Open Journal of Endocrine and Metabolic Diseases
Vol. 3 No. 3 (2013) , Article ID: 34472 , 11 pages DOI:10.4236/ojemd.2013.33026
Effectiveness of Telmisartan as an Adjunct to Metformin in Treating Type II Diabetes Mellitus in Rats
1Pharmacology Department, Faculty of Pharmacy, Misr International University (MIU), Cairo, Egypt
2Biochemistry Department, Faculty of Pharmacy, Misr International University (MIU), Cairo, Egypt
3Pharmacology Department, Faculty of Pharmacy, Sinai University, Sinai, Egypt
4Pharmacology Department, Faculty of Pharmacy, Ain Shams University, Cairo, Egypt
5Pharmacology Department, Faculty of Medicine, Assiut University, Cairo, Egypt
Email: rania.salama@miuegypt.edu.eg
Copyright © 2013 Rania M. Salama et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 17, 2013; revised May 17, 2013; accepted June 17, 2013
Keywords: Type II Diabetes Mellitus; Insulin Resistance; High-Fat High-Fructose Diet; Metformin; Telmisartan; Peroxisome-Proliferator Activated Receptor Gamma
ABSTRACT
The monitoring reports of most patients with type II diabetes mellitus (T2DM) revealed that monotherapy with metformin could not achieve long-term glycemic control. Thus, we designed this study aiming to investigate the effect of telmisartan, a unique AT1 receptor antagonist and a partial agonist of peroxisome-proliferator activated receptor gamma (PPARγ), individually and as an adjunct to metformin, on a rat model that simulates the metabolic characteristics of human T2DM. Adult male Wistar rats were fed high-fat, high-fructose diet (HFFD) for 8 weeks followed by a single low dose of streptozotocin (STZ) (35 mg/kg/day, i.p.) rendering them diabetic and insulin resistant. The effectiveness of both drugs and their combination was tested by assessing the changes in the levels of serum glucose, insulin, homeostasis model assessment of insulin resistance (HOMA-IR) index, lipid profile and adiponectin. In addition, the level of reduced glutathione (GSH) in the liver, was investigated. Results showed that the addition of telmisartan to metformin successfully restored serum glucose back to normal levels and corrected the altered serum total cholesterol (TC), triglycerides (TG) and adiponectin, emphasizing the potential role of telmisartan as an adjunct to metformin.
1. Introduction
The new millennium has witnessed the emergence of a modern worldwide epidemic, referred to as “Diabesity” [1], which describes the increasing incidence of diabetes in combination with obesity as a result of changes in human behavior, available nutrition and the adoption of more sedentary lifestyles. At the same time, increased consumption of fructose was found to disturb normal hepatic carbohydrate metabolism leading to two major consequences: perturbations in glucose metabolism and glucose uptake pathways; and a significantly enhanced rate of de novo lipogenesis and triglycerides (TG) synthesis that lead to adipocyte dysfunction. These metabolic disturbances appear to underlie the induction of insulin resistance commonly observed with type II diabetes mellitus (T2DM) [2].
Among many classes, a few drugs exert compelling effects upon features of the metabolic syndrome. The biguanide metformin is widely regarded as the standard first-line agent, in terms of efficacy and safety profile. The anti-diabetic effect of metformin owes to its ability to suppress hepatic glucose production [3], enhance peripheral glucose uptake [4] and improve peripheral insulin sensitivity [5]. However, achieving and maintaining good glycemic control has always been a challenge, implying the need of an adjunct therapy.
This led to the use of telmisartan, a structurally unique ARB [angiotensin II-type1 receptor (AT1R) blocker] that can function as a partial agonist of peroxisome-proliferator activated receptor gamma (PPARγ) [6]. In this regard, dual ARB [AT1R/PPAR] ligand and biguanides target insulin resistance via PPARγ activation by significantly decreasing pro-inflammatory adipokine release and reducing hepatic glucose output. Given these dual effects, as well as its structural similarity to pioglitazone, telmisartan is expected to be useful in the treatment of both hemodynamic and biochemical aspects of type 2 diabetes [7].
This has tempted us to evaluate the possible increased efficacy of metformin after its concurrent administration with telmisartan for 5 weeks, on a rat model that simulates the natural history and metabolic characteristics of human T2DM. This was assessed by detecting the changes in interrupted glucose metabolism via estimation of serum glucose, insulin, homeostasis model assessment of insulin resistance (HOMA-IR) index, serum total cholesterol (TC), TG and free fatty acid (FFA), as well as the adipocytokine “adiponectin”. In addition, the level of reduced glutathione (GSH) was determined in the liver and its association to the other parameters assessed.
2. Materials and Methods
2.1. Drugs and Chemicals
Sreptozotocin (STZ) and standard GSH were purchased from Sigma-Aldrich Chemical Company, USA. The feeding ingredients, such as casein, lard and cellulose were obtained from commercial sources and were of analytical grades. Fructose was purchased from Safety Misr Co., Egypt. Telmisartan (Micardis®) was purchased from Boehringer Ingelheim, Germany; and metformin hydrochloride (Glucophage®) was purchased from Merck, USA.
2.2. Animals
Adult male Wistar rats weighing 100 - 120 g (National Research Center Laboratory, Cairo, Egypt) were housed in the animal facility of Faculty of Pharmacy, Misr International University in standard polypropylene cages (four rats per cage). Prior to the dietary manipulation, they were fed normal pellet diet (NPD) (EL-Nasr chemical Co., Cairo, Egypt) and permitted a free access to tap water. The rats were kept under standard conditions of temperature (22˚C ± 2˚C) and relative humidity (55% ± 5%) with 12-light/12-dark cycle. Experimental design and animal handling were according to the guidelines of the Ethical Committee of the Faculty of Pharmacy, Ain Shams University, for Animal Use.
2.3. Dietary Model of Insulin Resistant Hyperglycemia (T2DM)
Sixty rats were divided into two dietary regimen groups that lasted for a period of 8 weeks. Twelve rats were fed NPD [3.15 kcal/g; fat (5%), protein (21%), carbohydrate as starch (60%), fibers (3%), vitamins and minerals (1%)] and this group served as normal control. Forty eight rats were placed on a special high-fat, high-fructose diet (HFFD) to induce insulin resistance; the formula was obtained from Harlan laboratories (Teklad Diet TD. 03293) [4.1 kcal/g; fructose (60%), lard (10%), Casein (20.7%), Cellulose (4.2%), Mineral Mix (3.5%), Vitamin Mix (1%), Calcium Carbonate (0.3%) and DL-Methionine (0.3%)]. Afterwards; hyperglycemia and overt diabetes were induced by an intraperitoneal (i.p.) injection of a single sub-diabetogenic dose of freshly prepared STZ (35 mg/kg) [8] in citrate buffer (0.09 M, pH 4.8) after an overnight fasting. Normal control rats received i.p. citrate buffer only and were fed normal pellet diet.
To overcome the hypoglycemia which follows STZ, during the first 24 hours after its injection; diabetic rats were given 5% glucose solution to drink instead of tap water. The animals were monitored by periodic estimation of body weight and biochemical testing for fasting serum glucose. Only animals with persistent blood glucose levels higher than 200 mg/dl for 7 days after STZ administration were considered diabetic/insulin resistant and were continued to be used in the study and started to receive treatment.
2.4. Groups under Investigation
One week after the STZ injection, rats that fulfilled the aforementioned criteria were randomly divided into 5 different groups, each of 12 rats as follows: Group 1 served as normal control rats, fed NPD (3.15 kcal/g) and received single dose of citrate buffer (0.09 M, pH 4.8) alone i.p. Group 2 served as diabetic/insulin-resistant (DIR) rats, fed HFFD and received 0.5% aqueous solution of carboxymethyl cellulose sodium (pH 7.0) as a vehicle. Group 3 served as DIR rats, fed HFFD and received metformin (250 mg/kg/day; p.o.) [9]. Group 4 served as DIR rats, fed HFFD and received telmisartan (5 mg/kg/day; p.o.) prepared in 0.5% aqueous solution of carboxymethyl cellulose sodium (pH 7.0) [10]. Group 5 served as DIR rats, fed HFFD and received metformin (250 mg/kg/day; p.o.) plus telmisartan (5 mg/kg/day; p.o.).
