesda, 2006.
- A. B. Fogo, “The Role of Angiotensin II and Plasminogen Activator Inhibitor-1 in Progressive Glomerulosclerosis,” American Journal of Kidney Diseases: The Official Journal of the National Kidney Foundation, Vol. 35, No. 2, 2000, pp. 179-188.
- U. C. Brewster and M. A. Perazella, “The Renin-AngioTensin-Aldosterone System and the Kidney: Effects on Kidney Disease,” American Journal of Medicine, Vol. 116, No. 4, 2004, pp. 263-272. doi:10.1016/j.amjmed.2003.09.034
- C. Ruster and G. Wolf, “Renin-Angiotensin-Aldosterone System and Progression of Renal Disease,” Journal of the American Society of Nephrology, Vol. 17, No. 11, 2006, pp. 2985-2991. doi:10.1681/ASN.2006040356
- G. Wolf, “Renal Injury Due to Renin-Angiotensin-Aldosterone System Activation of the Transforming Growth Factor-Beta Pathway,” Kidney International, Vol. 70, No. 11, 2006, pp. 1914-1919.
- R. M. Carey and H. M. Siragy, “The Intrarenal ReninAngiotensin System and Diabetic Nephropathy,” Trends Endocrinol Metab, Vol. 14, No. 6, 2003, pp. 274-281. doi:10.1016/S1043-2760(03)00111-5
- R. A. Santos, A. C. Simoes e Silva, C. Maric, et al., “Angiotensin-(1-7) is an Endogenous Ligand for the G Protein-Coupled Receptor Mas,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 14, 2003, pp. 8258-8263. doi:10.1073/pnas.1432869100
- O. Baltatu, J. A. Silva, Jr., D. Ganten and M. Bader, “The Brain Renin-Angiotensin System Modulates Angiotensin II-Induced Hypertension and Cardiac Hypertrophy,” Hypertension, Vol. 35, No. 1, 2000, pp. 409-412.
- L. J. Dell’Italia, Q. C. Meng, E. Balcells, et al., “Compartmentalization of Angiotensin II Generation in the Dog Heart. Evidence for Independent Mechanisms in Intravascular and Interstitial Spaces,” Journal of Clinical Investigation, Vol. 100, No. 2, 1997, pp. 253-258.
- G. Mazzocchi, L. K. Malendowicz, A. Markowska, et al., “Role of Adrenal Renin-Angiotensin System in the Control of Aldosterone Secretion in Sodium-Restricted Rats,” American Journal of Physiology, Endocrinology and Metabolism, Vol. 278, No. 6, 2000, pp. E1027-1030.
- K. K. Griendling, C. A. Minieri, J. D. Ollerenshaw and R. W. Alexander, “Angiotensin II Stimulates NADH and NADPH Oxidase Activity in Cultured Vascular Smooth Muscle Cells,” Circulation Research, Vol. 74, No. 6, 1994, pp. 1141-1148.
- L. G. Navar, L. M. Harrison-Bernard, A. Nishiyama and H. Kobori, “Regulation of Intrarenal Angiotensin II in Hypertension,” Hypertension, Vol. 39, No. 2, 2002, pp. 316-322.
- L. G. Navar, M. C. Prieto-Carrasquero and H. Kobori, “Chapter 1: Molecular Aspects of the Renal Renin-Angiotensin System,” Taylor & Francis Medical, Oxfordshine, 2006, pp. 3-14.
