Open Journal of Endocrine and Metabolic Diseases
Vol. 3 No. 2A (2013) , Article ID: 31427 , 13 pages DOI:10.4236/ojemd.2013.32A004
Mitochondrial Signaling in Hypoxia
Institute of General Pathology and Pathophysiology of the Russian Academia of Medical Science,
Moscow, Russia
Email: ldlukyanova@gmail.com
Copyright © 2013 Ludmila D. Lukyanova. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 21, 2013; revised March 20, 2013; accepted April 20, 2013
Keywords: Respiratory Chain Reprogramming; Mitochondrial Complexes I and II; Adaptation to Hypoxia; Succinate; HIF-1; Energotropic Drugs; Antihypoxic Activity
ABSTRACT
This paper focuses on a bioenergetic mechanism responding to hypoxia. This response involves hypoxia-induced reprogramming of respiratory chain function and switching from oxidation of complex I (NAD-related substrates) to complex II (succinate oxidation). Transient, reversible, compensatory activation of respiratory chain complex II is a major mechanism of urgent adaptation to hypoxia, which is necessary for 1) succinate-related energy synthesis in the conditions of oxygen shortage and formation of urgent resistance; 2) succinate-related stabilization of HIF-1α and initiation of its transcriptional activity related with formation of long-term adaptation; 3) succinate-dependent activation of the succinate-specific receptor GPR91. Thus, mitochondria perform a signaling function with succinate as a signaling molecule. Effects of succinate in hypoxia occur at three levels, intramitochondrial, intracellular and intercellular. In these settings, succinate displays antihypoxic activity. The review is focused on tactics and strategy for development of the antihypoxic defense and antihypoxants with energotropic properties.
1. Introduction
Mitochondria play a key role in oxygen homeostasis. This role is determined primarily by the trigger function of mitochondria in the cell, the function of a self-regulating biological machine that uses oxygen for generation of energy in the form of ATP and mitochondrial membrane potential. Inhaled oxygen eventually reflects the state and the oxygen demand of mitochondria because specifically mitochondria are the main oxygen consumer: up to 98% of oxygen delivered to the body works for mitochondrial respiration. This process provides up to 80% - 90% of ATP to cells of different mammalian tissues. Due to this function, which is responsible for vital capacity and function of aerobic organisms, evolution is able to produce most sophisticated physiological systems for oxygen delivery to mitochondria and maintenance of optimal cell oxygenation (act of respiration; lung system of oxygen transport; cardiovascular circulation; blood mass and transportation system; red cells; hemoglobin). Arrangement of the digestive system, including consumption and subsequent stepwise enzymic processing of food, is also determined primarily by the need for supplying substrates to reactions of mitochondrial oxidation and oxidative phosphorylation [1,2]. Mitochondria participate in regulation of various physiological functions by providing energy for most of intracellular processes necessary for vital functions of the body including, first of all, contractility of the heart and smooth muscles of the gastrointestinal tract, blood vessels and lungs, maintenance of ion gradients in excitable tissues, accumulation of secretable material in vesicles, and support of hormonal and neurotransmitter functions.
Nevertheless, the major function of mitochondria is their regulatory role in oxygen homeostasis, which is evident at both systemic and cellular levels.
At the systemic level, mitochondria determine the concentration gradient of oxygen arriving from the environment since specifically mitochondria represent the final step of the interaction with molecular oxygen [1,3, 4].
At the cell level, the mitochondrial respiratory chain performs as a signal-transforming metabolic system which activates the functional response and actualizes the physiological response to hypoxia.
Therefore, it is possible to say that mitochondria are involved in intraand extracellular signaling mechanisms and that mitochondria function as active signal organelles participating in transmission of information through various intracellular signaling pathways.
It is known that hypoxia is associated with disturbances of ATP synthesis resulting from depressed functions of electron transport and oxidative phosphorylation in the respiratory chain. The factor determining the hypoxic state is disorders of oxygen delivery from the environment to the cell. In the cell, oxygen participates in reactions of aerobic energy synthesis as a substrate for cytochrome oxidase (COX), the terminal enzyme of mitochondrial respiratory chain. Therefore, oxygen deficiency in the extracellular environment may lead either to reduction or total inhibition of aerobic energy synthesis involving decreased content of high-energy molecules (ATP and PC), the main marker of hypoxic conditions. This results in inhibition of multiple energy-dependent reactions, such as formation of membrane potential, ion transport, electrogenic cell function, muscle contraction, receptor function, etc. Mitochondrial dysfunction inevitably leads to various pathologies and even death.
Therefore, defense of the body against disorders of oxygens homeostasis is primarily related with prevention of disturbanced in the work of respiratory chain under oxygen shortage. The antihypoxic defence is a high priority for medicine as many abnormalities, such as ischemia, hypoxia, stroke, traumatic injury, inflammation, etc., involved impaired oxygen delivery to cells.
2. Performance of Respiratory Chain in Hypoxia
In normoxic conditions, performance of the respiratory chain is usually related with oxidation of NAD-related substrates, the major suppliers of reduction equivalents to the respiratory chain through mitochondrial complex I (MtC I—NADH: ubiquinone-oxidoreductase). This complex contribution as estimated by oxygen consumption may reach 55% - 65% in intact cells (Figure 1(a)). At the same time, 25% - 30% of mitochondrial respiration under these conditions is attributed to mitochondrial complex II (MtC II—succinate-ubiquinone reductase), which participates in the citric acid cycle by oxidizing succinate to fumarate. Its function is—transferring electrons to the ubiquinone pool without translocating protons. In normoxia, the succinate level in mitochondrial matrix is very low (0.2 - 0.4 mМ) [5]. The ratio between the two oxidation pathways depends primarily on properties of the principal enzyme complexes I and II. Kinetic characteristics of two major enzymes of these complexes, NAD-ubiquinon oxidoreductase (MtC I) and succinate dehydrogenase (SDH—MtC II), are tissue-specific and differ in intact animals with different sensitivity to hypoxia [3,6-10].
Hypoxic environments induce reversible depression of MtC I function. At the present time, a great wealth of experimental evidence confirms the inactivation of MtC I electron transport function in hypoxic conditions, which persists and even increases in the post-hypoxic period (first 30 min - 2 hours of reoxygenation) [6-26].
It has been demonstrated that the hypoxic inactivation of enzyme complex I is only one of three steps in a sophisticated phase process of respiratory chain reprogramming. This process starts during a hypoxic impact and continues in the first hours of post hypoxic period [6-10]. These three steps are the following:
Step 1: Activated electron transport function of MtC I, which provides enhancement of ATP synthesis in early hypoxia. This activation reflects a primary compensatory mechanism for mobilization of basic cell energy resources under the conditions of comparatively mild environmental impacts such as slightly decreased oxygen in the environment.
Step 2: Depressed MtC I activity and, correspondingly, oxidation of NAD-dependent substrates along with compensatory activation of MtC II and succinate oxidation, with increased contribution of succinate oxidation to respiration and energy production. This is an alternative pathway for respiratory chain substrates, which provides reduction equivalents to MtC III and MtC IV under progressive hypoxia and plays a special role. In hypoxic conditions, this pathway is thermodynamically superior to oxidation of NAD-dependent substrates and provides a high energetic efficiency of the process as a whole, although only two phosphorylation points are preserved in this process (Figure 1(b)).
Step 3: The step of decompensation differs from the first two steps. Decompensation develops under an acute or long-term hypoxic impact. Decompensation is associated with disturbed electron transport function of the respiratory chain in the region of MtC III (cytochromes b-c) and then MtC IV (cytochrome oxidase, COX), which leads to general de-energization.
