World Journal of Cardiovascular Diseases
Vol.05 No.08(2015), Article ID:59156,8 pages
10.4236/wjcd.2015.58025
Arrhythmogenic Ventricular Dysplasia/Cardiomyopathy: Insights from the Rationale of Disease Nomenclature and Clinical Perspectives
Aimé Bonny1,2*, Mohammed A. Talle3, Guy Fontaine4
1Douala Cardiovascular Research Center, University of Douala, Douala, Cameroon
2Clinique Paul Picquet, Sens, France
3Division of Cardiology, Department of Internal Medicine, University of Maiduguri Teaching Hospital/University of Maiduguri, Maiduguri, Nigeria
4Groupe Hospitalier Pitie Salpetriere, Unite de Rythmologie, Paris, France
Email: *aimebonny@yahoo.fr
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 29 May 2015; accepted 23 August 2015; published 26 August 2015
ABSTRACT
“Arrhythmogenic right ventricular dysplasia” (ARVD), a heart muscle disorder characterized by the presence of fibro-fatty tissue and ventricular electrical vulnerability related to sudden death, was first described in 1977 by a French team. Since then, other terms such as “arrhythmogenic right ventricular cardiomyopathy” (ARVC), “arrhythmogenic cardiomyopathy” (AC), “left-dominant arrhythmogenic cardiomyopathy” (LDAC), and “arrhythmogenic left ventricular dysplasia” (ALVD) have been introduced. These changes in nomenclature of the same disease entity are based on different explanations of pathomorphologic patterns. The dysplasia theory claims cardiac growth “maldevelopment” whereas the cardiomyopathy has been seen as an atrophy from acquired injury (myocyte death) and repair (fibrofatty replacement). The other area of divergent opinion is with regards to involvement of both ventricles rather than being an isolated right ventricular anomaly that may result in increased likelihood of diagnosing the concealed form manifesting with predominant left ventricular arrhythmias. Multiple line of evidences support common disease pathways: Presence of fibro-fatty and superimposed myocarditis, desmosome mutations and malfunction. These compelling data regarding the heart growth, and pathological, clinical, phenotype/ genotype correlates have advanced our understanding of arrhythmogenic ventricular dysplasia/ cardiomyopathy and increased the diagnostic accuracy as well as providing an avenue for future development of new mechanism-based therapies.
Keywords:
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy, Arrhythmogenic Cardiomyopathy, Ventricular Arrhythmia, Sudden Cardiac Death

1. Introduction
Arrhythmogenic right ventricular dysplasia (ARVD), an inherited arrhythmogenic disease of heart muscle that can lead to sudden cardiac death (SCD), has been variously named as arrhythmogenic right ventricular cardiomyopathy (ARVC), arrhythmogenic cardiomyopathy (AC), “left-dominant arrhythmogenic cardiomyopathy” (LDAC), “arrhythmogenic left ventricular dysplasia” (ALVD). The conflicting nomenclature associated with this disease entity is rather unparalleled [1] . After the discovery of the autosomal recessive pattern of transmission in the Greek island of Naxos, the first gene (plakoglobin) associated with the development of ARVD was identified. This was followed by the discovery of multiple other mutations affecting the desmosomal proteins. Defective desmosomal proteins may result in impaired mechanical coupling between individual myocardial cells, leading to myocyte uncoupling especially under conditions that increase myocardial strain [2] . The resulting non-specific inflammatory and fibrotic responses to injury, as well as adipocytosis may be similar to that seen in other forms of myocardial damage. Although it is widely agreed that the morphologic substrate of ARVD/C is the presence of fibro-fatty tissue predominantly involving predominantly involving the right ventricle (Figure 1), its pathogenic mechanisms are yet to be fully elucidated. The plethora of academic discourse about nomenclature of ARVD/C from its discovery to date is hardly reflected in any other disease entity in the medical literature. We discuss herein the rationale of this multiple nosology and its clinical impact. The broader term “arrhythmogenic ventricular dysplasia/cardiomyopathy” (AVD/C) will be used when appropriate to name ARVD, ARVC, AC, LDAC or ALVD.