- J. R. Ingelfinger, W. M. Zuo, E. A. Fon, et al., “In Situ Hybridization Evidence for Angiotensinogen Messenger RNA in the Rat Proximal Tubule. An Hypothesis for the Intrarenal Renin Angiotensin System,” Journal of Clinical Investigation, Vol. 85, No. 2, 1990, pp. 417-423. doi:10.1172/JCI114454
- Y. Terada, K. Tomita, H. Nonoguchi and F. Marumo, “PCR Localization of Angiotensin II Receptor and Angiotensinogen mRNAs in Rat Kidney,” Kidney International ernational, Vol. 43, No. 6, 1993, pp. 1251-1259. doi:10.1038/ki.1993.177
- J. P. Richoux, J. L. Cordonnier, J. Bouhnik, et al., “Immunocytochemical Localization of Angiotensinogen in Rat Liver and Kidney,” Cell and Tissue Research, Vol. 233, No. 2, 1983, pp. 439-451. doi:10.1007/BF00238309
- I. A. Darby and C. Sernia, “In Situ Hybridization and Immunohistochemistry of Renal Angiotensinogen in Neonatal and Adult Rat Kidneys,” Cell and Tissue Research, Vol. 281, No. 2, 1995, pp. 197-206. doi:10.1007/BF00583388
- H. Kobori, L. M. Harrison-Bernard and L. G. Navar, “Expression of Angiotensinogen mRNA and Protein in Angiotensin II-Dependent Hypertension,” Journal of the American Society of Nephrology, Vol. 12, No. 3, 2001, pp. 431-439.
- P. P. Leyssac, “Changes in Single Nephron Renin Release Are Mediated by Tubular Fluid Flow Rate,” Kidney International ernational, Vol. 30, No. 3, 1986, pp. 332-339. doi:10.1038/ki.1986.189
- N. Yanagawa, A. W. Capparelli, O. D. Jo, et al., “Production of Angiotensinogen and Renin-Like Activity by Rabbit Proximal Tubular Cells in Culture,” Kidney International, Vol. 39, No. 5, 1991, pp. 938-941. doi:10.1038/ki.1991.117
- W. L. Henrich, E. A. McAllister, A. Eskue, et al., “Renin Regulation in Cultured Proximal Tubular Cells,” Hypertension, Vol. 27, No. 6, 1996, pp. 1337-1340.
- O. W. Moe, K. Ujiie, R. A. Star, et al., “Renin Expression in Renal Proximal Tubule,” Journal of Clinical Investigation, Vol. 91, No. 3, 1993, pp. 774-779. doi:10.1172/JCI116296
- M. Sibony, J. M. Gasc, F. Soubrier, et al., “Gene Expression and Tissue Localization of the Two Isoforms of Angiotensin I Converting Enzyme,” Hypertension, Vol. 21, No. 6, 1993, pp. 827-835.
- W. W. Schulz, H. K. Hagler, L. M. Buja and E. G. Erdos, “Ultrastructural Localization of Angiotensin I-Converting Enzyme (EC 3.4.15.1) and Neutral Metalloendopeptidase (EC 3.4.24.11) in the Proximal Tubule of the Human Kidney,” Laboratory Investigation, Vol. 59, No. 6, 1988, pp. 789-797.
- C. P. Vio and V. A. Jeanneret, “Local Induction of Angiotensin-Converting Enzyme in the Kidney as a Mechanism of Progressive Renal Diseases,” Kidney International, No. 86, 2003, pp. S57-63.
- D. E. Casarini, M. A. Boim, R. C. Stella, et al., “Angiotensin I-Converting Enzyme Activity in Tubular Fluid along the Rat Nephron,” American Journal of Physiology, Heart and Circulatory Physiology, Vol. 272, No. 3, 1997, pp. F405-409.
- A. B. Gould and D. Green, “Kinetics of the Human Renin and Human Substrate Reaction,” Cardiovascular Research, Vol. 5, No. 1, 1971, pp. 86-89. doi:10.1093/cvr/5.1.86
- A. R. Brasier and J. Li, “Mechanisms for Inducible Control of Angiotensinogen Gene Transcription,” Hypertension, Vol. 27, No. 3, 1996, pp. 465-475.