Groups 3-5 continued the treatment for 5 weeks. The last dose of any treatment was given 24 hours before sacrificing the rats which fasted 14 hours before the time of sacrifice and blood samples were withdrawn, to minimize feeding-induced variations in lipid pattern and to measure fasting blood glucose level.
2.5. Oral Glucose Tolerance Test (OGTT)
All groups were subject to an oral glucose tolerance test (OGTT) after 8 hours fasting, during which animals were given an oral dose of aqueous glucose solution (2 g/kg) using oral gavage and blood samples were withdrawn at 0, 15, 30, 60, 90 and 120 minutes to evaluate the resulting glucose concentrations. Glucose was measured using Accucheck Compact (Roche Diagnostics, Almere, Netherlands).
2.6. Glucose Are under Curve (AUC)
The glucose AUC was calculated according to the following equation [11]:
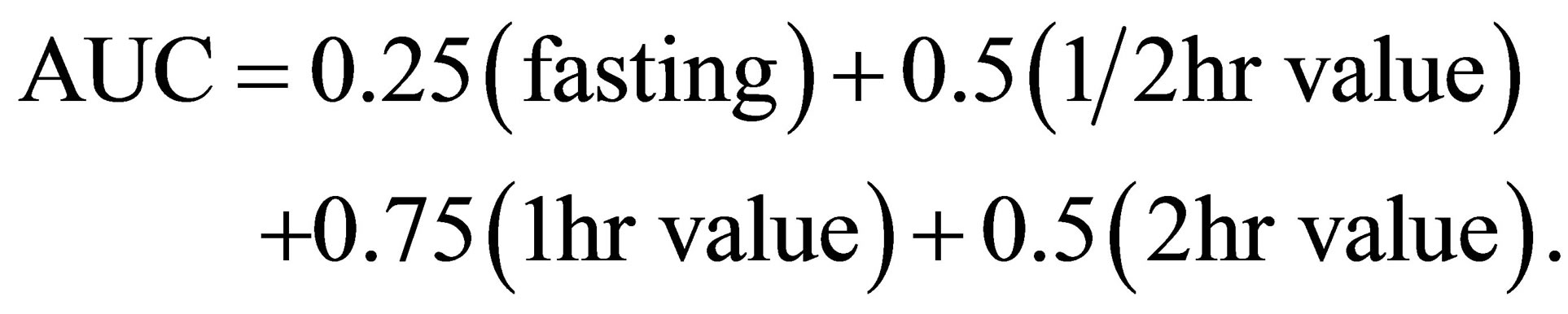
2.7. Serum Separation
Blood was withdrawn from the retro-orbital plexus of ether-anesthetized animals and centrifuged (3000 rpm, 4˚C, 30 min) for separation of serum that was analyzed for glucose, insulin, FFA, TC, TG as well as the adipocyte-secreted adiponectin.
Serum glucose was determined colorimetrically according to the glucose oxidase/peroxidase method [12], using a Stanbio Laboratories kit, USA. Sandwich type immunoassay technique was adopted to determine insulin content using an ELISA kit obtained from ALPCO Diagnostics, USA [13]. The HOMA-IR index was calculated according to the following equation [14]:

Serum TC was determined enzymatically according to the cholesterol oxidase/4-aminophenazone method [15], using a Stanbio Laboratories kit, USA. Serum TG was determined according to the Glucose oxidase/Glycerylphosphate oxidase method [16], using a Stanbio Laboratories kit, USA. Serum FFA was determined colorimetrically according to the method of enzymatic conversion to acetyl-CoA and subsequently to H2O2 [17], using a kit obtained from BioAssay Systems, USA.
The adipocytokine, adiponectin, was measured using enzyme-linked immunosorbent assay (ELISA) kit obtained from Chemicon International, USA, which employs the quantitative two-step sandwich enzyme immunoassay technique [18].
2.8. Preparation of Liver Tissue Homogenate
Immediately after sacrificing the rats, dissection was done for the isolation of the liver. Liver tissues (0.5 g) were excised and washed twice with phosphate buffered saline, dried between two filter papers, then homogenized in 5 ml phosphate buffered saline (10% w/v) using glass-Teflon Potter-Elvejhem device, divided into aliquots and frozen at −70˚C until assayed. Liver content of GSH was determined depending on the fact that both protein and non-protein SH-groups react with Ellman’s reagent [5,5’-dithiobis-2-nitrobenzoic acid]; (DTNB) to form a stable yellow color of 2-Nitro-5-thiobenzoic acid, which can be measured colorimetrically [19].
2.9. Statistical Analysis
Results are expressed as means ± S.E.M. of 8 animals, and differences between groups were tested for significance using analysis of variance (ANOVA), followed by LSD post hoc test. The level of statistical significance was taken at P < 0.05, P < 0.01 and P < 0.001. Statistical analysis of the experimental data was performed using the statistical package SPSS for Windows (version 13.0, USA) and the Microsoft excel program. The correlational analysis was performed using Pearson Correlation.
3. Results
The OGTT performed showed significant elevation in the glucose level in the DIR rats after oral administration of glucose (2 mg/kg, p.o.); an effect that leveled off significantly after treatment with each of metformin, telmisartan and their combination (Figure 1).
The data of OGTT was reflected on the glucose area under curve (AUC) (Figure 2), showing a 4.5-fold increase in the DIR group compared to the control group; an effect that was significantly decreased in DIR + metformin (DIR + Met) group (71%) and DIR + telmisartan (DIR + Tel) group (69%). The combination of metformin and telmisartan was able to successfully normalize the glucose AUC, showing superior results over telmisartan monotherapy (P < 0.05).
Rats fed HFFD for 8 weeks followed by a single sub-diabetogenic dose of STZ (35 mg/kg) resulted in 31% decline in their body weight, a 3.7-fold increase in their fasting serum glucose and 4-fold increase in HOMA-IR index, while serum insulin level showed 11% significant increase compared to the control group (Table 1). The combination therapy of metformin and telmisartan was able to successfully normalize the body weight and serum glucose level (P < 0.05). As for metformin monotherapy, it normalized the body weight at P < 0.01; while the telmisartan monotherapy normalized the body weight at P < 0.001. However, monotherapy with metformin or telmisartan was able to normalize the serum glucose at P < 0.01. Combination treatment with metformin and telmisartan managed to normalize the HOMA-IR index (P < 0.001). Also, this combination therapy showed significant elevation in body weight compared to telmisartan monotherapy (P < 0.05).
The data in Table 2 reveals a significant increase in serum level of TC (1.6 times), TG (2.5 folds) and FFA (1.8 times) in the DIR group compared to the control group. Telmisartan monotherapy significantly reduced TC serum level by 29%. While, metformin alone and in combination with telmisartan were able to normalize the TC serum level, where the combination of both drugs showed significant reduction compared to telmisartan monotherapy (P < 0.001).
