Open Journal of Organ Transplant Surgery
Vol.3 No.2(2013), Article ID:31868,6 pages DOI:10.4236/ojots.2013.32007
Role of Oxidative Stress in Cardiac Allograft Vasculopathy
1Department of Cardiology and Cardiothoracic Surgery, Scott & White Healthcare, Temple, USA
2Department of Cardiothoracic Surgery, Drexel University College of Medicine/Hahnemann University Hospital, Philadelphia, USA
Email: nanair@sw.org
Copyright © 2013 Nandini Nair et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 12, 2013; revised May 13, 2013; accepted May 20, 2013
Keywords: Cardiac Allograft Vasculopathy; Oxidative Stress; Asymmetric Dimethyl Arginine; Cyto Megalo Virus; Transplantation
ABSTRACT
Cardiac Allograft Vasculopathy, an accelerated form of arterial occlusive disease, is the major cause of death in the long-term after heart transplantation. Multiple factors influence the initiation and progression of CAV. These include ischemia-reperfusion, dyslipidemia, insulin resistance, and hypertension due to the use of immunosuppressive agents, the direct effects of immunosuppressive agents on endothelial function, and viruses (CMV). Impaired endothelial function reflects abnormalities in the production or activity of several vasoactive substances. Disruption of the nitric oxide synthase (NOS) pathway leads to changes in vascular reactivity, structure, and interaction with circulating blood components. Since endothelium-derived nitric oxide (NO) suppresses vascular cell proliferation and vascular inflammation, a deficit in vascular NO facilitates development of CAV. The link between oxidative stress and CAV largely exists in the balance between free radical superoxide 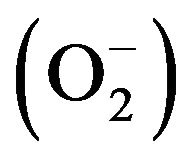 generation and NO production. This review focuses on identifying the oxidative stress factors affecting CAV.
generation and NO production. This review focuses on identifying the oxidative stress factors affecting CAV.
1. Introduction
The technique of successful heart transplantation has existed for more than 45 years now and so has Cardiac Allograft Vasculopathy (CAV) almost for the same number of years. However, CAV still continues to remain the major threat to long term survival of the cardiac allograft. CAV, an accelerated form of arterial occlusive disease, is the major cause of death in the long-term after heart transplantation. This review focuses on identification of factors of oxidative stress adversely influencing the initiation and progression of CAV.
CAV is considered as the result of a combination of immunological and non-immunological insults to the cardiac allograft status post transplantation [1]. Multiple factors can influence the initiation and progression of CAV. These include ischemia-reperfusion, dyslipidemia, insulin resistance, and hypertension due to the use of immunosuppressive agents, the direct effects of some immunosuppressive agents on endothelial function, and viral infections most commonly cytomegalovirus (CMV). It must be emphasized that the common denominator is endothelial dysfunction.
Impaired endothelial function reflects abnormalities in the production or activity of several vasoactive substances. Disruption of the nitric oxide synthase (NOS) pathway leads to changes in vascular reactivity, structure, and interaction with circulating blood components. Since endothelium-derived nitric oxide (NO) suppresses vascular cell proliferation and vascular inflammation, a deficit in vascular NO facilitates development of CAV. One of the molecules that have risen to prominence as an endogenous inhibitor of NOS is Asymmetric Dimethyl Argninine (ADMA) [2]. ADMA, first reported in 1992 [2], is a modified amino acid which is derived from the methylation of internal arginine residues in proteins and the subsequent hydrolysis of these proteins. ADMA is an endogenous inhibitor of NOS. Increased tissue and plasma levels of this molecule are consistent with development of intimal hyperplasia. Infections with cytomegalo virus (CMV) can cause elevations of plasma ADMA levels leading to impaired endothelial function and predisposition to CAV.