As the severity of hypoxia increases Step 1 becomes shorter and Step 2 becomes more pronounced. However when the inhaled O2 concentration falls below 8%, the capability for MtC II activation decreases.
Therefore the respiratory chain reprograming in response to hypoxia starts from its substrate region. The respiratory chain terminal region (cytochrome oxidase, COX) is not a limiting step of this process in a broad range of oxygen concentrations up to anoxia, due to COX kinetic properties (low values of Km (O2)), which determine high affinity of COX to oxygen.
Mitochondrial reprogramming induced by hypoxia correlates with phase changes in the content of adenine nucleotide pool components, ATP, ADP and AMP [3,9,10]. The first step is characterized by a relatively minor, mild (within 6% - 10%) increase in ATP level followed by transition to a somewhat reduced (by 10% - 15%), steady-state ATP level (second step), which re-
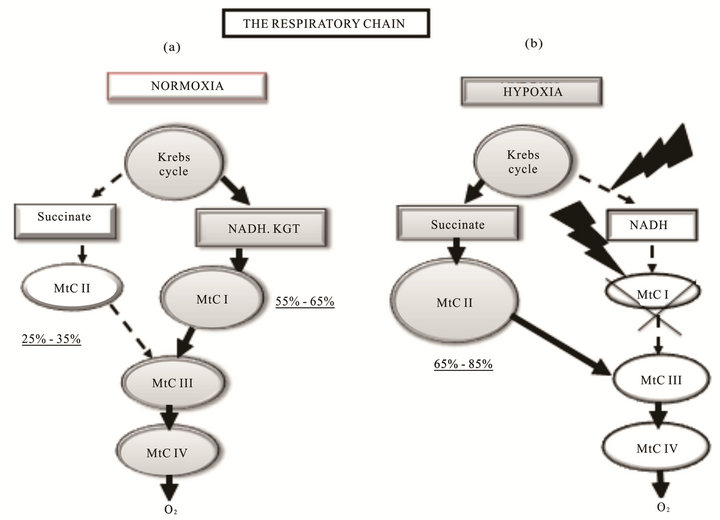
Figure 1. Reprogramming of the respiratory chain function and switching from oxidation of NAD-related substrates (MtC I) in normoxia (a) to succinate oxidation (MtC II) in hypoxia (b). (a) Normoxia; high activity of MtC I (55% - 65% of mitochondrial respiration), low activity of MtC II (25% - 35% of mitochondrial respiration); (b) Hypoxia; depression of MtC I, activation of MtC II (65% - 85% of mitochondrial respiration).
flects ATP hydrolysis. This reduction is accompanied by a rise of intracellular ADP content and a decrease in ATP/ADP ratio, respectively. However, in parallel with the ongoing slight reduction of intracellular ATP level, the intracellular AMP content gradually raises. The regulatory significance of ATP/ADP ratio gradually decreases and becomes replaced by the control of ATP synthesis through the ATP/AMP ratio. At the last stage of hypoxia, when a linear decrease in ATP content is observed, the regulatory role of ATP/ADP and ATP/AMP ratios becomes minimal, and total adenine nucleotides plus energetic charges decrease [3,9]. The hypoxia-induced changes in adenine nucleotides pool resulting from disturbed electron flow and oxidative phosphorylation reactions precede changes in other functional and metabolic parameters responsible for the cell vital activity. Contrastingly, the compensatory role of glycolysis as an ATP supplier is effective only in early hypoxia. During the terminal period, despite a sharp activation of glycolysis, glycolysis fails to prevent the decrease in ATP content [3,9].
Signs of hypoxic inactivation of MtC I include an initial increase and subsequent decrease in both the intensity of NAD-dependent substrate oxidation and efficiency of coupled oxidative phosphorylation, reduced sensitivity of respiration to specific inhibitors of the respiretory chain NAD-dependent region (ex., rotenone, amytal) in various tissues (brain, heart, liver), and reduction of MtC I respiratory carriers (pyridine nucleotides and flavins), which reflects suppression of reduction equivalent transport through this regions [3,4,6-9].
This process is associated with activation of MtC II (SDH) and increased contribution of succinate oxidation to cell respiration; this contribution may reach 70% - 80% [4,6-11,16-19,21,22,24-41].
Therefore, hypoxic changes are associated with succinate accumulation in tissues and blood. The accumulation of succinate in tissues during hypoxia and the reprogramming of respiratory enzyme performance develop fast and under different levels of oxygenation. For example, Goldberg et al., found that after 30 sec of global brain ischemia, succinate level increased 1.5 times in the setting of reduced concentrations of certain NAD-dependent substrates [19]. Similar data were also obtained by other researchers. In the first 30 minutes of anoxic incubation, formation of succinate increases by an order of magnitude, reaches 4 - 7 mM and continues to grow for the first 30 minutes of reoxygenation [17,35,41]. As early as during the first minutes of hypoxic exposure, succinate concentration in blood and tissues increases by an order of magnitude, which allows considering succinate a marker of hypoxia [3-5,10,16,17,31-33,35,41,42]. Any regimen of hypoxic exposure with any level of oxygenation results in pronounced changes in kinetic properties of MtC I and MtC II including increased Km values for complex I and decreased Km values for complex II. This process reflects impaired and enhanced efficiency of enzyme performance, respectively [3,4].
Note that phenoand genotypic peculiarities of the response to hypoxia are essential; these changes are relatively more pronounced in the brain of low-resistance rats and are not statistically significant for the neocortex of high-resistance rats [3,4,7-9].
Depending on tissue-specific peculiarities of energy metabolism, formation of endogenous succinate in hypoxia uses different pathways. However all these pathways are based on aspartateand glutamate-dependent aminotransferase reactions, substrate-level α-ketoglutarate phosphorylation during the conversion of α-ketoglutarate to succinate by α-ketoglutarate dehydrogenese, reversal of tricarbonic cycle and oxidative phosphorylation reactions (substrate-level phosphorylation in citric acid cycle via 2-oxoglutarate dehydrogenase and fumarate reductase reactions) [18,25,31,32,35,41,43-46].
Activation of succinate oxidation constitutes a regulatory and compensatory mechanism, which occurs in most tissues (brain, myocardium, liver, kidneys, lymphocytes) in the conditions of oxygen shortage [7,35]. This mechanism prevents or attenuates disorders in ATP synthesis, exerts normalizing effect on the adenylate pool and vital functions of the body, stabilizes and normalizes pH to eliminate hypoxia-characteristic acidosis, and increases the resistance to oxygen shortage. If such switch does not occur in hypoxia, MtC I dysfunction leads to severe de-energization (decreased membrane potential, ATP loss, and changes in the adenine nucleotide pool) and disturbed respiration due to oxidation of NAD-related substrates (electron donors for MtC I) [3,4,7,9,25, 26,31,32,41].
This regulatory reprogramming is designed for using the succinate oxidase pathway of substrate oxidation, which is an energetically more efficient pathway in hypoxic conditions. This reprogramming prevents or alleviates disorders of ATP synthesis, adenylate pool parameters, vitally significant functions of the body, and typical for hypoxia acidosis. As a result, the body resistance to oxygen shortage increases. Hypoxia-induced reprogrammming of the respiratory chain substrate region correlates with phase changes observed at the systemic level in hypoxia [9,10,36].
The switch between mitochondrial substrate oxidation pathways accompanies practically all types of hypoxia or ischemia and incorporates into these pathways as a mandatory unit, a basic molecular (bioenergetics) mechanism. Thus, the reversible reprogramming of respiratory chain during hypoxia (switching from oxidation of NAD-related substrates to succinate oxidation is an obligatory, evolutionary developed, urgent signaling compensatory mechanism. Due to this mechanism, the energy synthesizing function of respiratory chain is preserved under disturbed oxygen homeostasis.