- Y. Ding, R. L. Davisson, D. O. Hardy, et al., “The Kidney Androgen-Regulated Protein Promoter Confers Renal Proximal Tubule Cell-Specific and Highly Androgen-ReSponsive Expression on the Human Angiotensinogen Gene in Transgenic Mice,” The Journal of Biological Chemistry, Vol. 272, No. 44, 1997, pp. 28142-28148. doi:10.1074/jbc.272.44.28142
- S. Kimura, J. J. Mullins, B. Bunnemann, et al., “High Blood Pressure in Transgenic Mice Carrying the Rat Angiotensinogen Gene,” EMBO Journal, Vol. 11, No. 3, 1992, pp. 821-827.
- A. Fukamizu, K. Sugimura, E. Takimoto, et al., “Chimeric Renin-Angiotensin System Demonstrates Sustained Increase in Blood Pressure of Transgenic Mice Carrying Both Human Renin and Human Angiotensinogen Genes,” The Journal of Biological Chemistry, Vol. 268, No. 16, 1993, pp. 11617-11621.
- J. Bohlender, J. Menard, D. Ganten and F. C. Luft, “Angiotensinogen Concentrations and Renin Clearance: Implications for Blood Pressure Regulation,” Hypertension, Vol. 35, No. 3, 2000, pp. 780-786.
- O. Smithies, “Theodore Cooper Memorial Lecture. A Mouse View of Hypertension,” Hypertension, Vol. 30, No. 6, 1997, pp. 1318-1324.
- D. C. Merrill, M. W. Thompson, C. L. Carney, et al., “Chronic Hypertension and Altered Baroreflex Responses in Transgenic Mice Containing the human Renin and Human Angiotensinogen Genes,” Journal of Clinical Investigation, Vol. 97, No. 4, 1996, pp. 1047-1055. doi:10.1172/JCI118497
- H. Kobori, Y. Ozawa, R. Satou, et al., “Kidney-Specific Enhancement of ANG II Stimulates Endogenous Intrarenal Angiotensinogen in Gene-Targeted Mice,” American Journal of Physiology, Vol. 293, No. 3, 2007, pp. F938-945. doi:10.1152/ajprenal.00146.2007
- S. Sachetelli, Q. Liu, S. L. Zhang, et al., “RAS Blockade Decreases Blood Pressure and Proteinuria in Transgenic Mice Overexpressing Rat Angiotensinogen Gene in the Kidney,” Kidney International, Vol. 69, No. 6, 2006, pp. 1016-1023. doi:10.1038/sj.ki.5000210
- J. L. Lavoie, K. D. Lake-Bruse and C. D. Sigmund, “Increased Blood Pressure in Transgenic Mice Expressing Both Human Renin and Angiotensinogen in the renal Proximal Tubule,” American Journal of Physiology, Vol. 286, No. 5, 2004, pp. F965-971. doi:10.1152/ajprenal.00402.2003
- O. Smithies and H. S. Kim, “Targeted Gene Duplication and Disruption for Analyzing Quantitative Genetic Traits in Mice,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 9, 1994, pp. 3612-3615. doi:10.1073/pnas.91.9.3612
- H. S. Kim, J. H. Krege, K. D. Kluckman, et al., “Genetic Control of Blood Pressure and The Angiotensinogen Locus,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, No. 7, 1995, pp. 2735-2739. doi:10.1073/pnas.92.7.2735
- I. Inoue, T. Nakajima, C. S. Williams, et al., “A Nucleotide Substitution in the Promoter of Human Angiotensinogen is Associated with Essential Hypertension and Affects Basal Transcription in vitro,” Journal of Clinical Investigation, Vol. 99, No. 7, 1997, pp. 1786-1797.
- X. Jeunemaitre, F. Soubrier, Y. V. Kotelevtsev, et al., “Molecular Basis of Human Hypertension: Role of Angiotensinogen,” Cell, Vol. 71, No. 1, 1992, pp. 169-180.
- Y. Y. Zhao, J. Zhou, C. S. Narayanan, et al., “Role of C/A Polymorphism at -20 on the Expression of Human Angiotensinogen Gene,” Hypertension, Vol. 33, No. 1, 1999, pp. 108-115.