The link between oxidative stress and CAV largely exists in the balance between free radical (superoxide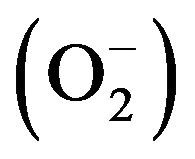 ) generation and NO production. An imbalance, favoring superoxide anion generation, activates oxidant-sensitive transcriptional pathways that induce genes such as MCP-1 and VCAM involved in atherogenesis (see Figures 1 and 2). This assumes importance as CAV and native atherogenesis share certain pathophysiological traits. Insult and injury secondary to oxidative stress occurs when an organ is separated from its native blood supply and then reperfused. Thus, graft ischemic time, quality of graft preservation during transport, hemodynamic status of the donor, catecholamines used for inotropic support, and reperfusion itself will influence ischemia perfusion effects. This injury continues during subsequent periods of immunological attacks on the donor organ by the recipient system. Damage to the endothelium following hypoxia, occurs via loss of the ability to release nitric oxide within minutes after reperfusion. This can be explained by the consumption of nitric oxide by superoxide radicals formed early during reperfusion. Endothelial damage can result from either denuding or nondenuding injury. In nondenuding injury, replacement of injured endothelial cells leads to endothelial dysfunction [3]. Both immune-related and nonimmune-related factors contribute to nondenuding injury. In contrast, denuding injury is caused by ischemia-reperfusion injury during transplantation or during episodes of acute cellular rejection.
) generation and NO production. An imbalance, favoring superoxide anion generation, activates oxidant-sensitive transcriptional pathways that induce genes such as MCP-1 and VCAM involved in atherogenesis (see Figures 1 and 2). This assumes importance as CAV and native atherogenesis share certain pathophysiological traits. Insult and injury secondary to oxidative stress occurs when an organ is separated from its native blood supply and then reperfused. Thus, graft ischemic time, quality of graft preservation during transport, hemodynamic status of the donor, catecholamines used for inotropic support, and reperfusion itself will influence ischemia perfusion effects. This injury continues during subsequent periods of immunological attacks on the donor organ by the recipient system. Damage to the endothelium following hypoxia, occurs via loss of the ability to release nitric oxide within minutes after reperfusion. This can be explained by the consumption of nitric oxide by superoxide radicals formed early during reperfusion. Endothelial damage can result from either denuding or nondenuding injury. In nondenuding injury, replacement of injured endothelial cells leads to endothelial dysfunction [3]. Both immune-related and nonimmune-related factors contribute to nondenuding injury. In contrast, denuding injury is caused by ischemia-reperfusion injury during transplantation or during episodes of acute cellular rejection.
Another important factor contributing to the development of CAV is CMV infections. The mechanisms underlying the process are poorly understood but there exists a definite link. As early as 1989, it was noted that CMV infection led to increased frequency of rejection and CAV [4]. Endothelial dysfunction has been described as the major force in CMV induced CAV
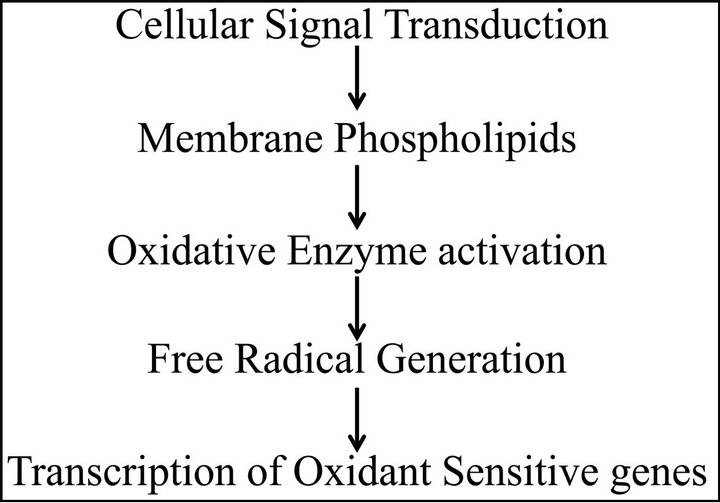
Figure 1. Molecular basis of oxidative stress.
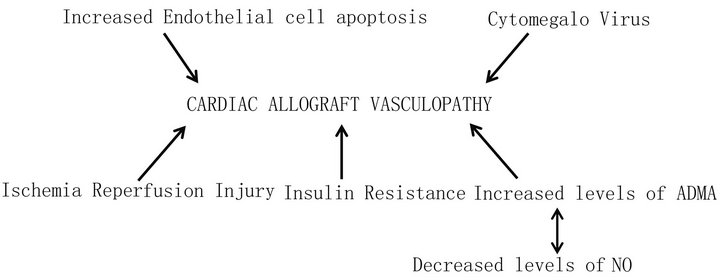
Figure 2. Cellular mechanisms contributing to cardiac allograft vasculopathy.
[4-6].