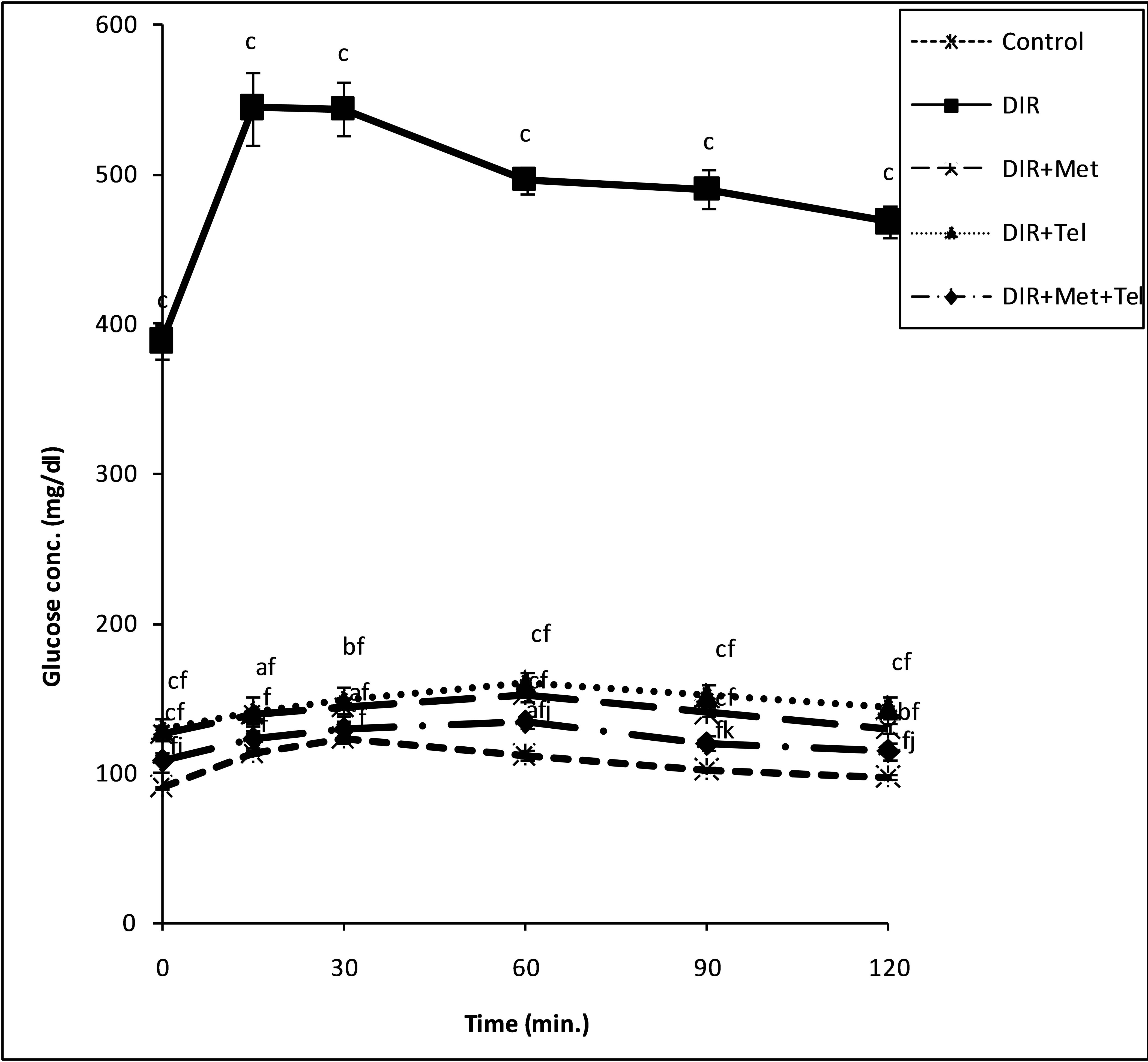
Figure 1. Glucose tolerance curve depicting the effect of glucose (2 g/kg, p.o.) in normal control, non-treated DIR group and treated groups with metformin, telmisartan and their combination. Values represent the mean of 6 rats ± S.E.M. (one-way ANOVA followed by LSD test). (a)P < 0.05, (b)P < 0.01, (c)P < 0.001 compared to the control group; (f)P < 0.001 compared to DIR group; (j)P < 0.05, (k)P < 0.01 compared to DIR + Tel group.
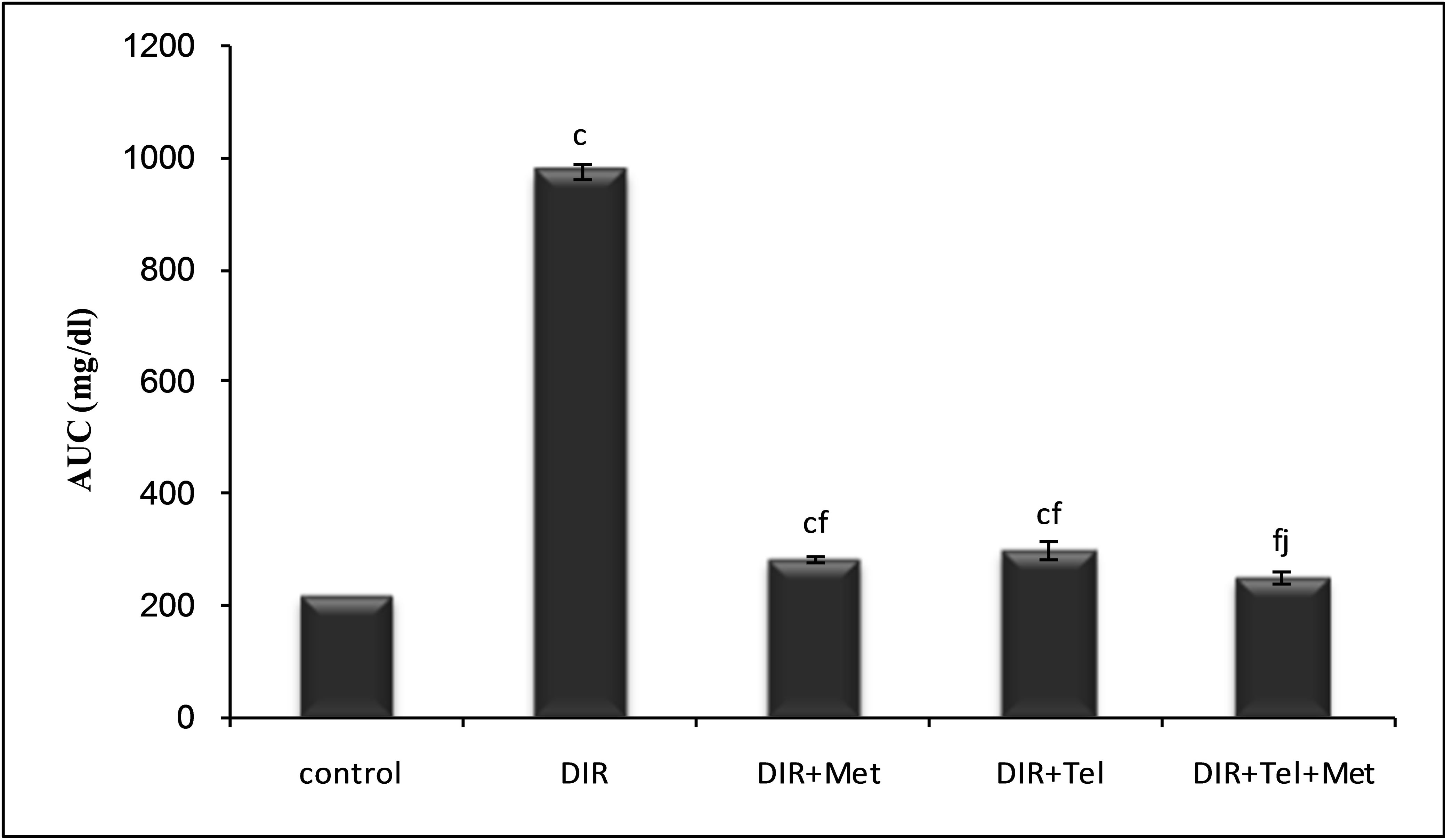
Figure 2. Changes in the area under the curve (AUC) as derived from the OGTT for normal control rats, non-treated and treated DIR rats by metformin, telmisartan and their combination at P < 0.05, P < 0.01 and P < 0.001. Values represent the mean of 6 rats ± S.E.M. (one-way ANOVA followed by LSD test). (b)P < 0.01, (c)P < 0.001 compared to control group; (f)P < 0.001 compared to DIR group; (j)P < 0.05 compared to DIR + Tel group.