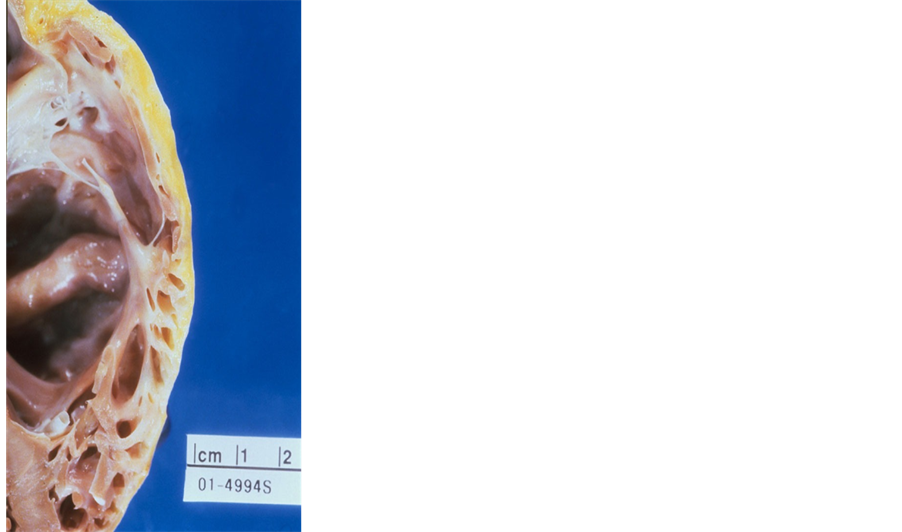
Figure 1. Gross pathology in ARVD/C patient. An evidence of fibro-fatty tissue with thinning and enlargement of the RV wall. A fatty infiltration is seen in a portion of the RV wall. ARVD/C = arrhythmogenic right ventricular dysplasia/cardiomyopathy, RV = right ventricle.
2. History of Naming the Disease
During a surgical procedure to map and treat ventricular tachycardia (VT) at the Hôpital de La Salpêtrière [3] , arrhythmogenic right ventricular dysplasia was first recognized as a “trouble in development” (dysontogenetic mechanism) by Fontaine’s team (1977 in a book chapter). Fontaine et al. first described VT originating from the right ventricle (RV) rather than the usual scarred regions of the left ventricular (LV) serving as the substrate/ origin for VT. They identified an interesting, but unexpected, low amplitude signals during epicardial ventricular mapping that consistently occurred after the end of each QRS complex on the surface electrocardiogram [3] . Later, a similar signal was also observed as a notch at the end of right precordial QRS complexes (Figure 2), which is now recognised as an epsilon wave [4] . In 1982, the same French team first described the clinical entity of this arrhythmogenic disease of the RV in 24 young patients with resistant VT and preserved LV function [5] . Among these patients, 6 had 2-D echocardiograms confirming enlarged RV dimensions. Since then, compelling data regarding pathological, clinical, diagnostic, phenotype/genotype correlates, and outcomes of this disease entity have been published.
Eleven years after the original report by Fontaine et al., a group of pathologist at Padua reported in the Veneto region, similar right ventricular pathologies in 12 of 49 hearts of young adults who died suddenly, with most death occuring during exertion [6] . They observed a progressive loss of myocardium; lipomatous or fibro-lipomatous replacement; and foci of inflammation, degeneration, and necrosis predominantly in the RV. Because cardiac pathologies were not previously reported in these young adults, the findings described were attributed to an unknown mechanism, and they used the term “right ventricular cardiomyopathy” to describe this disease entity. In 1990, these authors reported 6 competitive athletes presenting with fatal arrhythmias arising from the RV [7] . The term arrhythmogenic right ventricular cardiomyopathy (ARVC) was first used by Nava and his colleagues (an Italian group) in 1992 when they described the clinical profile of a concealed form of this disease entity presenting with apparently idiopathic ventricular arrhythmias [8] . Interestingly, the Italian group first used the term “cardiomyopathy” in January 1988 [6] , and thereafter they reverted to the term “dysplasia” in November 1988 [9] . Both reports included patients from the same disease cohort with fatty and fibro-fatty replacement of RV free wall being the hallmark of the disease.
In 2007, Sen-Chowdhry et al. from a London school introduce the term “arrhythmogenic cardiomyopathy” as they found a left-dominant arrhythmogenic cardiomyopathy (LDAC) where LV involvement may precede the onset of significant right ventricular dysfunction [10] . Importantly, they also demonstrated desmosomal mutations and the presence of fibrosis and fat in LDAC to be similar in proportion as obtained in ARVD/C [11] .
In 2015, Lopez-Ayala et al. proposed the term “arrhythmogenic left ventricular dysplasia” (ALVD) [12] , affirming that contrary to previous knowledge, the left ventricle is involved not only in the later stages, but also to a great extent in the “concealed” course or, as a distinct entity of the same disease.