2. Endothelial Cell Dysfunction and Molecular Aspects of Oxidative Stress
Understanding the mechanisms of endothelial dysfunction may lead to endothelial-targeted therapies for prevention of CAV. Endothelial dysfunction is characterized by vascular inflammation and intimal proliferation, leading to luminal stenosis of epicardial arteries, occlusion of smaller vessels, and myocardial infarction. Histological lesions may range from concentric, diffuse intimal hyperplasia to fibrofatty plaques indistinguishable from spontaneously occurring atherosclerosis [7]. Coronary endothelial cells of the allograft can serve as antigenresenting cells as well as targets of allogeneic lymphocyte reactivity. T-cell-interaction with graft endothelial cells, initiates and sustains the chronic immune response leading to injury [8]. At the molecular level endothelial cell dysfunction occurs with up regulation of expression of adhesion molecules and chemokines, which participate in the inflammatory process leading to a prothrombogenic state. Predisposing conditions include preservation injury, ischemia-reperfusion, acute rejection, T cell activation, antibody deposition and complement fixation, and viral infection. The ongoing inflammation is thought to accelerate the development of CAV.
Endothelial dysfunction of the epicardial arteries can be defined as a paradoxical vasoconstriction of >10% in response to intracoronary injections of acetylcholine and this may be manifested very early status post transplant. Endothelium dependent flow responses declined significantly in a 3 year follow-up study [9-12]. Homeostatic balance between endothelial vasodilators and vasoconstrictors contributes to adverse changes in vascular structure and reactivity. Endothelium derived NO is vasoprotective and reduction in the synthesis or bioactivity of endothelium-derived NO promotes processes favoring CAV.
Increase in reactive oxygen species (ROS) in cells within the graft or recruited to it and associated with a decline of antioxidant enzymes, can lead to covalent oxidative modifications of lipids, proteins, and DNA. Elevations in reactive nitrogen species (RNS) contribute to tissue modification and destruction. RNS are derived from nitric oxide (NO) via up regulation of inducible NO synthase (iNOS) noted in animal models [13-15] with excess NO reacting with superoxide anion to form peroxynitrite. The nitrating and oxidizing properties of peroxynitrite produce significant cellular toxicity in the setting of cardiac allograft rejection [16]. The formation of both ROS and RNS via the induction of a ROS-sensitive transcription factor, nuclear factor-κB (NF-κB), leads to iNOS upregulation [17]. In a murine heterotopic transplantation model, administration of antioxidants until postop day 3 produced a significant decrease in nitrosative and oxidative stress in cardiac allografts leading to better modulation of the immune system and in turn shielding the graft from CAV. In this model, a significant decrease of NF-κB and inducible NOS was noted [18].
Immunesuppressive therapy itself can cause increased levels of ROS leading to vascular dysfunction. ROS is produced mainly by the NADPH oxidase system in vascular endothelial cells. The NADPH oxidase subunits No×1, No×2 and No×4 are exclusively found in vascular cells. Activation of the GTP-dependent protein subunit Race 1 is required for this enzyme to function. Therefore inhibition of this protein subunit should reduce NADPH oxidase-induced oxidative stress. Oxidative stress is manifested as hypertension, hypertrophy, fibrosis, conduction abnormalities, endothelial dysfunction, and CAV. As opposed to cyclosporine and tacrolimus, the immunosuppressive drug mycophenolate inhibits the Rac 1 subunit thus inhibiting NADPH oxidase in the vasculature. Reducing oxidative stress leads to decrease in graft rejection and vascular damage and therefore appears to exert a protective effect against CAV [19].