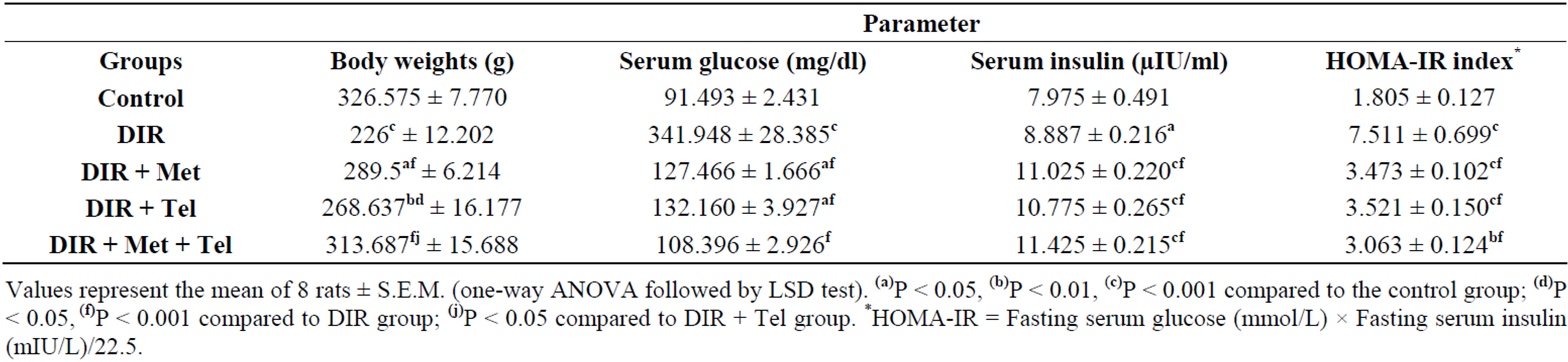
Table 1. Effect of oral daily administration of metformin (250 mg/kg) [DIR + Met], telmisartan (5 mg/kg) [DIR + Tel] and metformin (250 mg/kg) plus telmisartan (5 mg/kg) [DIR + Met + Tel] on their body weight, serum content of glucose, insulin and HOMA-IR index using DIR rats.
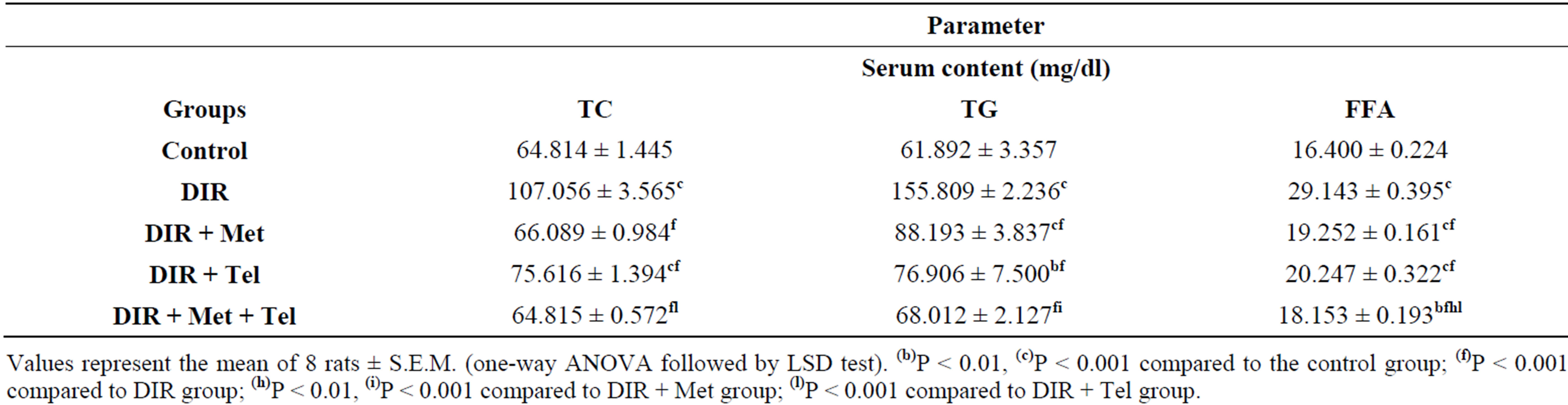
Table 2. Effect of oral daily administration of metformin (250 mg/kg) [DIR + Met], telmisartan (5 mg/kg) [DIR + Tel] and metformin (250 mg/kg) plus telmisartan (5 mg/kg) [DIR + Met + Tel] on serum content of TC, TG and FFA using DIR rats.
As for serum TG, treatment with metformin significantly reduced its level compared to the DIR group by 43%. However, treatment with telmisartan alone was able to normalize the serum TG level at P < 0.001, while both drugs taken together successfully normalized the serum TG level at P < 0.05, showing superior results over metformin monotherapy (P < 0.001).
Treatment with each of metformin and telmisartan significantly reduced the level of serum FFA by 34% and 31%; respectively. Combination of metformin and telmisartan was able to normalize the serum FFA level (P < 0.001), showing significant decrease compared to each of metformin (P < 0.01) and telmisartan (P < 0.001) when given alone.
As shown in Table 3, the HFFD/STZ induced significant decline in the serum adiponectin level and the hepatic GSH content in the DIR group by 64% and 75%; respectively compared to the control group.
Telmisartan when given alone or as an adjunct to metformin was able to normalize serum adiponectin level (P < 0.05), where the combination of both drugs showed significant elevation compared to metformin monotherapy (P < 0.001) which failed to produce any significant change alone.
On the other hand, metformin monotherapy managed to normalize the hepatic GSH level at P < 0.0. However, telmisartan when given alone and as an adjunct to metformin showed 2.9 and 3.1 folds increase compared to the DIR group.
The data in Table 4 shows the correlational analysis of glucose, HOMA-IR index, adiponectin and GSH. It is shown that HOMA-IR index is positively correlated with serum glucose level (0.901, P < 0.01). As for hepatic GSH content, it is negatively correlated with serum glucose level (–0.746, P < 0.01) and HOMA-IR index (–0.626, P < 0.01), but positively correlated with serum adiponectin level (0.248, P < 0.05). Moreover, serum adiponectin level shows negative correlation with serum glucose level (–0.466, P < 0.01) and HOMA-IR index (–0.530, P < 0.01).
4. Discussion
Simulation of T2DM was achieved by combining the feeding of HFFD which produced insulin resistance and low dose of STZ treatment that caused the initial β-cell dysfunction and subsequently the frank hyperglycemia and mild hyperinsulinemia, reflected as an increased HOMA-IR index [20]. The HFFD/STZ rat model is also known to stimulate lipogenesis and TG accumulation in adipocytes, resulting in their dysfunction. The perturbation of the lipid profile is reflected by prominent dyslipidemia; hypercholesterolemia, hypertriglyceridemia and excessive release of FFA into the blood. The FFA, in turn, induce insulin resistance as evidenced here and
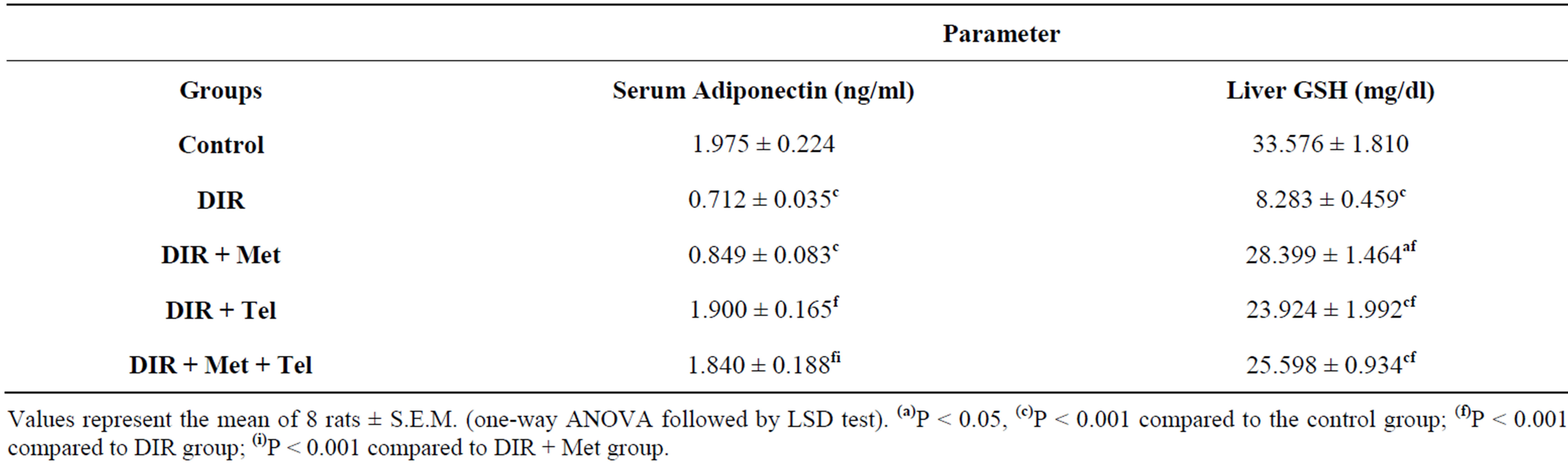
Table 3. Effect of oral daily administration of metformin (250 mg/kg) [DIR + Met], telmisartan (5 mg/kg) [DIR + Tel] and metformin (250 mg/kg) plus telmisartan (5 mg/kg) [DIR + Met + Tel] on serum content of adiponectin as well as hepatic content of GSH using DIR rats.