- T. Ishigami, S. Umemura, K. Tamura, et al., “Essential Hypertension and 5’ Upstream Core Promoter Region of Human Angiotensinogen Gene,” Hypertension, Vol. 30, No. 6, 1997, pp. 1325-1330.
- H. Kobori, L. M. Harrison-Bernard and L. G. Navar, “Enhancement of Angiotensinogen Expression in Angiotensin II-Dependent Hypertension,” Hypertension, Vol. 37, No. 5, 2001, pp. 1329-1335.
- H. Kobori, L. M. Harrison-Bernard and L. G. Navar, “Urinary Excretion of Angiotensinogen Reflects Intrarenal Angiotensinogen Production,” Kidney International, Vol. 61, No. 2, 2002, pp. 579-585. doi:10.1046/j.1523-1755.2002.00155.x
- H. Kobori, A. Nishiyama, L.M. Harrison-Bernard and L. G. Navar, “Urinary Angiotensinogen as an Indicator of Intrarenal Angiotensin Status in Hypertension,” Hypertension, Vol. 41, No. 1, 2003, pp. 42-49.
- H. Kobori, M. C. Prieto-Carrasquero, Y. Ozawa and L. G. Navar, “AT1 Receptor Mediated Augmentation of Intrarenal Angiotensinogen in Angiotensin II-Dependent Hypertension,” Hypertension, Vol. 43, No. 5, 2004, pp. 1126-1132.
- H. Schunkert, J. R. Ingelfinger, H. Jacob, et al., “Reciprocal Feedback Regulation of Kidney Angiotensinogen and Renin mRNA Expressions by Angiotensin II,” American Journal of Physiology, Vol. 263, No. 5, 1992, pp. E863-869.
- H. Kobori and A. Nishiyama, “Effects of Tempol on Renal Angiotensinogen Production in Dahl Salt-Sensitive Rats,” Biochemical and Biophysical Research Communications, Vol. 315, No. 3, 2004, pp. 746-750. doi:10.1016/j.bbrc.2004.01.120
- H. Kobori, A. Nishiyama, Y. Abe and L.G. Navar, “Enhancement of Intrarenal Angiotensinogen in Dahl SaltSensitive Rats on High Salt Diet,” Hypertension, Vol. 41, No. 3, 2003, pp. 592-597.
- H. Kobori, Y. Ozawa, Y. Suzaki and A. Nishiyama, “Enhanced Intrarenal Angiotensinogen Contributes to Early Renal Injury in Spontaneously Hypertensive Rats,” Journal of the American Society of Nephrology, Vol. 16, No. 7, 2005, pp. 2073-2080. doi:10.1681/ASN.2004080676
- S. Anderson, F. F. Jung and J. R. Ingelfinger, “Renal Renin-Angiotensin System in Diabetes: Functional, Immunohistochemical, and Molecular Biological Correlations,” American Journal of Physiology, Vol. 265, No. 4, 1993, pp. F477-486.