Figure 2. Epsilon wave in patient with ARVD/C. The deflection inscribed at the end of QRS wave in right precordial leads represent the area of slow ventricular conduction due to fibrofatty replacement. ECG = Electrocardiogram, ARVD/C = arrhythmogenic right ventricular dysplasia/cardiomyopathy.
Whether naming the same disease entity differently by each school is based on scientific evidence will be discussed herein.
3. Rationale of Each Terminology
3.1. The “Dysplasia” Theory
The aspect of the surgical cut of the RV free wall showing minor myocardium covered by extensive amount of fat led the Fontaine’s group to hypothesize that this structure was the result of a trouble in development and gave the name of “dysplasia”. Dysplasia is defined as an interruption in the ontogenetic development of any organ. The disontogenetic theory claims that the absence of myocardium, with the apposition of epicardium to endocardium, is considered to be the consequence of a congenital aplasia or hypoplasia of the RV wall, leading to a parchment-like appearance [13] -[17] . Accordingly, the disease should be regarded as a gross cardiac structural defect, a “trouble in development”, present at birth and considered a congenital heart disease [3] -[18] . Fontaine decided that the term dysplasia or dystrophy was most appropriate because these cardiac pathologies occurred mostly in young patients and were likely the result of abnormal postnatal development [1] . James et al. discussed this concept from a paediatric case with major absence of the RV myocardium and suggested a phenomenon of cardiomyocyte apoptosis [19] . The proliferation of cardiomyocytes may play an unrecognized role during the period of developmental heart growth between birth and adolescence. Mollova et al. tested this hypothesis by examining the cellular growth mechanisms of the left ventricle in a set of healthy hearts from humans aged 0 - 59 years [20] . The percentages of cardiomyocytes in mitosis and cytokinesis were highest in infants, decreasing to low levels by 20 years. This finding shows that cardiomyocyte proliferation contributes to developmental heart growth in young humans, and corroborates the results of Roberts JT et al. who in 1941, found that human cardiomyocytes continue to develop, mature, and regenerate postnatally, and only reach adult size at ages from 10 to 20 years [21] . Thus, presence of fibro-fatty tissue can be seen as the results of ontogenesis “maldevelopment” with superimposed virus invasion.
3.2. The “Cardiomyopathy” Theory
“Cardiomyopathy” is a heart muscle disorder which is usually associated with dilatation of a ventricle and an impairment of contractility. Usually, both ventricles are involved. In this regard, ARVC which can affect the LV in the vast majority of cases looks like a cardiomyopathy [22] -[26] . More importantly, histological examination commonly reveals islands of surviving myocytes interspersed with fibrous and fatty tissue [23] . The myocardial atrophy appears to be the consequence of acquired injury (myocyte death) and repair (fibrofatty invasion), mediated by patchy myocarditis [23] . Clusters of myocytes can also be seen to be dying at histology, providing evidence of the acquired nature of myocardial atrophy. These changes are frequently associated with inflammatory infiltrates, which probably plays a major part in triggering life-threatening arrhythmias [27] -[29] . Whether the inflammatory cells are a reaction to cell death (either necrosis or apoptosis) or the consequence of infective or immune mechanisms is not clear [30] . To support this infective pathogenesis, researchers emphasize that a genetic predisposition and/or susceptibility to viral infections and immune reactions, as well as a genetically determined spontaneous cell death (apoptosis), could be an explanation [31] [32] . The Padua group also argued that the acquired nature of the disease (possibly postnatal phenotype expression) is corroborated by the age range of the affected patients at 15 to 65 years [22] .
The London school adopted the broader term “arrhythmogenic cardiomyopathy” (AC) [10] . Of 200 probands and relatives satisfying task force or modified diagnostic criteria for ARVD/C, desmosomal mutations were identified in 39 individuals from 20 different families. In >80%, ECG, rhythm monitoring, and/or gadolinium- enhanced cardiovascular magnetic resonance were suggestive of LV involvement, the extent of which often was marked among individuals with chain-termination mutations and/or desmoplakin disease. They found 3 patterns of disease expression that included a left dominant, with early and prominent LV manifestations (arrhythmias originating from left ventricle) and relatively mild right-sided disease. For these authors, the prevalence of LV involvement is estimated at 15% to 93%, being a late complication of classic disease or occurring early in the other subtypes, either in parallel with or predominating over RV disease [33] .