Table 4. Correlational analysis of the studied parameters.
elsewhere [20,21], as they alter insulin signaling, diminish glucose uptake, and induce gluconeogenesis in the liver.
Apart from the increases in release of FFA, the HFFD/ STZ also resulted in a decrease in the secretion of adiponectin, supposedly due to adipocyte hypertrophy and dysfunction, events that are likely to result in metabolic pathologies. The relationship between adipocyte dysfunction, metabolic disturbances and inflammation was addressed in the study of [21] and concluded a clear association between the dysfunctional adipocyte-induced hypoadiponectinemia along with increases in the HOMAindex and in serum lipid levels. As it is solely produced by adipocytes, a low plasma adiponectin level is a good marker of adipocyte dysfunction that precedes insulin resistance. It has been suggested that Adiponectin might function as an adipostat with insulin-sensitizing and antiinflammatory activity [22].
Moreover, hypoadiponectinemia, resulting from obesity-induced insulin resistance in adipose tissue, especially the visceral ones, was found to mediate metabolic alterations in other peripheral tissues, especially liver by binding to its receptors that are abundant mainly in the liver (Adipo-R2) [23]. The induced hepatic insulin resistance manifested as hepatic steatosis is linked to hypoadiponectinemia, due to low levels of hepatic AdipoR2. The induced hepatic inflammatory state is always associated with a state of systemic and hepatic oxidative stress, added to the effect of angiotensin in generating free radicals, altogether cause consumption of hepatic GSH content and general imbalance in oxidant/antioxidant system [7]. Other remarkable findings were profound reduction in serum adiponectin, concomitant with decreased hepatic GSH content, implying increased oxidative stress and impaired antioxidant capacity in diabetes [24].
These negative outcomes were alleviated or reversed by both pharmacological approaches. The reported hypoglycemic effect of metformin [25-27] was further illustrated by OGTT [26]; showing that plasma glucose excursion after oral glucose loading was significantly improved; and that glucose AUC was significantly decreased owing to extra-pancreatic mechanisms such as the inhibition of hepatic glucose output in the liver and glucose absorption in the gut as well as enhancing peripheral glucose disposal. Confirming the current results; Matafome et al. (2011) illustrated the significant improvement in HOMA-IR index by metformin treatment reflecting correction of the induced insulin resistance [27]. Metformin produced significant elevation in insulin level which was supported by other studies [25,28]. While, activation of adenosine monophosphate activated protein kinase (AMPK) by metformin was proposed to be responsible for the markedly reduced glucosestimulated insulin release from primary pancreatic islets [29] and β-cell lines [30]. When compared with sitagliptin and telmisartan, Souza-Mello et al., (2010) [31] reported that metformin treatment was the most successful approach to reduce insulin resistance in a mice model (C57BL/6) fed on high fat diet. They explained that the results of metformin treatment bore a resemblance to those of sitagliptin treatment, which can be accounted for by dipeptidyl peptidase-4 (DPP-IV) inhibition or enhanced glucagon-like peptide-1 (GLP-1) secretion induced by metformin. Despite being controversial, these two pathways lead to the same effect: increased GLP-1 levels, which synergizes with reduced glucose hepatic output, promoting satiety and body fat loss and thus improving insulin sensitivity.
Contradictory to the current results, Benhaddou-Andaloussi et al. (2011) showed significant decrease in plasma insulin level after treatment with metformin [32]; which can be attributed to the different diet used to induce diabetes where hypercaloric diet was used leading to hyperinsulinemia in the diabetic rats. In addition, Ong et al. (2011) showed no significant change in serum insulin levels [33]; which is probably caused by the large dose of STZ applied (65 mg/kg) leading to this severe decline in insulin levels and the unresponsiveness towards the large dose of metformin given (500 mg/kg). The lowering effect of metformin on the lipid profile was witnessed in other studies where metformin significantly decreased the serum level of TC, TG [28,34] and FFA [27,33]. Contradictory results showed no significant change in TC [25,33], TG [33] and FFA levels [28]; where in these studies induction of T2DM was done either by i.p. injection of high dose of STZ (60 mg/kg) [25], (65 mg/kg) [33] or feeding the animals with high-lipid and high glucose chow preceeding the i.p. injection of STZ (60 mg/kg) [28]. Souza-Mello et al. (2010) referred metformin’s effect in reducing systemic FFA concentrations to the suppression of TNF-α-mediated lipolysis, alleviating thereby insulin resistance, and promoting body weight control [31].
Metformin monotherapy failed to improve the serum adiponectin level which is in agreement with a previous study [32]. Other studies supported the beneficial effect of metformin on GSH level where metformin increased the GSH content in the kidneys [35] and heart [34] of diabetic rats; which was attributed to the ability of metformin to modulate the expression of several oxidative and pro-inflammatory genes at the transcriptional levels. On the contrary, Ong et al. (2011) showed no significant change in both hepatic GSH and antioxidant enzymes in diabetic rats treated with metformin [33].
The hypoglycemic effect of telmisertan observed in the current study is in agreement with another study [36]; which was further confirmed by improved glucose AUC and glucose response curve following insulin challenge in insulin tolerance test [37] and decreased HOMA-IR index [38].
In accordance with our results, Goyal et al. (2011) [36] reported similar hyperglycemia and hyperinsulinemeia in STZ-induced diabetic rats, events that were ameliorated by telmisartan. They further explained that in the early components of the insulin signaling cascade, Ang-II negatively affects insulin signaling, hence, the inhibition of the AT1 receptor prevents the decline of glucose transporter-4 (GLUT-4). ARBs may also increase the protein expression of GLUT-4 in the skeletal muscle and myocardium. Furthermore, telmisartan acts as a partial agonist of PPARγ and influences the expression of PPARγ target gene. As telmisartan structurally resembles pioglitazone, the glucose lowering effect might be due to an improvement in insulin sensitivity, and the reduction in the serum glucose levels might be attributed to various mechanisms.
Fujisaka et al. (2011) confirmed the efficient improving effect of telmisartan to insulin sensitivity and glucose metabolism more than other ARBs and concluded their underlying molecular mechanisms: 1) telmisartan acts as a partial agonist of PPARγ in addition to its effects as an ARB; 2) telmisartan increases energy expenditure by increasing the expressions of some mitochondrial genes, including uncoupling protein-1 (UCP1), in brown adipose tissue; 3) telmisartan attenuates body weight gains and decreases the amount of fat tissues in addition to increasing the number of smaller adipocytes; and 4) telmisartan relies on a thiazolidinedione-like effect to restore insulin sensitivity, which encompasses changes in the recruitment and differentiation of adipocytes, and increased adiponectin levels after PPAR-γ activation [37].
The interplay between partial PPARγ activation and AT1R blockade allows telmisartan to retain only the beneficial effects of PPARγ activation, excluding fluid retention and weight gain due to total agonism [38].
The effect of telmisartan on insulin level was supported by a study that showed elevation in plasma insulin [39], which could be justified by attenuation of increased mRNA expression of rennin-angiotensin system and NADPH oxidase components, vascular endothelial growth factor and transforming growth factor beta 1 (TGF-β1) in the pancreas of diabetic rats; changes that are correlated with an improvement in insulin secretion [10].
Contradictory to our results, another study showed that plasma insulin was reduced by telmisartan treatment [36]; where the large dose of i.p. injection of STZ (90 mg/kg) produced a significant increase in glucose levels associated with compensatory hyperinsulinemia; thus, treatment with telmisartan significantly reduced the serum glucose and insulin.