3. Mitochondrial Signaling in Transcriptional Activity at Hypoxia
The reprogramming of respiratory chain performance in hypoxia is associated with the transcriptional activation of the hypoxic factor, HIF-1α. The process of adaptation to low oxygen level is known to be regulated mostly by a specific transcription protein factor induced in all tissues under hypoxia, hypoxia-inducible factor 1 (HIF-1) [47-53]. This factor is a heterodimeric transcription protein composed of HIF-1α and HIF-1β subunits, which functions as a master regulator of oxygen homeostasis. Regulation of HIF-1α activity is attributed mostly to α subunit.
In normoxic conditions, due to the regulatory interaction between mitochondria and cytosol, prolyl hydroxylase reactions producing proteasomal degradation of HIF-1α are well supplied with substrates required for activation of these reactions, including aspartate and α-ketoglutarate [20,53,54]. α-Ketoglutarate is synthesized in the tricarbonic acid cycle and is oxidized in substrate phosphorylation reactions and NAD-dependent reactions catalyzed by MtC I. The malate-aspartate bypass can supply α-ketoglutarate to cytosol for prolyl hydroxylase reactions. In normoxic conditions, the involvement of α-ketoglutarate in prolyl hydroxylase reactions is associated with formation of succinate, the prolyl hydroxylase reaction inhibitor in cytosol (α-ketoglutarate + О2 à succinate + СО2). Apparently this is necessary for slowing these reactions, maintaining the basic HIF-α level, and inducing basic levels of genes encoding the mitochondrial complex protein responsible for energy production.
Hypoxia inhibits both electron transport in the respiratory chain through MtC I and activity of the malateaspartate bypass. In result, cytosol becomes deficient in α-ketoglutarate and aspartate. At the same time, hypoxia is associated with activation of succinate dehydrogenase (MtC II), and increased formation of succinate in aminotransferase reactions. Succinate is an inhibitor of prolyl hydroxylases. These processes along with О2 and Fe2+ shortage create favorable conditions for inhibition of prolyl hydroxylase reactions, HIF-1α stabilization, induction of transcriptional processes, including HIF-1α translocation to the nucleus, HIF-1α heterodimerization with HIF-1β, conformational changes, formation of the transcription-active complex (hypoxia response element, HRE), expression of HIF-1-dependent target genes, and synthesis of protective, adaptive proteins in response to hypoxia [54-58].
By the present time, conclusive evidence has demonstrated a direct correlation between inactivation of MtC I and activation of MtC II and formation of HIF-1α (Figure 2). For instance, it was shown that even a partial (20%) suppression of MtC I activity almost completely inhibited the hypoxic induction of HIF-1a. However it recovered in the presence of succinate [12-14,58]. We also showed that activation of HIF-1α synthesis after a single hypoxic exposure in the regimen of preconditioning correlated with increased efficiency of MtC II activeity and was associated with a multiple increase in the resistance of animals to acute hypoxia. In the absence of activated succinate oxidation, like, for example, in the neocortex of high-resistant rats, HIF-1α was undetectable [10,37]. Therefore, induction of HIF-1α requires a low MtC 1 activity and a high MtC II activity, i.e., potentiation of succinate oxidase oxidation.
Earlier we have shown that on the first day after any hypoxic exposure, a correlation is observed between intensity of succinate oxidase oxidation, intensity of HIF-1α expression, and capability of the body for developing adaptive responses in these conditions [10,18, 36,37]. An inverse relationship also exists; HIF-1α can influence enzyme activities in the mitochondrial respiretory chain. HIF-1α suppresses pyruvate oxidation in the tricarbonic acid cycle, that is, facilitates inhibition of MtC I by activating pyruvate dehydrogenase kinase 1 (PDK-1). After phosphorylation of PDK-1, it inhibits pyruvate dehydrogenase [33,48,59].
Therefore, an important mechanism of urgent adaptation to hypoxia is the two-way regulatory interaction between the respiratory chain substrate region and expression of the protein factor, HIF-1α. After a hypoxic exposure, the switch of metabolic pathways in the respiratory chain to succinate oxidation provides HIF-1α accumulation and induces transcriptional processes. Simultaneously, the succinate-dependent accumulation of HIF- 1α potentiates inhibition of complex 1 and creates conditions for maintaining succinate oxidase oxidation and HIF-1α accumulation.
In summary, it is obvious that the reprogramming of mitochondrial respiratory chain during the initiation phase of urgent compensatory, adaptive responses is the most important regulatory mechanism. This mechanism provides 1) more efficient succinate-dependent energy production in the conditions of rapidly progressing oxygen shortage; 2) succinate-dependent stabilization of HIF-1α and subsequent initiation of HIF-1α transcriptional activity; 3) succinate-dependent formation of ur-
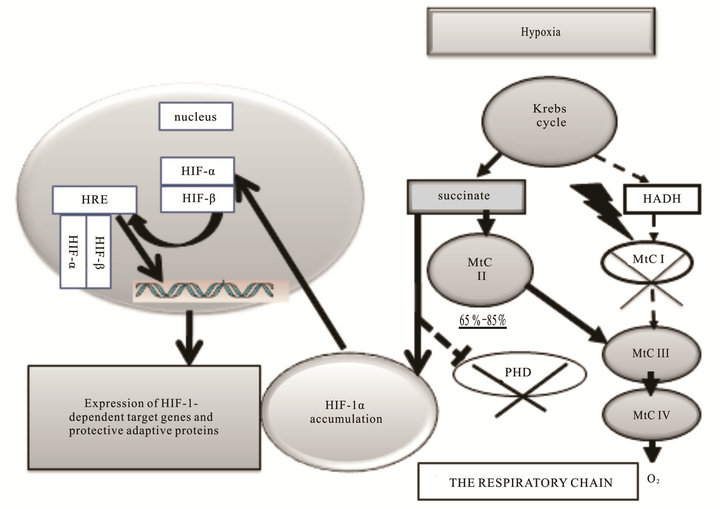
Figure 2. Role of mitochondrial signaling in transcriptional activity at hypoxia. Increased succinate synthesis in hypoxia can provide depression of prolyl hydroxylase activity (PHD), accumulation of HIF-1α in cytosol and expression of HIF-dependent target genes.
gent resistance to hypoxia in the body.
4. Role of Mitochondria in Cell-to-Cell Interactions
The increased level of succinate in blood and cytoplasm allows considering succinate a marker for hypoxic states [60]. It means that succinate may participate in cell-tocell interactions and regulatory, functional and metabolic processes at the systemic level. For instance, it has long been known that succinate contributes to regulation of blood pressure. Succinate stimulates renin formation in renal tubules and, thereby, produces a vasodilatory effect in hypoxia [61].
Studies have shown that a succinate-controlled electron bypass exists between peripheral cells and lungs. In hypoxia, blood transports succinate from peripheral hypoxic tissues to lungs, where succinate synthesis is depressed by intensive oxygenation. Delivered succinate is used in lungs as an energy substrate needed for pulmonary vasoconstriction. In this process, succinate is oxidized to fumarate which is again transported with blood to peripheral cells and again forms succinate through the fumarate oxidase reaction [43,62].
In the recent two decades, data on a receptor function of succinate have appeared. Reports have shown that MtC II contributes to gene expression in glutamateand dopaminergic signaling systems [53].