Nondesmosomal genes are also involved in a minority, but substantial cases (up to 60%) of AC, harbouring variants in gene mutations encoding desmosomal proteins (plakoglobin, desmoplakin, plakophilin-2, desmoglein-2, and desmocollin-2). Although histopathological and functional predominance of right ventricular abnormalities are frequently found, the left ventricle is affected in most patients as well (Figure 3). Moreover, at the molecular level, both ventricles are affected by altered expression and distribution of intercalated disk proteins [34] . Because of these considerations, AC has recently become the favored terminology of this group of frequently difficult-to-distinguish disorders.
4. Pathogenesis Revisited
Accumulating evidence suggests that gene mutation-related changes or distribution of desmosomal proteins give rise to altered expression and distribution of other intercalated disk proteins, including gap junction [35] and ion channel proteins, resulting in mechanical and electric uncoupling and slow conduction, and conduction block. These factors favor reentry in the early concealed stage of the disease. It is very likely that fibrofatty alteration responsible for altered tissue architecture follows in the later overt stage of the disease. This sequence of changes is strongly supported by identification of altered distribution of the desmosomal protein plakoglobin in histologically-unaffected left ventricular and septal tissue in patients with proven AC [34] . In addition, ventricular fibrillation and sudden death may occur in the early concealed stage of the disease even in the absence of identifiable fibrofatty changes [36] . In contrast, in the overt stage, the modified tissue architecture characterized by interconnecting vital myocardial bundles embedded in fibrotic tissue contributes importantly to further aggravation of activation delay by lengthening of conduction pathways and load mismatch at pivotal points [23] [37] - [39] . This highly arrhythmogenic substrate frequently is the cause of monomorphic ventricular tachycardia with left bundle branch block morphology.
The major metabolic differences between embryonic and adult cardiomyocytes are: 1) embryonic cardiomyocytes use mostly glycolysis for energy production, and 2) adult cardiomyocytes produce most energy via fatty acid oxidation (FAO) but retain capacity to readily switch to glucose or other substrate utilization when fatty acid is not available or FAO is compromised [40] [41] . Therefore, using a specific metabolic milieu, Kim et al. reproduced in vitro, exaggerated lipogenesis and apoptosis in mutant plakoglobin (PKP2) with induced pluripotent stem cells-derived cardiomyocytes (iPSC-CMs), thus revealing crucial pathogenic insights that metabolic derangement in adult-like metabolic milieu underlies ARVD/C pathologies [42] .




Figure 3. Histology of an AC patient. A young American top model died suddenly at age 17 years old (document provided by Dr Myeburg, Miami, FL, USA). The histology of her heart revealed abnormal architecture of both ventricles. Left ventricle (left side): Adipocytes infiltration and fibrosis interfere with normal cardiomyocytes. Right ventricle (right side): Trabeculations of endocardia and smooth epicardia constituting by adipocytes infiltration and islands of survived cardiomyocytes (top). A magnification of epicardia shows that survived cardiomyocytes are predominantly localised in the sub-epicardia layer. AC―Arrhythmogenic cardiomyopathy.
Arrhythmogenic ventricular dysplasia/cardiomyopathy (AVD/C) is characterize by the “malfunction” of des- mosomes which are cell-cell adhesion organelles. They are particularly abundant in heart and skin, tissues that normally experience mechanical stress. Patients with this arrhythmogenic ventricular disease often exhibit disease flares in response to stress or exercise, emphasizing the importance of biomechanical determinants of disease [43] . However, the relationship between changes in biomechanical properties of the myocardium produced by desmosomal mutations and heart rhythm abnormalities is poorly characterized. One of most recent and exciting avenue is the discovery of marked subcellular redistribution of plakoglobin, connexion 42, and Nav1.5 without changes in their total cellular content in a zebrafish model of AC [44] . These observations pinpoint aberrant trafficking of intercalated disc proteins as a central mechanism in AC myocyte injury and electrical abnormalities [44] . In general, membrane cellular abnormal trafficking is being recognised as a major arrhythmogenic mechanism of sudden cardiac death [45] .
Inflammation is a major factor in the pathogenesis of lethal arrhythmias in AVD/C. The deleterious effect of an inflammatory process has been originally suspected in 1995 from the study of pathological material of patients who experienced a worsening of AVD/C [46] . The presence of cardiotropic viruses has already been reported [47] , and a link between inflammation and ventricular arrhythmogenesis has been recently observed [48] . Pathologic myocardium displays a particular susceptibility to acute superimposed myocarditis [46] . Indeed, Lopez-Ayala et al. analysed the genetic basis of myocarditis in ARVD and ALVD affected patients and unaffected mutation-carrying relatives. Myocarditis clustered in families bearing certain mutations [12] .