3. ADMA an Endogenous Inhibitor of NOS
ADMA is a competitive inhibitor of NOS. This modified amino acid is derived from the methylation of internal arginine residues in proteins and the subsequent hydrolysis of these proteins. ADMA is never produced by methylation of free arginine, and is not affected by L-arginine intake [20,21]. ADMA is excreted by the kidney to a lesser extent and maximally metabolized by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) to citrulline and dimethylamine (Figures 3 and 4). Hence inhibitors of DDAH in turn increase ADMA levels. Endothelium-derived NO is a potent endogenous vasodilator and also prevents vascular inflammation and lesion formation in the vessel wall by inhibiting platelet and leukocyte adherence and by suppressing vascular smooth muscle cell proliferation [22]. In patients who have received cardiac allografts ADMA levels appear to be elevated in parallel with endothelial dysfunction. Therefore a notable link appears to exist. It has also been shown that infusion of L-arginine in patients who have received a cardiac transplant endothelial dysfunction is reversed. In animal models inhibition of the NOS pathway accel model of CAV overexpression of DDAH reduced plasma and tissue ADMA concentrations and thereby increased NO production [27]. In transgenic mice overexpressing DDAH an increase in plasma NO levels and a reduction in systemic vascular resistance were noted [28]. When donor hearts of wild-type mice were heterotopically transplanted into transgenic mice overexpressing human DDAH-I, or transplanted into wild-type (WT) littermates, the DDAH-I expressing mice
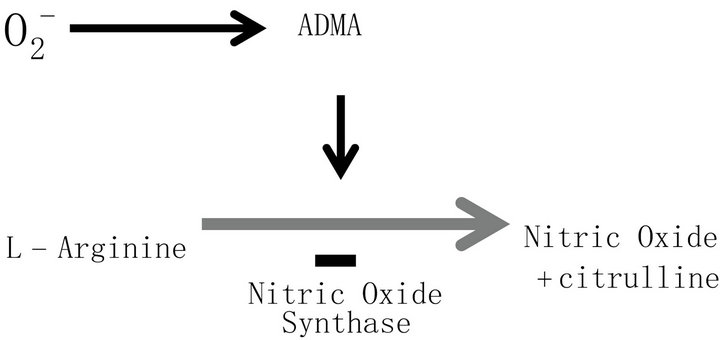
Figure 3. Inhibition of nitric oxide synthase by ADMA.
had less CAV incidence at erates atherosclerosis and enhancement of NO synthesis reduces vascular lesion formation [23-26]. In a murine 30 days post transplantation [29]. These animal models drive home the point that ADMA is an important regulator of NO synthesis and a genetic reduction in plasma or tissue. ADMA levels have a protective effect on vasculature.
4. Role of CMV
Viral pathogens such as CMV cause endothelial dysfunction via an activation of molecular mechanisms of inflammation and coagulation. In recipients of cardiac transplants CMV has been historically linked to CAV [4]. It is interesting that CMV levels correlate with development of CAV and plasma ADMA levels correlate with CMV viremia. In vitro endothelial cell cultures show CMV infection elevates ADMA levels and decreases DDAH activity and levels. Viral infection showed increased nitrite levels. Intracellular cyclic GMP levels reflect NO bioactivity. The levels of cGMP were reduced in CMV infected cells. In these experiments basal production of the superoxide radical was increased with CMV infection. Addition of superoxide dismutase to the culture medium showed decreased levels of superoxide molecules and this effect seemed to be accompanied by decreased ADMA production. The pathogenesis elucidated in vitro seems to exist in vivo and increased ADMA and decreased DDAH levels appear to correlate with CMV infection. CMV causes induction of TNF alpha which causes up regulation of inducible NOS (iNOS). iNOS in turn elicits generation of superoxide anions. TNF alpha is also known to down regulate vascular DDAH activity and therefore increases ADMA activity. Treatment of post-transplant patients with sirolimus for immunesuppression appears to decrease ADMA levels and incidence of CAV. Correlations of this nature deduced in small animal and human studies are interesting, but exact causation is difficult to extrapolate without larger studies [30-32].
4.1. Insulin Resistance in CAV
Metabolic syndrome and insulin resistance have known to be accelerating factors in development of CAV. In
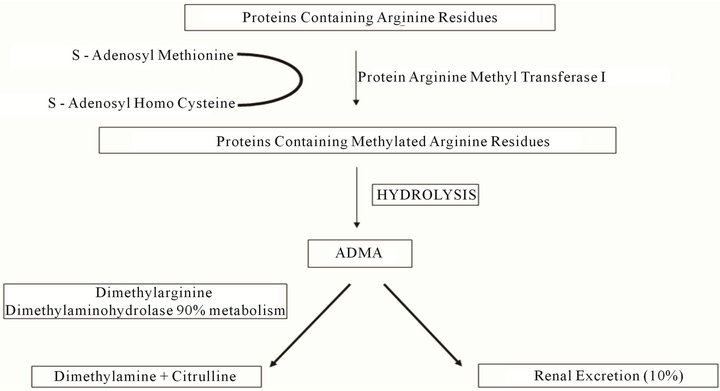
Figure 4. Synthesis and degradation of ADMA.
recent studies, metabolic syndrome has gained considerable importance in predisposing post-transplant patients to CAV. Insulin resistance, which is a part of the metabolic syndrome, is considered detrimental. In patients treated with pioglitazone, in-stent restenosis was found to be reduced in diabetics and non-diabetics. The mechanism has been postulated to be via a reduction of leptin and in insulin resistance, and therefore, improved endothelial function. Additionally, in small studies of patients’ status post transplants, a synergic interaction has been noted between markers of systemic inflammation such as CRP and insulin resistance (TG/HDL) [33-36].