A sensational discovery was made by a research group in San Francisco in 2004. This study demonstrated that succinate was a specific ligand of GPR91 [63]. This receptor belongs to a group of guanine nucleotide binding protein coupled receptors which form large and diverse gene families translating extracellular signals to intracellular ones. However the sequence similarity among the most distant GPCRs is minimal [64]. GPR91 is localized in plasma membranes. It is found in more than 20 tissue types, and the array of its localizations is continuously expanding. Kidneys, liver, spleen, small intestine, and bladder possess the best capability for expressing this receptor. Only succinate activates GPR91 in a recaptorspecific way and also induces GPR91 internalization [65].
Succinate-dependent receptor activation induces:
• accumulation of inositol phosphate and increases [Ca2+]i;
• extracellular signal-regulated kinase (Erk);
• mitogen-activated protein kinase, p38 and cyclooxygenase;
• synthesis and release of prostaglandin Е2 and NO.
Taken together, these results suggest that GPR91 activation by succinate couples to at least several signaling pathways [46,65]. However the succinate signal is apparently tissue-specific, and these pathways may not work in all tissues.
In kidneys, the succinate-induced expression of GPR91 associated with GPR91 internalization involves the renin-angiotensin system and precipitates renovascular hypertension, a disease closely related to atherosclerosis, diabetes mellitus and renal insufficiency [42,65]. This process is absent in GPR91-deficient rats [65]. It has been demonstrated recently that this activation is a part of kidney-specific paracrine signaling pathway initiated by high glucose levels. The signaling cascade includes local accumulation of succinate as well as expression and internalization of GPR91 receptors in endothelial cells of renal tubules. This GPR91 signaling cascade can modulate renal function and provide elimination of various metabolic products through renal hyperfiltration. The GPR91 cascade is apparently a connecting link to pathologies such as diabetes mellitus, syndrome of reninangiotensin system hyperactivity, systemic hypertension, and organ injury [46].
In liver ischemia associated with intensive succinate release, GPR91 is expressed only in hepatic stellate cells. However the fact that succinate does not increase hepatic perfusion pressure suggests that succinate activities in the liver do not contribute to its hypertensive effects. GPR91 transforms the signal related with increased extracellular levels of succinate into an intracellular signal providing activation of stellate cells in response to organ injury. Therefore the effect of succinate should be regarded as a paracrine signal in this instance as well.
Succinate-dependent expression of GPR91 in ischemia was observed in dendritic cells; it results in production of various angiogenic factors, including VEGF. In these cells, succinate triggers mobilization of intracellular calcium, induces migratory responses and acts in synergy with Toll-like receptor ligands to produce pro-inflamematory cytokines [66].
Succinate also enhanced antigen-specific activation of human and mouse helper T cells. GPR91-deficient mice showed less migration of Langerhans cells to draining lymph nodes and impaired tetanus toxoid-specific recall T cell responses [67].
Studies have shown that succinate accumulates in hypoxic retina of rodents and, via GPR91, acts as a potent mediator of vessel growth in the settings of both normal retina development and proliferative ischemic retinopathy. These effects of GPR91 are mediated by retinal ganglion neurons. These neurons respond to increased succinate levels and regulate production of numerous angiogenic factors, including VEGF. Accordingly, succinate did not display any angiogenic effects in RGC-deficient rats [68].
As a GPR91 ligand, succinate can regulate lipolysis in white adipose tissue suggesting that the succinate signaling may regulate energy homeostasis [69]
The receptor function along with the participation of succinate in cell-to-cell signaling and systemic regulation can explain many rapid effects of this metabolite, including the role of succinate in development of urgent adaptation. Data on the interaction of GPR91 with other receptors help to understand also the regulatory participation of catecholamines and acetylcholine in activation of succinate dehydrogenase.
Therefore, the response to hypoxia initially develops at the subcellular, mitochondrial level through compensatory potentiation of enzyme complex II and involves activation of a specific succinate-dependent receptor (Figure 3). This receptor initiates cytoplasmic signaling pathways which coordinate and facilitate formation of adaptive processes, as well as cell-to-cell interactions at the systemic level.
5. Use of Succinate-Containing Drugs as Energotropic Medicines in Clinical Practice
High prevalence of diseases bearing a hypoxic component defines the exceptional importance and social significance of protecting the body from oxygen shortage and related energy deficiency. The above data also show that understanding the role of succinate signaling in the electron transport function of respiratory chain under hypoxia is an essential practical challenge that has defined the tactics and strategy for antihypoxic defense and the development of antihypoxants with energotropic action.
The energotropic therapy implies using as medicines the substances that are able to interact with the mitochondrial respiratory chain and to prevent disorders of energy metabolism up to its complete recovery in pathological conditions. Principles of energotropic therapy are based on the concept of the phase nature of hypoxiaonduced mitochondrial dysfunction related with changes in activities of mitochondrial enzymes.
The central role among such medicines belongs to succinate-containing drugs which have been successfully used as effective antihypoxic and energotropic therapy [8, 9,18,35-37]. The most effective of these drugs are structural derivatives of vitamin В6, which belongs to 1-hydroxypyridine derivatives (ex., mexidole, proxipine).
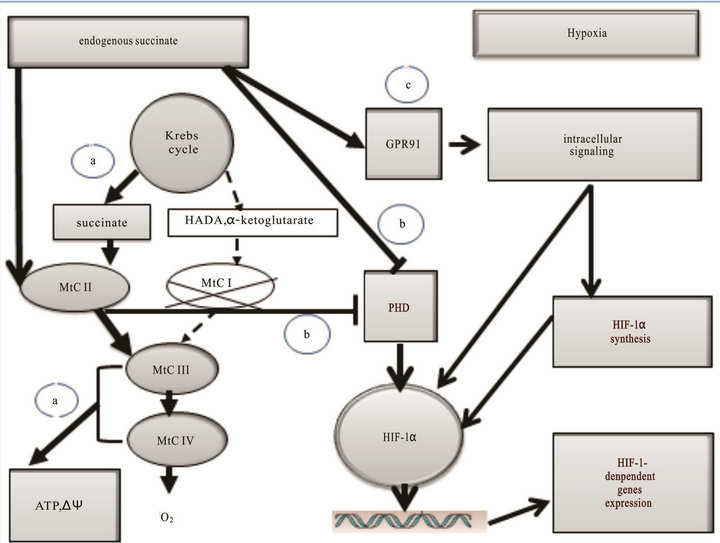
Figure 3. Role of mitochondrial signaling in cell-to-cell interactions at hypoxia. Reprogramming of the mitochondrial respiratory chain in hypoxia is a regulatory and compensatory mechanism, which provides 1) activation of succinate oxidation, a more efficient pathway of energy production in the conditions of rapidly progressing oxygen shortage (a); 2) succinate-dependent PDH inhibition, subsequent stabilization of HIF-1α, and initiation of HIF-1α transcriptional activity (b); 3) succinate-dependent activation of the GPR91 signaling pathway and related signaling pathways, stimulation of HIF-1α synthesis and transcriptional activity (c).
These drugs are used in the early period of acute disorders induced by oxygen shortage [8,10,18,37].
Special studies showed that the succinate moiety of these drugs is used by the respiratory chain as an energy substrate according to the classic schedule for succinate. In this process, respiration becomes stimulated, and the electron flow switches from complex I to complex II (succinate monopolization of the respiratory chain as demonstrated by the increased sensitivity of respiration to malonate and the decreased rotenone-sensitive respiretion due to oxidation of NAD-dependent substrates) [18, 37,39].