- Y. Nagai, L. Yao, H. Kobori, et al., “Temporary Angiotensin II Blockade at the Prediabetic Stage Attenuates the Development of Renal Injury in Type 2 Diabetic Rats,” Journal of the American Society of Nephrology, Vol. 16, No. 3, 2005, pp. 703-711. doi:10.1681/ASN.2004080649
- R. Singh, A. K. Singh and D. J. Leehey, “A Novel Mechanism for Angiotensin II Formation in Streptozotocin-Diabetic Rat Glomeruli,” American Journal of Physiology—Renal Physiology, Vol. 288, No. 6, 2005, pp. F1183-1190. doi:10.1152/ajprenal.00159.2003
- R. Singh, A. K. Singh and D. J. Leehey, “A Novel Mechanism for Angiotensin II Formation in Streptozotocin-Diabetic Rat Glomeruli,” American Journal of Physiology—Renal Physiology, Vol. 288, No. 6, 2005, pp. F1183-1190. doi:10.1152/ajprenal.00159.2003
- K. Miyata, N. Ohashi, Y. Suzaki, et al., “Sequential Activation of the Reactive Oxygen Species/ Angiotensinogen/ Renin-Angiotensin System Axis in Renal Injury of Type 2 Diabetic Rats,” Clinical and Experimental Pharmacology and Physiology, Vol. 35, No. 8, 2008, pp. 922- 927. doi:10.1111/j.1440-1681.2008.04938.x
- D. J. Leehey, A. K. Singh, J. P. Bast, et al., “Glomerular Renin Angiotensin System in Streptozotocin Diabetic and Zucker Diabetic Fatty Rats,” Translational Research, Vol. 151, No. 4, 2008, pp. 208-216. doi:10.1016/j.trsl.2008.01.003
- H. Kobori, A. Katsurada, Y. Ozawa, et al., “Enhanced Intrarenal Oxidative Stress and Angiotensinogen in IgA Nephropathy Patients,” Biochemical and Biophysical Research Communications, Vol. 358, No. 1, 2007, pp. 156- 163. doi:10.1016/j.bbrc.2007.04.105
- M. Takamatsu, M. Urushihara, S. Kondo, et al., “Glomerular Angiotensinogen Protein Is Enhanced in Pediatric IgA Nephropathy,” Pediatric Nephrology, Vol. 23, No. 8, 2008, pp. 1257-1267. doi:10.1007/s00467-008-0801-6
- H. Kobori, Y. Ozawa, Y. Suzaki, et al., “Young Scholars Award Lecture: Intratubular angiotensinogen in Hypertension and Kidney Diseases,” American Journal of Hypertension, Vol. 19, No. 5, 2006, pp. 541-550. doi:10.1016/j.amjhyper.2005.11.014
- A. Katsurada, Y. Hagiwara, K. Miyashita, et al., “Novel Sandwich ELISA for Human Angiotensinogen,” American Journal of Physiology—Renal Physiology, Vol. 293, No. 3, 2007, pp. F956-960. doi:10.1152/ajprenal.00090.2007
- H. Kobori, A. B. Alper, Jr., R. Shenava, et al., “Urinary Angiotensinogen as a Novel Biomarker of the Intrarenal Renin-Angiotensin System Status in Hypertensive Patients,” Hypertension, Vol. 53, No. 2, 2009, pp. 344-350.
- H. Kobori, M. Urushihara, J. H. Xu, et al., “Urinary Angiotensinogen Is Correlated with Blood Pressure in Men (Bogalusa Heart Study),” Hypertension, Vol. 28, No. 7, 2010, pp. 1422-1428. doi:10.1097/HJH.0b013e3283392673
- H. Kobori, N. Ohashi, A. Katsurada, et al., “Urinary Angiotensinogen as a Potential Biomarker of Severity of Chronic Kidney Diseases,” Journal of the American Society of Hypertension, Vol. 2, No. 5, 2008, pp. 349-354. doi:10.1016/j.jash.2008.04.008
- M. Urushihara, S. Kondo, S. Kagami and H. Kobori, “Urinary Angiotensinogen Accurately Reflects Intrarenal Renin-Angiotensin System Activity,” American Journal of Nephrology, Vol. 31, No. 4, 2010, pp. 318-325. doi:10.1159/000286037
- A. Nishiyama, Y. Konishi, N. Ohashi, et al., “Urinary Angiotensinogen Reflects the Activity of Intrarenal Renin-Angiotensin System in Patients with IgA Nephropathy,” Nephrology Dialysis Transplantation, Vol. 26, No. 1, 2011, pp. 170-177. doi:10.1093/ndt/gfq371
- H. R. Jang, S. M. Kim, Y. J. Lee, et al., “The Origin and the Clinical Significance of Urinary Angiotensinogen in Proteinuric IgA Nephropathy Patients,” Annals of Medicine, 2011, Ahead of Print.