4.2. Mia-Reperfusion Injury and Treatments with Antioxidants
Ischemia-reperfusion (IR) injury associated with oxidative stress is a major contributor to CAV initiation and progression. Microvascular dysfunction that ensues can result in primary graft failure or untreatable chronic rejection leading to CAV. At the molecular level, IR increased transendothelial PMN migration, NADPH oxidase activity, and SOD activity in PMNs and endothelial cells. Pioglitazone, a peroxisome proliferator-activated receptor (PPAR)-gamma agonist, could attenuate graft oxidant stress in cardiac transplantation in a murine model. It was able to achieve an anti-oxidant balance in a murine model. Seven days post transplantation, PPARgamma was significantly up-regulated by pioglitazone, but nuclear factor-kappaB and inducible nitric oxide synthase were significantly down-regulated in this model [37].
In a rat model, a peroral single-dose of simvastatin— the 3-hydroxy-3-methylglutaryl coenzyme A reductase and the Rho GTPase inhibitor given 2 hours before graft procurement attenuates the microvascular dysfunction. Simvastatin treatment inhibited microvascular endothelial cell and pericyte RhoA/Rho-associated protein kinase activation and endothelial cell-endothelial cell gap formation; decreased intragraft mRNA levels of hypoxiainducible factor-1α, inducible nitric oxide synthase, and endothelin-1. It increased heme oxygenase-1 reducing microvascular injury. In chronic rejection transforming growth, factor-β1-induced microvascular endothelial-tomesenchymal transition and led to myocardial fibrosis. This effect was abolished by simvastatin treatment via inhibition of transforming growth factor-β1 [38].
The discovery of the TLRs (Toll-like receptor) family has led to a better understanding of the innate immune system. TLR-2 and TLR 4 have been implicated in myocardial injury via ischemia-reperfusion. Murine models deficient in TLR -2 and TLR-4 confer protection from IR injury. TLR2 deficient models appear to have less LV dysfunction status post IR injury. TLR4 deficient mice show protection against myocardial injury via PI3K/Aktdependent signaling pathways [39,40]. Therefore modulation of TLRs and their downstream signaling pathways may help modulate IR injury.
5. Conclusion
CAV and oxidative stress are intrinsically linked via the NOS pathway and is mediated by a variety of factors. Preexisting arteriosclerotic disease in the graft, graft ischemia before transplantation, immunosuppressive agents post transplantation and the known risk factors (hyperpidemia, hypertension, diabetes, hyperhomocysteinemia), as well as CMV, all contribute to CAV initiation. Impairment of the NOS pathway and the role of the endogenous inhibitor of NOS (ADMA) are substantial detriments to graft survival due to oxidative stress. Identification and modulation of therapeutic targets that could impact the NOS pathway and endothelial function may be key to protection from CAV and enhancing long term graft survival.
6. Acknowledgements
This work was supported by an internal grant from the Division of Cardiology to Nandini Nair, MD, PhD.
REFERENCES
- J. D. Hosenpud, L. E. Benell, B. M. Keck, B. Fiol, M. M. Boucek and R. J. Novick, “The Registry of the International Society for Heart and Lung Transplantation: Sixteenth Official Report,” The Journal of Heart and Lung Transplantion, Vol. 18, No. 7, 1999, pp. 611-626. doi:10.1016/S1053-2498(99)00037-6
- P. Vallance, A. Leone, A. Calver, J. Collier and S. Moncada, “Endogenous Dimethlarginine as an Inhibitor of Nitric Oxide Synthesis,” Journal of Cardiovascular Pharmacology, Vol. 20, Suppl. 12, 1992, pp. S60-S62. doi:10.1097/00005344-199204002-00018
- M. E. Russell, “Cardiac Allograft Vasculopathy—A Changing Perspective,” Zeitschrift für Kardiologie, Vol. 89, Suppl. 9, 2000, pp. 6-10.