The obtained results suggest that the succinate-containing drug, proxipine, is a succinate donor for the respiratory chain. In hypoxic conditions, proxipine functions as an antihypoxant potentiating activation of the succinate oxidation pathway to facilitate recovery and normalization of aerobic energy production. Furthermore, the energotropic effect of succinate-containing drugs has been demonstrated to be due to oxidation of succinate incorporated into the structure of 3-oxypyridine by a classic pathway for succinate in the respiratory chain [18]. All succinate-containing drugs are very rapidly absorbed in various types of hypoxia/ischemia. They exert stabilizing or restoring effects on intracellular ATP already 15 minutes after administration.
Energotropic and antihypoxic effects of succinate-containing compounds are associated with 1) modification and resynthesis of phospholipids; 2) normalizing effect on calcium metabolism; 3) catecholaminemimetic, antiteratogenic, antitoxic, hepatoprotective, antiketogenic, and anticholesterolemic effects; 4) removal of excessive acetyl-Co-A associated with decreases in excessive lipids and their metabolites; 5) reduction and normalization of pH and elimination of metabolic acidosis [18,37,70].
Proxipine is characterized by a broad range of physicological actions. Succinate-containing drug therapies used during the phase of formation of urgent compensatory adaptive mechanisms, i.e., in the first 1 - 3 days of global cerebral ischemia, stroke, myocardial infarction, acute heart failure, traumatic shock, resuscitation after heart arrest, early postoperative period, after anesthesia, etc., exert pronounced protective, antihypoxic effects and increase ATP in tissues (Figures 4(a)-(c)). Along with the energotropic and antihypoxic effects, proxipine beneficially influence multiple vital functional parameters in hypoxia and ischemia. These drug reduce the death rate, recover the ability of the body for gaining weight, decrease severity of neurological disorders (Figure 3(d)) and aggression typical of hypoxia, exert antistress and normalizing effects on locomotor, exploratory and emotional activities of animals [10,18,36,37].
Timely use of this drug decreases mortality and provides faster and more complete regression of general and focal cerebral symptoms in the majority of patients. Therefore, a single preventive or therapeutic administration of succinate-containing drugs in the setting of hypoxic therapy will improve the development of urgent resistance in the body and, therefore, it may be used in practice for activation of urgent adaptive mechanisms [10, 37]. However a longer use of succinate-containing drugs loses its protective effects because it began to hamper the formation of long-term mechanisms of adaptation. Therefore the optimal therapeutic effect of succinate can be achieved with no more than three injections at early stages of hypoxic exposure.
6. Conclusions
The role of mitochondria as a cell “power-station” is well known. Participation of mitochondria in essential regulatory processes determining intracellular, intercellular and systemic interactions is presently discussed [19-21, 29,30,32,33,35,49-56,58,60,64,71-75]. However, mitochondria also play an important regulatory role in the conditions of hypoxia.
In hypoxia, the mitochondrial respiratory chain not only directly participates in formation of both early and late adaptive signs, but it is also involved in the sophisticated system of intraand inter-cellular signaling. This system provides systemic responses to oxygen shortage. At least three regulatory functions of this system can be distinguished: 1) compensatory function, which is responsible for development of urgent adaptive responses and resistance to hypoxia; this function is associated with changes in kinetic properties of mitochondrial enzymes and pathways of energy substrate oxidation; 2) transcriptional function, which provides HIF-1α dependent expression of genes responsible for development of mechanisms of long-term adaption to low рО2; 3) receptor function, which is related with participation of mitochondria in the system of intercellular signaling (Figure 3).
The tricarbonic acid cycle intermediate, succinate, plays a special role in these processes. Succinate is a signaling molecule that is involved in this process during various kinds and regimens of hypoxic exposure. In early hypoxia, for example, the ability of respiratory chain for reprogramming (switching from NAD-dependent substrate oxidation to succinate oxidation, which is energetically more efficient in these conditions) maintains both aerobic energy production and development of urgent resistance in the body (Figure 1). If such switch fails urgent mechanisms of adaptation cannot develop.
Increased intracellular succinate provides succinatedependent stabilization of HIF-1α and initiation of its transcriptional activity. Therefore, long-term adaptation to hypoxia is a succinate-dependent process, during which HIF-1α dependent formation of adaptive genes and generation of a new enzyme spectrum (including mito-
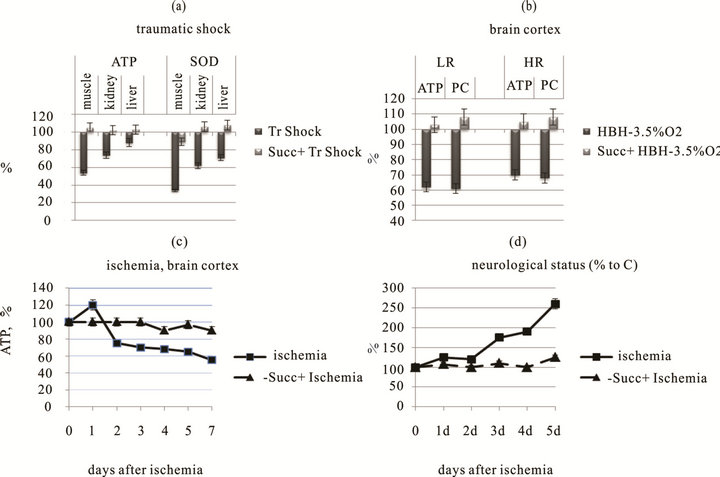
Figure 4. Energotropic and antihypoxic effects of the succinate-containing compound proxipin. Normalizing effect of the succinate-containing drug proxipin (3-hydroxypyridine, 40 mg/kg, i.p.) on the following parameters, (a) ATP level and SOD activity (% of control) in tissues of rats exposed to traumatic shock (Tr Shock). Tr Shock,without proxipin; Succ + Tr Shock, with proxipin. (b) ATP and phosphocreatine (PC) levels (% of control) in brain cortex of rats exposed to altitude chamber hypoxia (acute hypobaric hypoxia, HBH, 3.5% O2). HBH, without proxipin, Succ + HBH, with proxipin; LR, low-resistance rats, HR, high-resistance rats. (c) ATP level (% of control) in brain cortex of rats exposed to global ischemia. (d) Neurological status of rats.
chondrial enzymes) can ensure the vital activity of and the energy supply to cells in conditions of high reduction and low oxygen concentration (Figure 2).
Another, no less important signaling mechanism in hypoxia is succinate activation of a succinate-specific and very common tissue GPR91 receptor. The receptor signaling function provides initiation and coordination of a broad spectrum of other signaling pathways which may not only facilitate formation of adaptive processes but also provide cell-to-cell interactions at the systemic level (Figure 3).
Understanding of biological mechanisms for adaptation to hypoxia has substantiated the development of a new strategy for antihypoxic defense. A single preventive or therapeutic administration of succinate-containing drugs in the setting of hypoxic therapy enhances development of urgent resistance in the body and, therefore, it may be used in practice for activation of urgent adaptive mechanisms.
REFERENCES
- U. T. Brunk and A. Terman, “The Mitochondrial-Lysosomal Axis Theory of Aging,” European Journal of Biochemistry, Vol. 269, No. 8, 2002, pp. 1996-2002. doi:10.1046/j.1432-1033.2002.02869.x
- K. Michiels, “Physiological and Pathological Responses to Hypoxia,” American Journal of Pathology, Vol. 164, No. 6, 2004, pp. 1875-1882. doi:10.1016/S0002-9440(10)63747-9
- L. D. Lukyanova , A. M. Dudchenko, E. L. Germanova, T. A. Tsybina, R. A.Kopaladse, I. V. Ehrenbourg and E. N. Tkatchouk, “Mitochondria Signaling in Formation of Body Resistance to Hypoxia,” In: L. Xi and T. Serebrovskaya, Eds., Intermitten Hypoxia: From Molecular Mechanisms to Clinical Applications, Nova Science Publishers, 2009, pp. 423-450.