5. Clinical Implications and Perspectives
Inhibition of abnormal peroxisome proliferator activated receptor gamma (PPARγ) activation can prevent all AVD/C pathologies, and the elimination of reactive oxygen species can reduce apoptosis of AVD/C cardiomyocytes [42] . Aberrant membrane cell trafficking of intercalated disc proteins is a central mechanism of cardiomyocyte injury and related arrhythmogenesis in AVD/C [44] . These two observations highlights potential new therapeutic targets (pharmacologic or gene therapy) to either prevent or forestall disease progression that can be exploited in future researches [46] .
Since the discovery of the disease in 1977 and its full clinical description in 1988, there are many advances in the understanding of the disease morphology, diagnostic criteria, phenotype/genotype correlates, and risk prediction for its catastrophic complication of SCD. Naming the disease as ARVD, ARVC, AC, LDAC or ALVD did not substantially change our current management approaches. However, the knowledge of early LV involvement with minimal structural changes together with overt arrhythmic manifestations will reinforce our diagnostic decision-making especially in teasing out the protean aetiologies for left ventricular arrhythmias.
6. Conclusion
AVD/C which is an inherited desmosomal and nondesmosomal pathology affecting both ventricles is characterized by life-threatening arrhythmias and premature death. The diagnosis remains challenging and requires a multifaceted approach for patient evaluation. The multiplicity of nomenclature for the disease entity might be confusing but also shows that our knowledge on the morphology and pathophysiology of this disease is far from being completely elucidated. However, compelling existing data which underlies introduction of new nosologies have advanced our understanding of the pathophysiology and molecular basis of the disease, yielding an avenue for developing new mechanism-based therapies that is yet to be exploited.
Cite this paper
AiméBonny,Mohammed A.Talle,GuyFontaine,11, (2015) Arrhythmogenic Ventricular Dysplasia/Cardiomyopathy: Insights from the Rationale of Disease Nomenclature and Clinical Perspectives. World Journal of Cardiovascular Diseases,05,218-226. doi: 10.4236/wjcd.2015.58025
References
- 1. Fontaine, G. and Chen, V. (2014) Arrhythmogenic Right Ventricular Dysplasia Back in Force. American Journal of Cardiology, 113, 1735-1739.
http://dx.doi.org/10.1016/j.amjcard.2014.03.001 - 2. Calkins, H. (2015) Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. The Three Decades of Progress. Circulation Journal, 79, 901-913.
http://dx.doi.org/10.1253/circj.CJ-15-0288 - 3. Fontaine, G., Guiraudon, G., Frank, R., Vedel, J., Grosgogeat, Y., Cabrol, C., et al. (1977) Stimulation Studies and Epicardial Mapping in Ventricular Tachycardia: Study of Mechanisms and Selection for Surgery. In: Kulbertus, H.E., Ed., Reentrant Arrhythmias, MTP Pub, Lancaster, 334-350.
- 4. Fontaine, G., Guiraudon, G. and Frank, R. (1978) Intramyocardial Conduction Defects in Patients Prone to Ventricular Tachycardia. I—The Postexcitation Syndrome in Sinus Rhythm p39e55, II—A Dynamic Study of the Postexcitation Syndrome p56-66, III—The Post-Excitation Syndrome during Ventricular Tachycardia. In: Sandoe, E., Julian, D.G., Bell, J.W., Eds., Management of Ventricular Tachycardia, Role of Mexiletine, Excerpta Medica Pub, Amsterdam, 67- 79.
- 5. Marcus, F.I., Fontaine, G., Guiraudon, G., Frank, R., Laurenceau, J.L., Malergue, M.C., et al. (1982) Right Ventricular Dysplasia: A Report of 24 Cases. Circulation, 65, 384-399.
http://dx.doi.org/10.1161/01.CIR.65.2.384 - 6. Thiene, G., Nava, A., Corrado, D., Rossi, L. and Pennelli, N. (1988) Right Ventricular Cardiomyopathy and Sudden Death in Young People. New England Journal of Medicine, 318, 129-133.
http://dx.doi.org/10.1056/NEJM198801213180301 - 7. Corrado, D., Thiene, G., Nava, A., Rossi, L. and Pennelli, N. (1990) Sudden Death in Young Competitive Athletes: Clinicopathologic Correlation in 22 Cases. American Journal of Medicine, 89, 588-596.