- T. Saito, M. Urushihara, Y. Kotani, et al., “Increased urinary Angiotensinogen Is Precedent to Increased Urinary Albumin in Patients with Type 1 Diabetes,” The American Journal of the Medical Sciences, Vol. 338, No. 6, 2009, pp. 478-480. doi:10.1097/MAJ.0b013e3181b90c25
- S. Ogawa, H. Kobori, N. Ohashi, et al., “Angiotensin II Type 1 Receptor Blockers Reduce Urinary Angiotensinogen Excretion and the Levels of Urinary Markers of Oxidative Stress and Inflammation in Patients with Type 2 Diabetic Nephropathy,” Journal of Biomarker Insights, Vol. 4, 2009, pp. 97-102.
- H. R. Brunner, “ACE Inhibitors in Renal Disease,” Kidney Internationa, Vol. 42, No. 2, 1992, pp. 463-479.
- D. E. Kohan, “Angiotensin II and Endothelin in Chronic Glomerulonephritis,” Kidney International, Vol. 54, No. 2, 1998, pp. 646-647. doi:10.1046/j.1523-1755.1998.00038.x
- R. A. Lafayette, G. Mayer, S. K. Park and T. W. Meyer, “Angiotensin II Receptor Blockade Limits Glomerular Injury in Rats with Reduced Renal Mass,” Journal of Clinical Investigation, Vol. 90, No. 3, 1992, pp. 766-771. doi:10.1172/JCI115949
- I. Giatras, J. Lau and A. S. Levey, “Effect of Angiotensin-Converting Enzyme Inhibitors on the Progression of Nondiabetic Renal Disease: A Meta-Analysis of Randomized trials. Angiotensin-Converting-Enzyme Inhibition and Progressive Renal Disease Study Group,” Annals of Internal Medicine, Vol. 127, No. 5, 1997, pp. 337-345.
- B. G. Hudson, K. Tryggvason, M. Sundaramoorthy and E.G. Neilson, “Alport’s Syndrome, Goodpasture’s Syndrome, and Type IV Collagen,” New England Journal of Medicine, Vol. 348, No. 25, 2003, pp. 2543-2556. doi:10.1056/NEJMra022296
- K. K. Jindal, “Management of Idiopathic Crescentic and Diffuse Proliferative Glomerulonephritis: Evidence-Based Recommendations,” Kidney International, Vol. 70, No., 1999, pp. S33-40.
- T. Wada, H. Yokoyama, K. Furuichi, et al., “Intervention of Crescentic Glomerulonephritis by Antibodies to Monocyte Chemotactic and Activating Factor (MCAF/MCP- 1),” FASEB Journal, Vol. 10, No. 12, 1996, pp. 1418- 1425.
- T. Wada, K. Furuichi, N. Sakai, et al., “A New Anti-InFlammatory Compound, FR167653, Ameliorates Crescentic Glomerulonephritis in Wistar-Kyoto Rats,” Journal of the American Society of Nephrology, Vol. 11, No. 8, 2000, pp. 1534-1541.
- H. Y. Lan, D. J. Nikolic-Paterson, M. Zarama, et al., “Suppression of Experimental Crescentic Glomerulonephritis by the Interleukin-1 Receptor Antagonist,” Kidney International , Vol. 43, No. 2, 1993, pp. 479-485. doi:10.1038/ki.1993.70
- M. Le Hir, C. Haas, M. Marino and B. Ryffel, “Prevention of Crescentic Glomerulonephritis Induced by AntiGlomerular Membrane Antibody in Tumor Necrosis FactorDeficient Mice,” Laboratory Investigation, Vol. 78, No. 12, 1998, pp. 1625-1631.