- L. D. Lukyanova, A. M. Dudchenko, T. A. Tsybina, E. L. Germanova, E. N. Tkatchouk and V. Ehrenbourg, “Effect of Intermittent Normobaric Hypoxia on Kinetic Properties of Mitochondrial Enzymes,” Bulletin of Experimental Biology and Medicine, Vol. 144, No. 6, 2007, pp. 795-801. doi:10.1007/s10517-007-0434-y
- D. A. Hems and J. T. Brosnan, “Effects of Ischaemia on Content of Metabolites in Rat Liver and Kidney in Vivo,” Biochemical Journal, Vol. 120, 1970, pp. 105-111.
- L. D. Lukyanova, “Limiting Steps of Energy Metabolism in Brain in Hypoxia,” Neurochemistry International, Vol. 13, No. 1, 1988, pp. 146-147.
- L. D. Lukyanova, “Molecular, Metabolic and Functional Mechanisms of Individual Resistance to Hypoxia,” In: B. K. Sharma, N. Takeda, et al., Eds., Adaptation Biology and Medicine, Vol. I, Narosa Publishing House, New Dehli, 1997, pp. 261-272.
- L. D. Lukyanova, “Сellular Mechanism Responsible for Beneficial Effects of Hypoxic Therapy,” In: Moraveč, et al., Eds., Adaptation Biology and Medicine, Vol. 3, Narosa Publishing House, New Dehli, 2002, pp. 290-303.
- L. D. Lukyanova and A. M. Dudchenko, “Regulatory Role of the Adenylate Pool in the Formation of Hepatocyte Resistance to Hypoxia,” In: K. B. Pandolf, N. Takeda, P. K. Singal, Eds., Adaptation Biology and Medicine, Vol. 2, Narosa Publishing House, New Dehli, 1999, pp. 139-150.
- L. D. Lukyanova, E. L. Germanova and Yu. I. Kirova, “The Signal Function of Succinate and Free Radicals in Mechanisms of Preconditioning and Long-term Adaptation to Hypoxia,” In: P. Wang, C.-H. Kuo, N. Takeda and P. K. Singal, Eds., Adaptation Biology and Medicine, Vol. 6, Cell Adaptations and Challenges, Chapter 19, 2011, pp. 251-277.
- H. N. Aithal and T. Ramasarma, “Activation of Liver Succinate Dehydrogenase in Rats Exposed to Hypobaric Conditions,” Biochemical Journal, Vol. 115, No. 1, 1969, pp. 77-83.
- F. H. Agani, P. Pichiule, J. C. Chavez and J. C. La Manna, “The Role of Mitochondria in the Regulation of HypoxiaInducible Factor 1 Expression during Hypoxia,” JBC, Vol. 275, No. 46, 2000, pp. 35863-35867. doi:10.1074/jbc.M005643200
- F. H. Agani, M. Puchowicz, J. C. Chavez, P. Pichiule and J. LaManna, “Inhibitors of Mitochondrial Complex I Attenuate the Accumulation of Hypoxia-Inducible Factor-1 during Hypoxia in Hep3B Cells,” Comparative Biochemistry and Physiology, Vol. 132, No. 1, 2000, pp. 107-109.
- J. C. Chavez, F. Agani, P. Pichule and J. C. LaManna, “Expression of Hypoxia-Inducible Factor-1α in the Brain of Rats during Chronic Hypoxia,” Journal of Applied Physiology, Vol. 89, No. 5, 2000, pp. 1937-1942.
- M. M. Da Silva, F. Sartori, E. Belisle and A. J. Kowaltowsky, “Ischemic Preconditioning Inhibits Mitochondrial Respiration, Increase H2O2 Release, and Enhances K+ Transport,” American Journal of Physiology: Heart and Circulatory Physiology, Vol. 285, 2003, pp. 154-162.
- M. I. Genova, R. Casteluccio, G. Fato, M. Parenti-Castelli, G. Merlo-Pich, C. Formiggini, M. Bovina and G. M. Lenaz, “Major Changes in Complex I Activity in Mitochondrian from Aged Rats May Not be Detected by Direct Assay of NADH-Coenzyme Q Reductase,” Biochemical Journal, Vol. 311, 1995, pp. 105-109.
- N. D. Goldberg, J. V. Passonneau and O. H. Lowry, “Effects of Changes in Brain Acid Cycle Intermediates,” Journal of Biological Chemistry, Vol. 241, No.17, 1966, pp. 3997-4003.
- L. D. Lukyanova, E. L. Germanova, T. A. Tsybina and G. N. Chernobaeva, “Energotropic Effect of Succinate-Containing Derivatives of 3-Hydroxypyridine,” Bulletin of Experimental Biology and Medicine, Vol. 148, No. 4, 2009, pp. 587-591. doi:10.1007/s10517-010-0771-0
- E. Maklashinas, E. Sher, H.-Z. Zhou and M. Gray, “Effect of Anoxia/Reperfusion on the Reversible Active/deActive Transition of Complex I in Rat Hear,” Bioenergetics, Vol. 1556, No. 1, 2002, pp. 6-12. doi:10.1016/S0005-2728(02)00280-3
- L. Mela., C. W. Goodwin and L. D. Miller, “In Vivo Control of Mitochondrial Enzyme Concentrations and Activity by Oxygen,” American Journal of Physiology, Vol. 231, No. 6, 1976, pp. 1811-1816.
- S. Pitkänen and B. H. Robinso, “Mitochondrial Complex I Deficiency Leads to Increased Production of Superoxide Radicals and Induction of Superoxide Dismutase,” Journal of Clinical Investigation, Vol. 98, No. 2, 1996, pp. 345-351. doi:10.1172/JCI118798
- B. H. Robinson, “Human Comlex I Deficiency: Clinical Spectrum and Involvement of Oxygen Free Radicals in the Pathogenicity of the Defect,” Biochimica et Biophysica Acta, Vol. 1364, No. 2, 1998, pp. 271-286. doi:10.1016/S0005-2728(98)00033-4
- W. Rouslin and R. W. Millard, “Canine Myocardial Ischemia: Defect in Mitochondrial Electron Transfer Complex I,” Journal of Molecular and Cellular Cardiology, Vol. 12, No. 6, 1980, pp. 639-645. doi:10.1016/0022-2828(80)90021-8
- H. A. Sadek, P. A. Sweda and L. I Sweda, “Modulation of Mitochondrial Complex I Activity by Reversible Ca2+ and NADH Mediated Superoxide Anion Dependent Inhibition,” Biochemistry, Vol. 43, No. 26, 2004, pp. 8494- 8502. doi:10.1021/bi049803f
- J. M. Weinberg, M. A. Venkatachalm and F. Nancy, “Anaerobic and Aerobic Pathways for Salvage of Ptoximal Tubules from Hypoxia-Induced Mitochondrial Injury,” American Journal of Physiology: Renal Physiology, Vol. 279, 2000, pp. F927-F943.