- M. T. Grattan, C. E. Moreno-Cabral, V. A. Starnes, P. E. Oyer, E. B. Stinson and N. E. Shumway. “Cytomegalovirus Infection Is Associated with Cardiac Allograft Rejection and Atherosclerosis,” Journal of the American Medical Association, Vol. 261, No. 42, 1989, pp. 3561- 3566. doi:10.1001/jama.1989.03420240075030
- M. C. van Dam-Mieras, A. D. Muller, V. W. van Hinsbergh, W. J. Mullers, P. H. Bomans and C. A. Bruggeman, “The Procoagulant Response of Cytomegalovirus Infected Endothelial Cells,” Thrombosis and Haemostasis, Vol. 68, No. 3, 1992, pp. 364-370.
- M. Billstrom Schroeder and G. S. Worthen, “Viral Regulation of RANTES Expression During Human Cytomegalovirus Infection of Endothelial Cells,” Journal of Virology, Vol. 75, No. 7, 2001, pp. 3383-3390. doi:10.1128/JVI.75.7.3383-3390.2001
- M. E. Billingham, “Histopathology of Graft Coronary Disease,” Journal of Heart and Lung Transplantation, Vol. 11, No. 3, 1992, pp.S38-S44.
- M. Weis and W. VonScheidt, “Coronary Atherosclerosis in the Transplanted Heart,” Annual Review of Medicine, Vol. 51, 2000, pp. 81-100. doi:10.1146/annurev.med.51.1.81
- M, Weis, S. Pehlivali and W. von Scheidt, “Heart Allograft Endothelial Cell Dysfunction. Cause, Course, and Consequences,” Zeitschrift für Kardiologie, Vol. 89, Suppl. 9, 2000, pp. 58-62.
- W. F. Fearon, M. Nakamura, D. P. Lee, et al., “Simultaneous Assessment of Fractional and Coronary Flow Reserves in Cardiac Transplant Recipients: Physiologic Investigation for Transplant Arteriopathy (PITA Study),” Circulation, Vol. 108, No, 2003, pp. 1605-1610. doi:10.1161/01.CIR.0000091116.84926.6F
- M. Weis, W. P. Wolf, N. Mazilli, et al., “Variations of Segmental Endothelium-Dependent and EndotheliumIndependent Vasomotor Tone after Cardiac Transplantation (Qualitative Changes in Endothelial Function),” American Heart Journal, Vol. 134 No. 2, 1997, pp. 306- 315. doi:10.1016/S0002-8703(97)70139-X
- C. B. Treasure, J. A. Vita, P. Ganz, et al., “Loss of the Coronary Microvascular Response to Acetylcholine in Cardiac Transplant Patients,” Circulation, Vol. 86, No. 4, 1992, pp. 1156-1164. doi:10.1161/01.CIR.86.4.1156
- J. Koglin, D. J. Granville, T. Glysing-Jensen, J. S. Mudgett, C. M. Carthy, B. M. McManus and M. E. Russell, “Attenuated Acute Cardiac Rejection in NOS2−/− Recipients Correlates with Reduced Apoptosis,” Circulation, Vol. 99, No. 1999, pp. 836-842. doi:10.1161/01.CIR.99.6.836
- M. E. Russell, A. F. Wallace, L. R. Wyner, J. B. Newell and M. J. Karnovsky, “Upregulation and Modulation of Inducible Nitric Oxide Synthase in Rat Cardiac Allografts with Chronic Rejection and Transplant Arteriosclerosis,” Circulation. Vol. 92, No. 3, 1995, pp. 457-464. doi:10.1161/01.CIR.92.3.457
- N. K. Worrall, W. D. Lazenby, T. P. Misko, et al., “Modulation of in Vivo Alloreactivity by Inhibition of Inducible Nitric Oxide Synthase,” The Journal of Experimental Medicine, Vol. 181, No. 1, 1995, pp. 63-70. doi:10.1084/jem.181.1.63
- E. Akizuki, T. Akaike, S. Okamoto, et al., “Role of Nitric Oxide and Superoxide in Acute Cardiac Allograft Rejection in Rats,” Proceeding of the Society of Experimental Biology and Medicine, Vol. 225, No. 2, 2000, pp. 151- 159. doi:10.1046/j.1525-1373.2000.22519.x
- G. Hall, J. D. Hasday and T. B. Rogers, “Regulating the Regulator: NF-KappaB Signaling in Heart,” Journal of Molecular and Cellular Cardiology, Vol. 41, No. 4, 2006, pp. 580-591. doi:10.1016/j.yjmcc.2006.07.006
- T. Hasegawa, K. Iwanaga, D. E. Hultquist, H. Liao, S. H. Visovatti and D. J. Pinsky. “Suppression of Nitrosative and Oxidative Stress to Reduce Cardiac Allograft Vasculopathy,” American Journal of Physiology Heart and Circulatory Physiology, Vol. 296, No. 4, 2009, pp. H1007-H1016. doi:10.1152/ajpheart.00498.2008
- A. Iyer and L. Brown, “Is Mycophenolate More than Just an Immunosuppressant? An Overview,” Indian Journal of Biochemisty and Biophysics, Vol. 46, No. 1, 2009, pp. 25-30.