- Y. Ozawa, H. Kobori, Y. Suzaki and L. G. Navar, “Sustained Renal Interstitial Macrophage Infiltration Following Chronic Angiotensin II Infusions,” American Journal of Physiology—Renal Physiology, Vol. 292, No. 1, 2007, pp. F330-339. doi:10.1152/ajprenal.00059.2006
- M. Shimizu, S. Kondo, M. Urushihara, et al., “Role of Integrin-Linked Kinase in Epithelial-Mesenchymal Transition in Crescent Formation of Experimental Glomerulonephritis,” Nephrology Dialysis Transplantation, Vol. 21, No. 9, 2006, pp. 2380-2390. doi:10.1093/ndt/gfl243
- B. G. Hudson, R. Kalluri, S. Gunwar, et al., “Molecular Characteristics of the Goodpasture Autoantigen,” Kidney International, Vol. 43, No. 1, 1993, pp. 135-139. doi:10.1038/ki.1993.22
- M. Urushihara, N. Ohashi, K. Miyata, et al., “Addition of Angiotensin II Type 1 Receptor Blocker to CCR2 Antagonist Markedly Attenuates Crescentic Glomerulonephritis,” Hypertension, Vol. 57, No. 3, 2011, pp. 586-593.
- A. A. Elmarakby, J. E. Quigley, J. J. Olearczyk, et al., “Chemokine Receptor 2b Inhibition Provides Renal Protection in Angiotensin II—Salt Hypertension,” Hypertension, Vol. 50, No. 6, 2007, pp. 1069-1076.
- H. Koike, T. Sada and M. Mizuno, “In vitro and in vivo Pharmacology of Olmesartan Medoxomil, an Angiotensin II type AT1 Receptor Antagonist,” Journal of Hypertension—Supplement, Vol. 19, No. 1, 2001, pp. S3-14.
- J. R. Ingelfinger, F. Jung, D. Diamant, et al., “Rat Proximal Tubule Cell Line Transformed with Origin-Defective SV40 DNA: Autocrine ANG II Feedback,” American Journal of Physiology, Vol. 276, No. 2, 1999, pp. F218- 227.
- A. Nishiyama, D. M. Seth and L. G. Navar, “Angiotensin II Type 1 Receptor-Mediated Augmentation of Renal Interstitial Fluid Angiotensin II in Angiotensin II-Induced Hypertension,” Journal of Hypertension, Vol. 21, No. 10, 2003, pp. 1897-1903. doi:10.1097/00004872-200310000-00017
- Y. Hisada, T. Sugaya, M. Yamanouchi, et al., “Angiotensin II Plays a Pathogenic Role in Immune-Mediated Renal Injury in Mice,” Journal of Clinical Investigation, Vol. 103, No. 5, 1999, pp. 627-635. doi:10.1172/JCI2454
- Y. Suzuki, I. Shirato, K. Okumura, et al., “Distinct contribution of Fc Receptors and Angiotensin II-Dependent Pathways in Anti-GBM Glomerulonephritis,” Kidney International, Vol. 54, No. 4, 1998, pp. 1166-1174. doi:10.1046/j.1523-1755.1998.00108.x
- N. Joss, K. R. Paterson, C. J. Deighan, et al., “Diabetic Nephropathy: How Effective Is Treatment in Clinical Practice?” QJM, Vol. 95, No. 1, 2002, pp. 41-49.