- J. M. Weinberg, M. A. Venkatachalm and F. Nancy, “Mitochondrial Disfunction during Hypoxia/ Reoxigenation and Its Correction by Anaerobic Metabolism of Citric Acid Cycle Intermediates,” Proceedings of the National Academy of Science of USA, Vol. 97, No. 6, 2000, pp. 2826-2831. doi:10.1073/pnas.97.6.2826
- P. R. Correa, E. A. Kruglov, M. Thompson, M. F. Leite, J. A. Dranoff and M. H. Nathanson, “Succinate Is a Paracrine Signal for Liver Damage,” Journal of Hepatology, Vol. 47, No. 2, 2007, pp. 262-269. doi:10.1016/j.jhep.2007.03.016
- T. Feldkamp, A. Kribben, N. F. Roeser , R. A. Senter, S. Kemner, M. A. Venkatachalam, I. Nissim and J. M. Weinberg, “Preservation of Complex I Function during Hypoxia-Reoxygenation-Induced Mitochondrial Injury in Proximal Tubules,” American Journal of Physiology: Renal Physiology, Vol. 286, No. 4, 2004. pp. 749-759. doi:10.1152/ajprenal.00276.2003
- M. Gutman, “Modulation of Mitochondrial Succinate Dehydrogenese Activity, Mechanism and Function,” Molecular and Cellular Biochemistry, Vol. 20, No. 1, 1978, pp. 41-60. doi:10.1007/BF00229453
- W. He, F. J. Miao and D. C. Lin, “Citric Acid Cycle Intermediates as Ligands for Orphan-G-Protein-Coupled Receptors,” Nature, Vol. 429, No. 6988, 2004, pp. 188- 193. doi:10.1038/nature02488
- P. W. Hochachka, T. G. Owen, J. F. Allen and G. C. Whittow, “Multiple End Products of Anaerobiosis in Diving Vertebrates,” Comparative Biochemistry and Physiology, Vol. 50, No. 1, 2001, pp. 17-22.
- P. W. Hochachka and G. N. Somero, “Biochemical Adaptation—Mechanism and Process in Physiological Evolution,” Oxford University Press, New York, 2001.
- C. Hohl, R. Oestreich, P. Rösen, R. Wiesner and M. Grieshaber, “Evidence for Succinate Production by Reduction of Fumarate during Hypoxia Inisolat Adult Rat Heart Cells,” Archives of Biochemistry and Biophysics, Vol. 259, No. 2, 1987, pp. 527-535. doi:10.1016/0003-9861(87)90519-4
- A. King, M. A. Selak and E. Gottlieb, “Succinate Dehydrogenase and Fumarate Hydratase: Linking Mitochondrial Dysfunction and Cancer,” Oncogene, Vol. 25, No. 34, 2006, pp. 4675-4682. doi:10.1038/sj.onc.1209594
- M. N. Kondrashova, “The Formation and Utilization of Succinate in Mitochondria as a Control Mechanism of Energization and Energy State of Tissue,” In: B. Chance, Ed., Biological and Biochemical Oscillators, Academy Press, New York, 1993, pp. 373-397.
- L. D. Lukyanova , E. L. Germanova and R. A. Kopaladze, “Development of Resistance of an Organism under Various Conditions of Hypoxic Preconditioning: Role of the Hypoxic Period and Reoxygenation,” Bulletin of Experimental Biology and Medicine, Vol. 147, No. 4, 2009, pp. 400-404. doi:10.1007/s10517-009-0529-8
- L. D. Lukyanova, Yu. I. Kirova and E. L. Germanova, “Energotropic Effects of Intermittent Hypoxia and a Possibility of Their Optimization by Modulatory Action of Mitochondrial Substrates,” In: L. Xi and T. V. Serebrovskaya, Eds., Intermittent Hypoxia and Human Diseases, Springer, London, 2012, pp. 239-252.
- L. Mela, C. W. Goodwin and L. D. Miller, “In Vivo Control of Mitochondrial Enzyme Concentrations and Activity by Oxygen,” The American Journal of Physiology, Vol. 231, No. 6, 1976, pp. 1811-1816.
- G. Nowak, G. L. Clifton and D. Bakajsova, “Succinate Ameliorates Energy Deficits and Prevents Dysfunction of Complex I in Injured Renal Proximal Tubular Cells,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 324, No. 3, 2008, pp. 1155-1162. doi:10.1124/jpet.107.130872
- F. Zoccarato, L. Cavallini, S. Bortolami and A. Alexandre, “Succinate Modulation of H2O2 Release at NADH: Ubiquinone Oxidoreductase (Complex I) in Brain Mitochondria,” Biochemical Journal, Vol. 406, No. 1, 2007, pp. 125-129. doi:10.1042/BJ20070215
- H. Taegmeyer, “Metabolic Responses to Cardiac Hypoxia. Increased Production of Succinate by Rabbit Papillary Muscles,” Circulation Research, Vol. 43, 1978, pp. 808- 815. doi:10.1161/01.RES.43.5.808
- N. Sadagopan, S. L. Roberds, T. Major, G. M. Preston, Yu. Y and M. A. Tones, “Circulating Succinate Is Elevated in Rodent Models of Hypertension and Metabolic Disease,” American Journal of Hypertension, Vol. 20, No. 11, 2007, pp.1209-1215.
- J. Cascarano, I. Z. Ades and J. D. O'Conner, “Hypoxia: A Succinate-Fumerate Electron Shuttle between Peripheral Cells and Lung,” Journal of Experimental Zoology, Vol. 198, No. 2, 1976, pp. 149-153. doi:10.1002/jez.1401980204
- D. A. Hems and J. T. Brosnan, “Effects of Ischaemia on Content of Metabolites in Rat Liver and Kidney in Vivo,” The Biochemical Journal, Vol. 120, No. 1, 1970, pp. 105- 111.
- T. Sanborn, W. Gavin, S. Berkowitz, T. Perille and M. Lesch, “Augmented Conversion of Aspartate and Glutamate to Succinate during Anoxia in Rabbit Heart,” The American Journal of Physiology, Vol. 237, No. 5, 1979, pp. H535-H541.
- I. Toma, J. J. Kang, A. Sipos, S. Vargas, E. Bansal, F. Hanner, E. Meer and J. Peti-Peterdi, “Succinate Receptor GPR91 Provides a Direct Link between High Glucose Levels and Renin Release in Murine and Rabbit Kidney,” The Journal of Clinical Investigation, Vol. 118, No. 7, 2008, pp. 2526-2534.
- G. L. Semenza, “Signal Transduction to Hypoxia-Inducible Factor 1,” Biochemical Pharmacology, Vol. 64, No. 5-6, 2002, pp. 993-998. doi:10.1016/S0006-2952(02)01168-1
- G. L. Semenza, “Oxygen-Dependent Regulation of Mitochondrial Respiration by Hypoxia-Inducible Factor 1,” The Biochemical Journal, Vol. 405, No. 1, 2007, pp. 1-9.
- G. L. Semenza, “Involvement of Oxygen-Sensing Pathways in Physiologic and Pathologic Erythropoiesis,” Blood, Vol. 114, No. 10, 2009, pp. 2015-2019. doi:10.1182/blood-2009-05-189985
- G. L. Semenza and G. Wang, “A Nuclear Factor Induced by Hypoxia via de Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation,” Molecular and Cellular Biology, Vol. 12, No. 12, 1992, pp. 5447-5454.
- G. Wang and G. L. Semenza, “Characterization of Hypoxia-Inducible Factor 1 and Regulation of DNA Binding Activity by Hypoxia,” The Journal of Biological Chemistry, Vol. 268, No. 29, 1993, pp. 21513-21518.
- L. A. Shimoda and G. L. Semenza, “Role of HypoxiaInducible Factors in Pulmonary Development and Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 183, No. 2, 2011, pp. 152-156. doi:10.1164/rccm.201009-1393PP
- M. Napolitano, D. Centoze, P. Gubellini, S. Rossi, S. Spiezia, G. Bernardi, F. Gulino and P. Calabresi, “Inhibition of Mitochondrial Complex II Alters Strial Expression of Genes Involved in Glutamatergi Signaling: Possible Implications for Huginton’s Diease,” Neurobiology of Disease, Vol. 15, No. 2, 2004, pp. 407-414. doi:10.1016/j.nbd.2003.11.021
- D. M. Stroka, T. Burkhardt and I. Desballerts, “HIF-1 Is Expressed in Normoxia Tissue and Displays an OrganSpecific Regulation under Systemic Hypoxia,” FASEB Journal, Vol. 15, No. 13, 2001, pp. 2445-2453.