In agreement with the current results; recent studies proved the effectiveness of telmisartan treatment in reducing TC, TG [36,40] and FFA level [40]. However, another study showed no significant change in plasma TG and FFA level [41] in which mice were fed high-fat diet (HFD) only. Benson et al. (2004) have also reported that telmisartan administration causes a significant reduction in TG levels in rats that are fed a high-fat, highcarbohydrate diet compared to treatment with losartan [42].
Activation of PPARγ by telmisartan enhances lipolysis of circulating TG and their storage in adipose tissue, thereby reducing FFA release from adipocytes. This reduction in FFA alleviates lipotoxicity in skeletal muscle, liver and pancreas, leading to a reduction in hepatic glucose production and improved glucose utilization in skeletal muscles [43]. Although PPARγ regulates lipid metabolism and telmisartan exerts partial agonistic activity on PPARγ, the control of dyslipidemia might also be due to a direct AT1 inhibition [36].
Goyal et al. (2011) reported that FFA may impair endothelial function through several mechanisms, including the increased production of oxygen-derived free radicals, the activation of PKC, and the exacerbation of dyslipidemia [36]. Ang-II modulates the effects of oxidized LDL on endothelial cell function. Our study revealed a decrease in the hepatic input of non-esterified “free” fatty acids (NEFA) that directly correlates with higher adiponectin levels and lower deposits of adipose tissue [44], both of which were observed in all of the treated groups.
As for adiponectin, a previous study agreed with our results showing increased adiponectin level [40]. Conversely, another study showed no significant increase in adiponectin level after 6-month treatment with telmisartan [45], where patients with metabolic syndrome were used in this study having impaired glucose tolerance but no overt hyperglycemia.
Anti-inflammatory effects, such as reduced TNF-α level, concomitant with increased adiponectin level in animals treated with telmisartan can be accounted for by AT1R inhibition and PPARγ activation. These adipokines are largely produced by adipose tissue and their profile after telmisartan use might be related to beneficial adipose tissue remodeling [21]. Not only did telmisartan normalize adipocyte size, but it also interfered with the pattern of adipose tissue storage. This finding correlates with PPARγ activation, which leads to the up-regulation of adiponectin and the reversal of insulin resistance.
Ang-II may directly contribute to oxidative stress by increasing the production of superoxide radicals that, by reacting with nitric oxide to form peroxynitrite, reduces the levels of free nitric oxide [36].
Oxidative stress induced by angiotensin generates free radicals that consume GSH; thus, telmisartan by blocking angiotensin action could ameliorate oxidative stress and consequently increase GSH levels [46]; which was witnessed in another study [36] and supported by the current results.
Furthermore increased GSH biosynthesis may be a direct effect of temisartan [47]. Another possible explanation for the antioxidant effect is that the lipophilicity of telmisartan is so high that it may provide more useful protection against oxidative stress than other ARBs. Thus, the increases in adiponectin and insulin sensitivity can also be explained by this effect of telmisartan [7].
It is noteworthy that telmisartan-induced increase in plasma adiponectin and binding to its hepatic receptors caused prominent improvement of insulin sensitivity and hepatic metabolic functions, in terms of decrease in hepatic output, depression of circulating levels of FFA, TG, glucose and elevation of GSH hepatic content. These associations were confirmed by our correlational studies that provided positive correlations between adiponectin levels and GSH hepatic content and a negative one with HOMA-IR index.
The current study highlighted that the combination of metformin and telmisartan managed to restore serum glucose back to normal levels, making it superior over monotherapy with each of these two drugs that failed to achieve the same goal alone. Previous studies have been conducted to test the combination of metformin and PPARγ agonists. The combination of metformin with rosiglitazone, the full agonist of PPARγ, showed a superior reduction of fasting plasma glucose levels, compared to monotherapy with metformin [48]. However, another study showed that using the combination of rosiglitazone and metformin (Avandamet®) produced reduction in fasting plasma glucose that was significantly greater than the reductions achieved individually with metformin and rosiglitazone [49]. The combination therapy used in our study showed significant reduction in HOMA-IR indicating enhancement of insulin sensitivity, although there was no superior effect of such combination treatment over single drug treatment. This was also shown through the elevation of serum insulin level caused by such combination treatment that did not show significant difference from using each drug alone. Furthermore, this combination showed improvement in glucose tolerance as revealed by the pattern of the OGTT response and the resulting significant decrease in glucose AUC that was superior over that produced by telmisartan monotherapy. One study using the same combination showed significant reduction in OGTT compared to untreated mice fed HFD alone [31]. However, this significant reduction was not superior over single therapy with each of these two drugs. This study also showed significant decrease in HOMA-IR index and insulin level achieved by the combination treatment that was not different from using each drug alone. This decrease in insulin level is most probably due to the different induction method of T2DM used, where it lacked STZ, which explains the induced hyperinsulinemia in untreated mice and the resulting decrease in insulin level produced by these drugs. As for adiponectin level, this study showed significant increase compared to untreated mice. However, this combination treatment did not achieve better results than using each of these two drugs alone. As for the lipid profile, our study showed that these two drugs when taken together managed to restore TC back to normal values, which was not achieved by telmisartan treatment alone. Interestingly, this combined treatment managed to restore serum TG back to normal levels making it superior over using each of these drugs alone. However, this combination therapy failed to return serum FFA back to normal values although it managed to reduce its level significantly compared to single therapy by each of these two drugs. Serum adiponectin level was restored back to normal values, which is similar to the effect of using telmisartan alone, proving that metformin has no effect on serum adiponectin level. Concerning the hepatic GSH content, this combination therapy produced significant increase in this anti-oxidant factor but it didn’t add any beneficial effect to monotherapy by each of these two drugs.
5. Conclusion
In this study we demonstrated the effectiveness of telmisartan as an adjunct to metformin in restoring serum glucose as well as other metabolic disturbances back to normal levels. Therefore, this combinational therapy could be recommended to poorly controlled diabetic patients on metformin monotherapy. However, further studies are warranted to investigate the efficacy of other antidiabetic combinations including telmisartan, as well as enable the assessment of other cytokines/adipocytokines that could serve as new indicators of anti-diabetic efficacy. This may result in better and more efficient management of DM and its related complications.