- P. Fioretto and M. Mauer, “Histopathology of Diabetic Nephropathy,” Seminars in Nephrology, Vol. 27, No. 2, 2007, pp. 195-207. doi:10.1016/j.semnephrol.2007.01.012
- Y. Taguma, Y. Kitamoto, G. Futaki, et al., “Effect of Captopril on Heavy Proteinuria in Azotemic Diabetics,” New England Journal of Medicine, Vol. 313, No. 26, 1985, pp. 1617-1620. doi:10.1056/NEJM198512263132601
- E.J. Lewis, L.G. Hunsicker, R.P. Bain, and R.D. Rohde, “The Effect of Angiotensin-Converting-Enzyme Inhibition on Diabetic Nephropathy. The Collaborative Study Group,” New England Journal of Medicine, Vol. 329, No. 20, 1993, pp. 1456-1462. doi:10.1056/NEJM199311113292004
- B. M. Brenner, M. E. Cooper, D. de Zeeuw, et al., “Effects of Losartan on Renal and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Nephropathy,” New England Journal of Medicine, Vol. 345, No. 12, 2001, pp. 861-869. doi:10.1056/NEJMoa011161
- H. Haller, S. Ito, J. L. Izzo, Jr., et al., “Olmesartan for the Delay or Prevention of Microalbuminuria in Type 2 Diabetes,” New England Journal of Medicine, Vol. 364, No. 10, 2011, pp. 907-917. doi:10.1056/NEJMoa1007994
- J. R. Ingelfinger, “Preemptive Olmesartan for the Delay or Prevention of Microalbuminuria in Diabetes,” New England Journal of Medicine, Vol. 364, No. 10, 2011, pp. 970-971. doi:10.1056/NEJMe1014147
- P. Jacobsen, S. Andersen, K. Rossing, et al., “Dual Blockade of the Renin-Angiotensin System versus Maximal Recommended Dose of ACE Inhibition in Diabetic Nephropathy,” Kidney International, Vol. 63, No. 5, 2003, pp. 1874-1880. doi:10.1046/j.1523-1755.2003.00940.x
- C. E. Mogensen, S. Neldam, I. Tikkanen, et al., “Randomised Controlled Trial of Dual Blockade of ReninAngiotensin System in Patients with Hypertension, Microalbuminuria, and Non-Insulin Dependent Diabetes: The Candesartan and Lisinopril Microalbuminuria (CALM) Study,” BMJ, Vol. 321, No. 7274, 2000, pp. 1440-1444.
- S. Hoshi, Y. Shu, F. Yoshida, et al., “Podocyte Injury Promotes Progressive Nephropathy in Zucker Diabetic Fatty Rats,” Laboratory Investigation, Vol. 82, pp. 25-35, No. 1, 2002.
- M. Mizuno, T. Sada, M. Kato and H. Koike, “Renoprotective Effects of Blockade of Angiotensin II AT1 Receptors in an Animal Model of Type 2 Diabetes,” Hypertension Research, Vol. 25, No. 2, 2002, pp. 271-278.
- J. P. Vora, S. M. Zimsen, D. C. Houghton and S. Anderson, “Evolution of Metabolic and Renal Changes in the ZDF/ Drt-fa Rat Model of Type II Diabetes,” Journal of the American Society of Nephrology, Vol. 7, No. 1, 1996, pp. 113-117.
- G. J. Etgen and B. A. Oldham, “Profiling of Zucker Diabetic Fatty Rats in Their Progression to the Overt Diabetic State,” Metabolism, Vol. 49, No. 5, 2000, pp. 684- 688.
- N. Ohashi, M. Urushihara, R. Satou and H. Kobori, “Glomerular Angiotensinogen is Induced in Mesangial Cells in Diabetic Rats via Reactive Oxygen Species-ERK/ JNK Pathways,” Hypertension Research, Vol. 33, No. 11, 2010, pp. 1174-1181. doi:10.1038/hr.2010.143
- R. Satou, R. A. Gonzalez-Villalobos, K. Miyata, et al., “Costimulation with Angiotensin II and Interleukin 6 augments Angiotensinogen Expression in cultured Human Renal Proximal Tubular Cells,” American Journal of Physiology—Renal Physiology, Vol. 295, No. 1, 2008, pp. F283-289. doi:10.1152/ajprenal.00047.2008
- T. J. Hsieh, S. L. Zhang, J. G. Filep, et al., “High Glucose Stimulates Angiotensinogen Gene Expression via Reactive Oxygen Species Generation in Rat Kidney Proximal Tubular Cells,” Endocrinology, Vol. 143, No. 8, 2002, pp. 2975-2985.