http://dx.doi.org/10.1016/0002-9343(90)90176-E - 8. Nava, A., Thiene, G., Canciani, B., et al. (1992) Clinical Profile of Concealed form of Arrhythmogenic Right Ventricular Cardiomyopathy Presenting with Apparently Idiopathic Ventricular Arrhythmias. International Journal of Cardiology, 35, 195-206.
http://dx.doi.org/10.1016/0167-5273(92)90177-5 - 9. Nava, A., Thiene, G., Canciani, B., et al. (1988) Familial Occurrence of Right Ventricular Dysplasia: A Study Involving Nine Families. Journal of the American College of Cardiology, 12, 1222-1228.
http://dx.doi.org/10.1016/0735-1097(88)92603-4 - 10. Sen-Chowdhry, S., Syrris, P., Ward, D., Asimaki, A., Sevdalis, E. and McKenna, W.J. (2007) Clinical and Genetic Characterization of Families with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Provides Novel Insights into Patterns of Disease Expression. Circulation, 115, 1710-1720.
http://dx.doi.org/10.1161/CIRCULATIONAHA.106.660241 - 11. Sen-Chowdhry, S., Syrris, P., Prasad, S.K., Hughes, S.E., Merrifield, R., Ward, D., Pennell, D.J. and McKenna, W.J. (2008) Left-Dominant Arrhythmogenic Cardiomyopathy: An Under-Recognized Clinical Entity. Journal of the American College of Cardiology, 52, 2175-2187.
http://dx.doi.org/10.1016/j.jacc.2008.09.019 - 12. Lopez-Ayala, J.M., Pastor-Quirante, F., Gonzalez-Carrillo, J., Lopez-Cuenca, D., Sanchez-Munoz, J.J., Oliva-Sandoval, M.J. and Gimeno, J.R. (2015) Genetics of Myocarditis in Arrhythmogenic Right Ventricular Dysplasia. Heart Rhythm, 12, 766-773.
http://dx.doi.org/10.1016/j.hrthm.2015.01.001 - 13. McKenna, W.J., Thiene, G., Nava, A., Fontaliran, F., Blomstrom-Lundqvist, C., Fontaine, G., et al. (1994) Diagnosis of Arrhythmogenic Right Ventricular Dyspla-sia/Cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. British Heart Journal, 71, 215-218.
http://dx.doi.org/10.1136/hrt.71.3.215 - 14. Marcus, F.I., McKenna, W.J., Sherrill, D., Basso, C., Bauce, B., Bluemke, D.A., et al. (201o) Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: Proposed Modification of the Task Force Criteria. Circulation, 121, 1533-1541.
http://dx.doi.org/10.1161/CIRCULATIONAHA.108.840827 - 15. Frank, R., Fontaine, G., Vedel, J., Mialet, G., Sol, C., Guiraudon, G., et al. (1978) Electrocardiologie de quatre cas de dysplasie ventriculaire droite arythmogene. Archives des Maladies du Coeur, 71, 963-972.
- 16. Osler, W.M. (1905) The Principles and Practice of Medicine. 6th edition, D’Appleton & Co., New York, 820.
- 17. Castleman, B. and Towne, V.W. (1952) Presentation of Case 38201. The New England Journal of Medicine, 246, 785- 790.
http://dx.doi.org/10.1056/NEJM195205152462007 - 18. Vecht, R.J., Carmichael, J.S., Gopal, R. and Philip, G. (1979) Uhl’s anomaly. British Heart Journal, 41, 676-682.
http://dx.doi.org/10.1136/hrt.41.6.676 - 19. James, T.N., Nichols, M.M., Sapire, D.W., DiPatre, P.L. and Lopez, S.M. (1996) Complete Heart Block and Fatal Right Ventricular Failure in an Infant. Circulation, 93, 1588-1600.
http://dx.doi.org/10.1161/01.CIR.93.8.1588 - 20. Mollova, M., Bersell, K., Walsh, S., Savla, J., Das, L.T., et al. (2013) Cardiomyocyte Proliferation Contributes to Heart Growth in Young Humans. Proceedings of the National Academy of Sciences of the United States of America, 110, 1446-1451.
http://dx.doi.org/10.1073/pnas.1214608110 - 21. Roberts, J.T. and Wearn, J.T. (1941) Cell Proliferation during Cardiac Growth. American Journal of Cardiology, 21, 617-633.