- J. Najbauer, B. A. Johnson, A. L. Young and D. W. Aswad, “Peptides with Sequences Similar to Glycine, Arginine-Rich Motifs in Proteins Interacting with RNA Are Efficiently Recognized by Methyltransferase(s) Modifying Arginine in Numerous Proteins,” The Journal of Biological Chemistry, Vol. 268, No. 14, 1993, pp. 10501- 10509.
- S. Bode-Boger, R. H. Boger, S. Kienke, Junker and J. C. Frolich, “Elevated L-Arginine/Dimethylarginine Ratio Contributes to Enhanced Systemic NO Production by Dietary L-Arginine in Hypercholesterolemic Rabbits,” Biochemical and Biophysical Research Communications, Vol. 219, No. 1996, pp. 598-603. doi.10.1006/bbrc.1996.0279
- J. Cooke and V. Dzau, “Derangements of the Nitric Oxide Synthase Pathway, L-Arginine, and Cardiovascular Disease,” Circulation, Vol. 96, No. 2, 1997, pp. 379-382.
- H. Drexler, T. A. Fischell, F. J. Pinto, et al., “Effect of L-arginine on Coronary Endothelial Function in Cardiac Transplant Recipients: Relation to Vessel Wall Morphology,” Circulation. Vol. 89, No. 4, 1994, pp.1615-1623. doi:10.1161/01.ATV.14.5.753
- A. J. Cayatte, J. J. Palacino, K. Horten and R. A. Cohen, “Chronic Inhibition of Nitric Oxide Production Accelerates Neointima Formation and Impairs Endothelial Function in Hypercholesterolemic Rabbits,” Arteriosclerosis and Thrombosis, Vol. 14, No. 5, 1994, pp. 753-759. doi:10.1161/01.ATV.14.5.753
- P. L. Huang, “Disruption of the Endothelial Nitric Oxide Synthase Gene: Effect on Vascular Response to Injury,” The American Journal of Cardiology, Vol. 82, No. 10A, 1998, pp. 57S-59S. doi:10.1016/S0002-9149(98)00679-1
- J. Cooke, A. Singer, P. Tsao, P. Zera, R. A. Rowan and M. E. Billingham, “Anti-Atherogeneic Effects of L-Arginine in the Hypercholesterolemic Rabbit,” The Journal of Clinical Investigation, Vol. 90, No. 3, 1992, pp. 1168- 1172. doi:10.1172/JCI115937
- H. E. von der Leyen, G. H. Gibbons, R. Morishita, et al., “Gene Therapy Inhibiting Neointimal Vascular Lesion: In Vivo Transfer of Endothelial Cell Nitric Oxide Synthase Gene,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, No. 4, 1995, pp. 1137-1141. doi:10.1073/pnas.92.4.1137
- H. Dayoub, V. Achan, S. Adimoolam, et al., “Dimethylarginine Dimethylaminohydrolase Regulates Nitric Oxide Synthesis: Genetic and Physiological Evidence,” Circulation, Vol. 108, No. 24, 2003, 2042-3047. doi:10.1161/01.CIR.0000101924.04515.2E
- M. Tanaka, K. Sydow, F. Gunawan, et al., “Dimethylarginine Dimethylaminohydrolase Overexpression Suppresses Graft Coronary Artery Disease in Murine Cardiac Allografts,” Circulation, Vol. 112, No. 11, 2005, pp. 1549-1556. doi:10.1161/CIRCULATIONAHA.105.537670
- A. Ito, P. S. Tsao, S. Adimoolam, M. Kimoto, T. Ogawa and J. P. Cooke, “Novel Mechanism for Endothelial Dysfunction: Dysregulation of Dimethylarginine Dimethylaminohydrolase,” Circulation, Vol. 99, No. 24, 1999, pp. 3092-3095. doi:10.1161/01.CIR.99.24.3092