REFERENCES
- A. Astrup and N. Finer, “Redefining Type 2 Diabetes: ‘Diabesity’ or ‘Obesity Dependent Diabetes Mellitus’?” Obesity Reviews, Vol. 1, No. 2, 2000, pp. 57-59. doi:10.1046/j.1467-789x.2000.00013.x
- H. Basciano, L. Federico and K. Adeli, “Fructose, Insulin Resistance and Metabolic Dyslipidemia,” Nutrition & Metabolism (London), Vol. 2, No. 1, 2005, p. 5. doi:10.1186/1743-7075-2-5
- S. E. Inzucchi, D. G. Maggs, G. R. Spollett, S. L. Page, F. S. Rife, V. Walton and G. I. Shulman, “Efficacy and Metabolic Effects of Metformin and Troglitazone in Type II Diabetes Mellitus,” The New England Journal of Medicine, Vol. 338, No. 13, 1998, pp. 867-872. doi:10.1056/NEJM199803263381303
- K. Cusi, A. Consoli and R. A. DeFronzo, “Metabolic Effects of Metformin on Glucose and Lactate Metabolism in Noninsulin-Dependent Diabetes Mellitus,” The Journal of Clinical Endocrinology & Metabolism, Vol. 81, No. 11, 1996, pp. 4059-4067. doi:10.1210/jc.81.11.4059
- C. R. Sirtori and C. Pasik, “Re-Evaluation of a Biguanide, Metformin: Mechanism of Action and Tolerability,” Pharmacological Research, Vol. 30, No. 3, 1994, pp. 187-228. doi:10.1016/1043-6618(94)80104-5
- S. Z. Duan, M. G. Usher and R. M. Mortensen, “Peroxisome Proliferator-Activated Receptor-Gamma-Mediated Effects in the Vasculature,” Circulation Research, Vol. 102, No. 3, 2008, pp. 283-294. doi:10.1161/CIRCRESAHA.107.164384
- M. Watanbe, K. Inukai, T. Sumita, K. Ikebukuro, D. Ito, S. Kurihara, H. Ono, T. Awata and S. Katayama, “Effects of Telmisartan on Insulin Resistance in Japanese Type 2 Diabetic Patients,” Internal Medicine, Vol. 49, No. 17, 2010, pp. 1843-1847. doi:10.2169/internalmedicine.49.3189
- K. Srinivasan, B. Viswanad, L. Asrat, C. L. Kaul and P. Ramarao, “Combination of High-Fat Diet-Fed and LowDose Streptozotocin-Treated Rat: A Model for Type 2 Diabetes and Pharmacological Screening,” Pharmacological Research, Vol. 52, No. 4, 2005, pp. 313-320. doi:10.1016/j.phrs.2005.05.004
- S. Matthaei, J. P. Reibold, A. Hamann, H. Benecke, H. U. Häring, H. Greten and H. H. Klein, “In Vivo Metformin Treatment Ameliorates Insulin Resistance: Evidence for Potentiation of Insulin-Induced Translocation and Increased Functional Activity of Glucose Transporters in Obese (Fa/Fa) Zucker Rat Adipocytes,” Endocrinology, Vol. 133, No. 1, 1993, pp. 304-311. doi:10.1210/en.133.1.304
- G. Hasegawa, M. Fukui, H. Hosoda, M. Asano, I. Harusato, M. Tanaka, E. Shiraishi, T. Senmaru, K. Sakabe, M. Yamasaki, J. Kitawaki, A. Fujinami, M. Ohta, H. Obayashi and N. Nakamura, “Telmisartan, an Angiotensin II Type 1 Receptor Blocker, Prevents the Development of Diabetes in Male Spontaneously Diabetic Torii Rats,” European Journal of Pharmacology, Vol. 605, No. 1-3, 2009, pp. 164-169. doi:10.1016/j.ejphar.2009.01.001
- A. Psyrogiannis, V. Kyriazopoulou, A. Symeonidis, M. Leotsinidis and V. A. G. Vagenakis, “Relative Iron Overload: in Offspring of Patients with Type 2 Diabetes Mellitus: A New Component in the Conundrum of Insulin Resistance Syndrome?” Hormones (Athens), Vol. 2, No. 3, 2003, pp. 161-168.
- P. Trinder, “Determination of Blood Glucose Using 4- Amino Phenazone as Oxygen Acceptor,” Journal of Clinical Pathology, Vol. 22, No. 2, 1969, p. 246. doi:10.1136/jcp.22.2.246-b
- C. R. Morgan and A. Lazarow, “Immunoassay of Insulin Using a Two-Antibody System,” Proceedings of the Society for Experimental Biology and Medicine, Vol. 110, 1962, pp. 29-32. doi:10.3181/00379727-110-27411
- D. R. Matthews, J. P. Hosker, A. S. Rudenski, B. A. Naylor, D. F. Treacher and R. C. Turner, “Homeostasis Model Assessment: Insulin Resistance and Beta-Cell Function from Fasting Plasma Glucose and Insulin Concentrations in Man,” Diabetologia, Vol. 28, No. 7, 1985, pp. 412- 419. doi:10.1007/BF00280883
- C. C. Allain, L. S. Poon, C. S. Chan, W. Richmond and P. C. Fu, “Enzymatic Determination of Total Serum Cholesterol,” Clinical Chemistry, Vol. 20, No. 4, 1974, pp. 470-475.
- A. W. Wahlefeld, “Triglycerides Determination after Enzymatic Hydrolysis,” In: H. U. Bergmeyer, Ed., Methods of Enzymatic Analysis, Academic Press, New York, 1974, pp. 1831-1835.
- C. Matsubara, Y. Nishikawa, Y. Yoshida and K. Takamura, “A Spectrophotometric Method for the Determination of Free Fatty Acid in Serum Using Acyl-Coenzyme A Synthetase and Acyl-Coenzyme A Oxidase,” Analytical Biochemistry, Vol. 130, No. 1, 1983, pp. 128-133. doi:10.1016/0003-2697(83)90659-0
- Y. Nakano, S. Tajima, A. Yoshimi, H. Akiyama, M. Tsushima, T. Tanioka, T. Negoro, M. Tomita and T. Tobe, “A Novel Enzyme-Linked Immunosorbent Assay Specific for High-Molecular-Weight Adiponectin,” The Journal of Lipid Research, Vol. 47, No. 7, 2006, pp. 1572- 1582. doi:10.1194/jlr.D600010-JLR200
- E. Beutler, O. Duron and B. M. Kelly, “Improved Method for the Determination of Blood Glutathione,” Journal of Laboratory and Clinical Medicine, Vol. 61, No. 3, 1963, pp. 882-888.
- M. Schaalan, H. S. El-Abhar, M. Barakat and E. S. ElDenshary, “Westernized-Like-Diet-Fed Rats: Effect on Glucose Homeostasis, Lipid Profile, and Adipocyte Hormones and Their Modulation by Rosiglitazone and Glimepiride,” Journal of Diabetes and Its Complications, Vol. 23, No. 3, 2009, pp. 199-208. doi:10.1016/j.jdiacomp.2008.02.003
- M. F. Schaalan, “Effects of Pioglitazone and/or Simvastatin on Circulating TNFα and Adiponectin Levels in Insulin Resistance,” Journal of Immunotoxicology, Vol. 9, No. 2, 2012, pp. 201-209. doi:10.3109/1547691X.2012.660998
- E. Esteve, W. Ricart and J. M. Fernández-Real, “Adipocytokines and Insulin Resistance: The Possible Role of Lipocalin-2, Retinol Binding Protein-4, and Adiponectin,” Diabetes Care, Vol. 32, Supplement 2, 2009, pp. S362-S367.
- H. S. El-Abhar and M. Schaalan, “Topiramate-Induced Modulation of Hepatic Molecular Mechanisms: An Aspect for Its Anti-Insulin Resistant Effect,” PloS One, Vol. 7, No. 5, 2012, p. e37757. doi:10.1371/journal.pone.0037757
- E. Y. Sözmen, B. Sözmen, Y. Delen and T. Onat, “Catalase/Superoxide Dismutase (SOD) and Catalase/Paraoxonase (PON) Ratios May Implicate Poor Glycemic Control,” Archives of Medical Research, Vol. 32, No. 4, 2001, pp. 283-287. doi:10.1016/S0188-4409(01)00285-5
- O. O. Erejuwa, S. A. Sulaiman, M. S. Wahab, K. N. Sirajudeen, M. S. Salleh and S. Gurtu, “Glibenclamide or Metformin Combined with Honey Improves Glycemic Control in Streptozotocin-Induced Diabetic Rats,” International Journal of Biological Sciences, Vol. 7, No. 2, 2011, pp. 244-252. doi:10.7150/ijbs.7.244
- P. Bansal, P. Paul, J. Mudgal, P. G Nayak, S. Thomas Pannakal, K. I. Priyadarsini and M. K. Unnikrishnan, “Antidiabetic, Antihyperlipidemic and Antioxidant Effects of the Flavonoid Rich Fraction of Pilea microphylla (L.) in High Fat Diet/Streptozotocin-Induced Diabetes in Mice,” Experimental and Toxicologic Pathology, Vol. 64, No. 6, 2012, pp. 651-658.
- P. Matafome, T. Louro, L. Rodrigues, J. Crisóstomo, E. Nunes, C. Amaral, P. Monteiro, A. Cipriano and R. Seiça, “Metformin and Atorvastatin Combination Further Protect the Liver in Type 2 Diabetes with Hyperlipidaemia,” Diabetes/Metabolism Research and Reviews, Vol. 27, No. 1, 2011, pp. 54-62. doi:10.1002/dmrr.1157
- F. Hu, X. Li, L. Zhao, S. Feng and C. Wang, “Antidiabetic Properties of Purified Polysaccharide from Hedysarum Polybotrys,” Canadian Journal of Physiology and Pharmacology, Vol. 88, No. 1, 2010, pp. 64-72. doi:10.1139/Y09-098
- G. da Silva Xavier, I. Leclerc, A. Varadi, T. Tsuboi, S. K. Moule and G. A. Rutter, “Expression. Role for AMPActivated Protein Kinase in Glucose-Stimulated Insulin Secretion and Preproinsulin Gene Expression,” Biochemical Journal, Vol. 371, No. Pt 3, 2003, pp. 761-774.