- K .S. Hewitson, B. M. Lienard, M. A. McDonough, I. J. Clifton, D. Butler, A. S. Soares, N. J. Oldham, L. A. McNeill and C. J. Schofield, “Structural and Mechanistic Studies on the Inhibition of the Hypoxia-Inducible Transcription Factor Hydroxylases by Tricarbonic Acid Cycle Intermediates,” The Journal of Biological Chemistry, Vol. 282, No. 5, 2007, pp. 3293-3230. doi:10.1074/jbc.M608337200
- P. Kolvunen, M. Hirsila, A. M. Remes, I. E. Hassinen, K. I. Kivirikko and J. Myllyharju, “Inhibition of HypoxiaInducible Factor (HIF) Hydroxylases by Citric Acid Cycle Intermediates: Possible Links between Cell Metabolism and Stabilization of HIF,” The Journal of Biological Chemistry, Vol. 282, No. 7, 2007, pp. 4524-4532. doi:10.1074/jbc.M610415200
- M. A. Selak, S. M. Armour and E. D. McKenzie, “Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase,” Cancer Cell, Vol. 7, No. 1, 2005, pp. 77-85. doi:10.1016/j.ccr.2004.11.022
- E. C. Vaux, E. Metzen, K. M. Yeates, P. J. Ratcliffe, “Regulation of Hypoxia-Inducible Factor Is Reserved in the Absence of a Functioning Respiratory Chain,” Blood, Vol. 98, No. 2, pp. 296-302. doi:10.1182/blood.V98.2.296
- J. -W. Kim, I. Tchernyshyov, G. L. Semenza and C. V. Dang, “HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia,” Cell Metabolism, Vol. 3, No. 3, 2006, pp. 177-185. doi:10.1016/j.cmet.2006.02.002
- G. Komaromy-Hiller, P. D. Sundquist, L. J. Jacobsen, K. L. Nuttall, “Serum Succinate by Capillary Zone Electrophoresis: Marker Candidate for Hypoxia,” Annals of Clinical Laboratory Science, Vol. 27, No. 2, 1997, pp. 163-168.
- L. Baumbach, P. P. Leyssac, S. l. Skinner, “Studies on Renin Release from Isolated Superfused Glomeruli: Effects of Temperature, Urea, Ouabain and Ethacrynic Acid,” The journal of physiology, Vol. 258, No. 1, 1976. pp.243-256.
- R. Paddenberg, B. Ishak, A. Goldenberg, P. Faulhammer, F. Rose, N. Weissmann, R. Braun-Dullaeus and W. Kummer, “Essential Role of Complex II of the Respiraitory Chain in Hypoxia-Induced ROS Generation in Pulmonary Vasculature,” American Journal of Physilogy, Lung Cellular and Molecular Physiology, Vol. 284, 2003, pp. L710-L719.
- W. He, F. J. Miao and D. C. Lin, “Citric Acid Cycle Intermediates as Ligands for Orphan-G-Protein-Coupled Receptors,” Nature, Vol. 429, 2004, pp.188-193. doi:10.1038/nature02488
- C. Peers and P. J. Kemp, “Acute Oxygen Sensing: Diverse but Convergent Mechanisms in Airway and Arterial Chemoreceptors,” Respiratory Research, Vol. 2, No. 3, 2001, pp. 145-149. doi:10.1186/rr51
- Y. Hakak, K. Lehmann-Bruinsma, S. Phillips, Le T. Llaw, D. T. Connolly and D. P. Behan, “The Role of GPR91 Ligand Succinate in Hematopoiesis,” Journal of Leukocyte Biology, Vol. 85, No. 5, 2009, pp. 837-843. doi:10.1189/jlb.1008618
- T. Rubic, G. Lametschwandtner, S. Hinteregger and J. Kund, “Triggering the Succinate Receptor GPR91 on Dendritic Cells Enhances Immunity,” Nature Immunology, Vol. 9, No. 11, 2008, pp. 1261-1269.
- G. Serviddio, N. Di Venosa, D. Agostino, T. Rollo, F. Priggigalo, E. Altomare and T. Fiore, “Brief Hypoxia before Normoxic Reperfusion (Postconditioning) Protects the Heart against Ischemia-Reperfusion Injury by Preventing Mitochondria Peroxide Production and Glutathion Depletion,” The Journal of the Federation of American Societies for Experimental Biology, Vol. 19, No. 3, 2005, pp. 354-361. doi:10.1096/fj.04-2338com
- P. Sapieha, M. Sirinyan, D. Hamel, K. Zaniolo, J. S. Joyal, J. C. Honore, E. Kermorvant-Duchemin and D. R. Varma, “The Succinate Receptor GPR91 in Neurons Has a Major Role in Retinal Angiogenesis,” Nature Medicine, Vol. 14, No. 10, 2008, pp. 1067-1076.
- J. B. Regard, I. T. Sato and S. R. Coughlin, “Anatomical Profiling of G Protein-Coupled Receptor Expression,” Cell, Vol. 135, No. 3, 2008, pp. 561-571. doi:10.1016/j.cell.2008.08.040
- M. N. Kondrashova and N. M. Doliba, “Polarografiphic Observation of Substrate-Level Phosphorylation and Its Stimulation by Acetylcholine,” FEBS Letters, Vol. 243, No. 2, 1989, pp. 153-155. doi:10.1016/0014-5793(89)80119-X
- R. A. Butow and N. G. Avadhani, “Mitochondria Signaling. The Retrograde Response,” Molecular Cell, Vol. 14, No. 1, 2004, pp. 1-15.
- E .L. Bell, T. A. Klimova, J. Eisenbart, C. T. Shumacker and N. S. Chandel, “Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Inducible Factor-Dependent Extension of the Replicative Life Span during Hypoxia,” Molecular and Cellular Biology, Vol. 27, No. 16, 2007, pp. 5737-5745. doi:10.1128/MCB.02265-06
- N. S. Chandel and P. T. Schumacker, “Cellular Oxygen Sensing by Mitochondria: Old Questions, New Insight,” The American Journal of Physiology, Vol. 88, No. 5, 2000, pp. 1880-1889.
- J. Das, “The Role of Mitochondrial Respiration in Physiological and Evolutionary Adaptation,” BioEssays, Vol. 28, No. 9, 2006, pp. 890-901. doi:10.1002/bies.20463
- M. R. Duchen, “Roles of Mitochondria in Health and Disease,” Diabetes, Vol. 53, No. S1, 2009, pp. S96-S102. doi:10.2337/diabetes.53.2007.S96
Abbreviations
ATP, adenosine triphosphate;
ADP, adenosine diphosphate;
AMP, adenosine monophosphate;
COX, cytochrome oxidase;
MtC, mitochondrial complex;
GPR91, succinate-related guanine nucleotide - binding protein-coupled receptor;
HBH, acute hypobaric hypoxia;
HIF, hypoxia-inducible factor;
HRE, hypoxia response element;
HR, high-resistance rats;
LR, low-resistance rats;
NAD, NADH, nicotine adenine nucleotides;
PC, phosphocreatine;
PDK, pyruvate dehydrogenase kinase;
SDH, succinate dehydrogenase.