- 22. Basso, C., Corrado, D., Marcus, F.I., Nava, A. and Thiene, G. (2009) Arrhythmogenic Right Ventricular Cardiomyopathy. The Lancet, 373, 1289-1300.
http://dx.doi.org/10.1016/S0140-6736(09)60256-7 - 23. Basso, C., Thiene, G., Corrado, D., Angelini, A., Nava, A. and Valente, M. (1996) Arrhythmogenic Right Ventricular Cardiomyopathy: Dysplasia, Dystrophy or Myocarditis? Circulation, 94, 983-991.
http://dx.doi.org/10.1161/01.CIR.94.5.983 - 24. Thiene, G. and Basso, C. (2001) Arrhythmogenic Right Ventricular Cardiomyopathy. An Update. Cardiovascular Pathology, 10, 109-111.
http://dx.doi.org/10.1016/S1054-8807(01)00067-9 - 25. Corrado, D., Basso, C., Thiene, G., et al. (1997) Spectrum of Clinicopathologic Manifestations of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: A Multicenter Study. Journal of the American College of Cardiology, 30, 1512-1520.
http://dx.doi.org/10.1016/S0735-1097(97)00332-X - 26. Groeneweg, J.A. and Hauer, R.N. (2012) Noninvasive Parameters for Evaluation of Activation Delay in Arrhythmogenic Cardiomyopathy. Circulation: Arrhythmia and Electrophysiology, 5, 453-455.
http://dx.doi.org/10.1161/CIRCEP.112.971895 - 27. Panidis, I.P., Greenspan, A.M., Mintz, G.S. and Ross, J. (1985) Inducible Ventricular Fibrillation in Arrhythmogenic Right Ventricular Dysplasia. American Heart Journal, 110, 1067-1069.
http://dx.doi.org/10.1016/0002-8703(85)90213-3 - 28. De Bakker, J.M., van Capelle, F.J., Janse, M.J., Wilde, A.A., Coronel, R., Becker, A.E., et al. (1988) Reentry as a Cause of Ventricular Tachycardia in Patients with Chronic Ischemic Heart Disease: Electrophysiologic and Anatomic Correlation. Circulation, 77, 589-606.
http://dx.doi.org/10.1161/01.CIR.77.3.589 - 29. James, T.N. (1994) Normal and Abnormal Consequences of Apoptosis in the Human Heart: From Postnatal Morphogenesis to Paroxysmal Arrhythmias. Circulation, 90, 556-573.
http://dx.doi.org/10.1161/01.CIR.90.1.556 - 30. Tamaru, Y., Miyawaki, T., Iwai, K., Tsuji, T., Nibu, R., Yachie, A., et al. (1993) Absence of Bcl-2 Expression by Activated CD45RO+T Lymphocytes in Acute Infectious Mononucleosis Supporting Their Susceptibility to Programmed Cell Death. Blood, 82, 521-527.
- 31. Mallat, Z., Tedjui, A., Fontaliran, F., Frank, R., Durigon, M. and Fontaine, G. (1996) Evidence of Apoptosis in Arrhy- thmogenic Right Ventricular Dysplasia. The New England Journal of Medicine, 335, 1190-1196.
http://dx.doi.org/10.1056/NEJM199610173351604 - 32. Sen-Chowdhry, S. and McKenna, W.J. (2010) Reconciling the Protean Manifestations of Arrhythmogenic Cardiomy- opathy. Circulation: Arrhythmia and Electrophysiology, 3, 566-570.
http://dx.doi.org/10.1161/CIRCEP.110.960237 - 33. Asimaki, A., Tandri, H., Huang, H., Halushka, M.K., Gautam, S., Basso, C., Thiene, G., Tsatsopoulou, A., Protonotarios, N., McKenna, W.J., Calkins, H. and Saffitz, J.E. (2009) A New Diagnostic Test for Arrhythmogenic Right Ventricular Cardiomyopathy. The New England Journal of Medicine, 360, 1075-1084.
http://dx.doi.org/10.1056/NEJMoa0808138 - 34. Kaplan, S.R., Gard, J.J., Protonotarios, N., Tsatsopoulou, A., Spiliopoulou, C., Anastasakis, A., Squarcioni, C.P., McKenna, W.J., Thiene, G., Basso, C., Brousse, N., Fontaine, G. and Saffitz, J.E. (2004) Remodeling of Myocyte Gap Junctions in Arrhythmogenic Right Ventricular Cardiomyopathy Due to a Deletion in Plakoglobin (Naxos Disease). Heart Rhythm, 1, 3-11.