- S. Zhang and K. H. Kim, “Glucose Activation of AcetylCoa Carboxylase in Association with Insulin Secretion in a Pancreatic Beta-Cell,” Journal of Endocrinology, Vol. 147, No. 1, 1995, pp. 33-41. doi:10.1677/joe.0.1470033
- V. Souza-Mello, B. M. Gregório, F. S. Cardoso-de-Lemos, L. de Carvalho, M. B. Aguila and C. A. Mandarimde-Lacerda, “Comparative Effects of Telmisartan, Sitagliptin and Metformin Alone or in Combination on Obesity, Insulin Resistance, and Liver and Pancreas Remodelling in C57BL/6 Mice Fed on a Very High-Fat Diet,” Clinical Science (London), Vol. 119, No. 6, 2010, pp. 239-250. doi:10.1042/CS20100061
- A. Benhaddou-Andaloussi, L. Martineau, T. Vuong, B. Meddah, P. Madiraju, A. Settaf and P. S. Haddad, “The in Vivo Antidiabetic Activity of Nigella sativa Is Mediated through Activation of the AMPK Pathway and Increased Muscle Glut4 Content,” Evidence-Based Complementary and Alternative Medicine, Vol. 2011, 2011, Article ID: 538671. doi:10.1155/2011/538671
- K. W. Ong, A. Hsu, L. Song, D. Huang and B. K. Tan, “Polyphenols-Rich Vernonia Amygdalina Shows AntiDiabetic Effects in Streptozotocin-Induced Diabetic Rats,” Journal of Ethnopharmacology, Vol. 133, No. 2, 2011, pp. 598-607. doi:10.1016/j.jep.2010.10.046
- G. B. Behera, A. N. Kurnool, M. Baidya, B. S. Kumar and S. Bilal, “Anti-Hyperglyceamic, Anti-Hyperlipidemic and Antioxidant Activity of the Stem of Glinus oppositifolius (L.) AUG. DC,” International Journal of Pharmacy and Pharmaceutical Sciences, Vol. 3, No. 1, 2011, pp. 1080-1090.
- O. O. Erejuwa, S. A. Sulaiman, M. S. Wahab, S. K. Salam, M. S. Salleh and S. Gurtu, “Comparison of Antioxidant Effects of Honey, Glibenclamide, Metformin, and Their Combinations in the Kidneys of StreptozotocinInduced Diabetic Rats,” International Journal of Molecular Sciences, Vol. 12, No. 1, 2011, pp. 829-843. doi:10.3390/ijms12010829
- B. R. Goyal, K. Parmar, R. K. Goyal and A. A. Mehta, “Beneficial Role of Telmisartan on Cardiovascular Complications Associated with Stz-Induced Type 2 Diabetes In Rats,” Pharmacological Reports, Vol. 63, No. 4, 2011, pp. 956-966.
- S. Fujisaka, I. Usui, Y. Kanatani, M. Ikutani, I. Takasaki, K. Tsuneyama, Y. Tabuchi, A. Bukhari, Y. Yamazaki, H. Suzuki, S. Senda, A. Aminuddin, Y. Nagai, K. Takatsu, M. Kobayashi and K. Tobe, “Telmisartan Improves Insulin Resistance and Modulates Adipose Tissue Macrophage Polarization in High-Fat-Fed Mice,” Endocrinology, Vol. 152, No. 5, 2011, pp. 1789-1799. doi:10.1210/en.2010-1312
- A. Zanchi, A. G. Dulloo, C. Perregaux, J. P. Montani and M. Burnier, “Telmisartan Prevents the Glitazone-Induced Weight Gain without Interfering with Its Insulin-Sensitizing Properties,” Endocrinology and Metabolism: American Journal of Physiology, Vol. 293, No. 1, 2007, pp. E91-E95. doi:10.1152/ajpendo.00024.2007
- A. A. Fouad, A. I. Al-Sultan, M. T. Yacoubi and W. Gomaa, “Ameliorative Effects of Telmisartan in Diabetic Rats with Indomethacin-Induced Gastric Ulceration,” European Journal of Pharmacology, Vol. 637, No. 1-3, 2010, pp. 162-170. doi:10.1016/j.ejphar.2010.04.007
- Z. Guo, C. Zheng, Z. Qin and P. Wei, “Effect of Telmisartan on the Expression of Cardiac Adiponectin and Its Receptor 1 in Type 2 Diabetic Rats,” Journal of Pharmacy and Pharmacology, Vol. 63, No. 1, 2011, pp. 87-94. doi:10.1111/j.2042-7158.2010.01157.x
- X. Rong, Y. Li, K. Ebihara, M. Zhao, J. Naowaboot, T. Kusakabe, K. Kuwahara, M. Murray and K. Nakao, “Angiotensin II Type 1 Receptor-Independent Beneficial Effects of Telmisartan on Dietary-Induced Obesity, Insulin Resistance and Fatty Liver in Mice,” Diabetologia, Vol. 53, No. 8, 2010, pp. 1727-1731. doi:10.1007/s00125-010-1744-6
- S. C. Benson, H. A. Pershadsingh, C. I. Ho, A. Chittiboyina, P. Desai, M. Pravenec, N. Qi, J. Wang, M. A. Avery and T. W. Kurtz, “Identification of Telmisartan as a Unique Angiotensin II Receptor Antagonist with Selective PPARgamma-Modulating Activity,” Hypertension, Vol. 43, No. 5, 2004, pp. 993-1002. doi:10.1007/s00125-010-1744-6
- B. Gross and B. Staels, “PPAR Agonists: Multimodal Drugs for the Treatment of Type-2 Diabetes,” Best Practice & Research: Clinical Endocrinology & Metabolism, Vol. 21, No. 4, 2007, pp. 687-710. doi:10.1016/j.beem.2007.09.004
- G. Svegliati-Baroni, C. Candelaresi, S. Saccomanno, G. Ferretti, T. Bachetti, M. Marzioni, S. De Minicis, L. Nobili, R. Salzano, A. Omenetti, D. Pacetti, S. Sigmund, A. Benedetti and A. Casini, “A Model of Insulin Resistance and Nonalcoholic Steatohepatitis in Rats: Role of Peroxisome Proliferator-Activated Receptor-Alpha and n-3 Polyunsaturated Fatty Acid Treatment on Liver Injury,” American Journal of Pathology, Vol. 169, No. 3, 2006, pp. 846-860. doi:10.1016/j.beem.2007.09.004
- T. Kakuma, K. Gotoh, T. Masaki, E. Itateyama, N. Abe and H. Yoshimatsu, “Telmisartan Reduced Abdominal Circumference and Body Weight with Decreasing Triglyceride Level in Patients with Type 2 Diabetes and Metabolic Syndrome,” Obesity Research & Clinical Practice, Vol. 4, No. 2, 2009, pp. e145-e152. doi:10.1016/j.orcp.2009.12.003
- P. Pacher, I. G. Obrosova, J. G. Mabley and C. Szabó, “Role of Nitrosative Stress and Peroxynitrite in the Pathogenesis of Diabetic Complications. Emerging New Therapeutical Strategies,” Current Medicinal Chemistry, Vol. 12, No. 3, 2005, pp. 267-275. doi:10.2174/0929867053363207
- A. A. Hamed and H. A. Malek, “Effect of Telmisartan in Experimentally Induced Diabetetes Mellitus in Rats,” International Journal of Health Sciences, Vol. 1, No. 2, 2007, pp. 249-256.
- K. Wellington, “Rosiglitazone/Metformin,” Drugs, Vol. 65, No. 11, 2005, pp. 1581-1592. doi:10.2165/00003495-200565110-00013
- J. Rosenstock, J. Rood, A. Cobitz, N. Biswas, H. Chou and A. Garber, “Initial Treatment with Rosiglitazone/ Metformin Fixed-Dose Combination Therapy Compared with Monotherapy with Either Rosiglitazone or Metformin in Patients with Uncontrolled Type 2 Diabetes,” Diabetes, Obesity and Metabolism, Vol. 8, No. 6, 2006, pp. 650-660. doi:10.1111/j.1463-1326.2006.00659.x