- M. Weis, T. N. Kledal, K.Y. Lin, et al., “Cytomegalovirus Infection Impairs the Nitric Oxide Synthase Pathway: Role of Asymmetric Dimethylarginine in Transplant Arteriosclerosis,” Circulation, Vol. 109, No. 4, 2004, pp. 500-505. doi:10.1161/01.CIR.0000109692.16004.AF
- L. Potena, W. F. Fearon, K. Sydow, et al., “Asymmetric Dimethylarginine and Cardiac Allograft Vasculopathy Progression: Modulation by Sirolimus,” Transplantation, Vol. 85, No. 6, 2008, pp. 827-833. doi:10.1097/TP.0b013e318166a3a4
- K. Nishio, M. Sakurai, T. Kusuyama, et al., “A Randomized Comparison of Pioglitazone to Inhibit Restenosis after Coronary Stenting in Patients with Type 2 Diabetes,” Diabetes Care, Vol. 29, No. 1, 2006, pp. 101-106. doi:10.2337/diacare.29.01.06.dc05-1170
- N. Marx, J. Wohrle, T. Nusser, et al., “Pioglitazone Reduces Neointima Volume after Coronary Stent Implantation: A Randomized, Placebo-Controlled, Double-Blind Trial in Nondiabetic Patients,” Circulation, Vol. 112, No. 18, 2005, pp. 2792-2798. doi:10.1161/CIRCULATIONAHA.105.535484
- J. M. Sánchez-Gómez, L. Martínez-Dolz, I. SánchezLázaro, et al., “Influence of Metabolic Syndrome on Development of Cardiac Allograft Vasculopathy in the Transplanted Heart,” Transplantation, Vol. 93, No. 1, 2012, pp. 106-111. doi:10.1097/TP.0b013e3182398058
- O. Biadi, L. Potena, W. F. Fearon, et al., “Interplay between Systemic Inflammation and Markers of Insulin Resistance in Cardiovascular Prognosis after Heart Transplantation,” The Journal of Heart and Lung Transplantation, Vol. 26, No. 4, 2007, pp. 324-330. doi:10.1016/j.healun.2007.01.020
- T. Hasegawa ,K. Okada, Y. Okita and D. J. Pinsky, “Antioxidant Properties of Pioglitazone Limit Nicotinamide Adenine Dinucleotide Phosphate Hydrogen Oxidase and Augment Superoxide Dismutase Activity in Cardiac Allotransplantation,” The Journal of Heart and Lung Transplantation, Vol. 30, No. 10, 2011, pp. 1186-1196. doi:10.1016/j.healun.2011.07.006
- R. Tuuminen, S. Syrjälä, R. Krebs, et al., “Donor Simvastatin Treatment Abolishes Rat Cardiac Allograft Ischemia/Reperfusion Injury and Chronic Rejection through Microvascular Protection,” Circulation, Vol. 124, No. 10, 2011, pp. 1138-1150. doi:10.1161/CIRCULATIONAHA.110.005249
- F. Hua, T. Ha, J. Ma, et al., “Protection Against Myocardial Ischemia/Reperfusion Injury in TLR4-Deficient Mice is Mediated through a Phosphoinositide 3-Kinase-Dependent Mechanism,” Journal of Immunology, Vol. 178, No. 11, 2007, pp. 7317-7324.
- Y. Sakata, J. W. Dong, J. G. Vallejo, et al., “Toll-Like Receptor 2 Modulates Left Ventricular Function Following Ischemia-Reperfusion Injury,” American Journal of Physiology. Heart and Circulatory Physiology, Vol. 292, No. 1, 2007, pp. H503-H509. doi:10.1152/ajpheart.00642.2006