http://dx.doi.org/10.1016/j.hrthm.2004.01.001 - 35. Cox, M.G., van der Zwaag, P.A., van der Werf, C., van der Smagt, J.J., Noorman, M., Bhuiyan, Z.A., Wiesfeld, A.C., Volders, P.G., van Langen, I.M., Atsma, D.E., Dooijes, D., van den Wijngaard, A., Houweling, A.C., Jongbloed, J.D., Jordaens, L., Cramer, M.J., Doevendans, P.A., de Bakker, J.M., Wilde, A.A., van Tintelen, J.P. and Hauer, R.N. (2011) Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Pathogenic Desmosome Mutations in Index-Patients Predict Outcome of Family Screening: Dutch Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Genotype-Phenotype Follow-Up Study. Circulation, 123, 2690-2700.
http://dx.doi.org/10.1161/CIRCULATIONAHA.110.988287 - 36. De Bakker, J.M., van Capelle, F.J., Janse, M.J., Tasseron, S., Vermeulen, J.T., de Jonge, N. and Lahpor, J.R. (1993) Slow Conduction in the Infarcted Human Heart. “Zig-zag” Course of Activation. Circulation, 88, 915-926.
http://dx.doi.org/10.1161/01.CIR.88.3.915 - 37. Cabo, C., Pertsov, A.M., Baxter, W.T., Davidenko, J.M., Gray, R.A. and Jalife, J. (1994) Wave-Front Curvature as a Cause of Slow Conduction and Block in Isolated Cardiac Muscle. Circulation Research, 75, 1014-1028.
http://dx.doi.org/10.1161/01.RES.75.6.1014 - 38. Fast, V.G. and Kléber, A.G. (1997) Role of Wavefront Curvature in Propagation of Cardiac Impulse. Cardiovascular Research, 33, 258-271.
http://dx.doi.org/10.1016/S0008-6363(96)00216-7 - 39. Onay-Besikci, A. (2006) Regulation of Cardiac Energy Metabolism in Newborn. Molecular and Cellular Biochemistry, 287, 1-11.
http://dx.doi.org/10.1007/s11010-006-9123-9 - 40. Lopaschuk, G.D., Ussher, J.R., Folmes, C.D., Jaswal, J.S. and Stanley, W.C. (2010) Myocardial Fatty Acid Metabolism in Health and Disease. Physiological Reviews, 90, 207-258.
http://dx.doi.org/10.1152/physrev.00015.2009 - 41. Kim, C., Wong, J., Wen, J., Wang, S., Wang, C., Spiering, S., Kan, N.G., Forcales, S., Puri, P.L., Leone, T.C., Marine, J.E., Calkins, H., Kelly, D.P., Judge, D.P. and Chen, HS. (2013) Studying Arrhythmogenic Right Ventricular Dysplasia with Patient-Specific iPSCs. Nature, 494, 105-110.
http://dx.doi.org/10.1038/nature11799 - 42. Asimaki, A. and Saffitz, J.E. (2012) Gap Junctions and Arrhythmogenic Cardiomyopathy. Heart Rhythm, 9, 992-995.
http://dx.doi.org/10.1016/j.hrthm.2011.11.024 - 43. Asimaki, A., Kapoor, S., Plovie, E., Karin Arndt, A., Adams, E., Liu, Z., James, C.A., Judge, D.P., Calkins, H., Churko, J., Wu, J.C., MacRae, C.A., Kléber, A.G. and Saffitz, J.E. (2014) Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Science Translational Medicine, 6, 240-274.
http://dx.doi.org/10.1126/scitranslmed.3008008 - 44. Delisle, B.P., Anson, B.D., Rajamani, S. and January, C.T. (2004) Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circulation Research, 94, 1418-1428.
http://dx.doi.org/10.1161/01.RES.0000128561.28701.ea - 45. Fontaine, G.H., Andreoletti, L. and Redheuil, A. (2015) Genetics of Myocarditis in Cardiomyopathies. Heart Rhythm, 12, 774-775.
http://dx.doi.org/10.1016/j.hrthm.2015.02.011 - 46. Bowles, N.E., Ni, J., Marcus, F. and Towbin, J.A. (2002) The Detection of Cardiotropic Viruses in the Myocardium of Patients with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Journal of the American College of Cardiology, 39, 892-895.
http://dx.doi.org/10.1016/S0735-1097(02)01688-1 - 47. Bonny, A., Lellouche, N., Ditah, I., Hidden-Lucet, F., Yitemben, M.T., Granger, B., Larrazet, F., Frank, R. and Fontaine, G. (2010) C-Reactive Protein in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy and Relationship with Ventricular Tachycardia. Cardiology Research and Practice, 2010, Article ID: 919783.
http://dx.doi.org/10.4061/2010/919783
NOTES

*Corresponding author.