Open Journal of Biophysics
Vol.07 No.01(2017), Article ID:73592,17 pages
10.4236/ojbiphy.2017.71004
Molecular Docking of Selective Binding Affinity of Sulfonamide Derivatives as Potential Antimalarial Agents Targeting the Glycolytic Enzymes: GAPDH, Aldolase and TPI
Neville Forlemu, Porshaye Watkins, Joseph Sloop
School of Science & Technology (Chemistry), Georgia Gwinnett College, 1000 University Center Lane, Lawrenceville, GA, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: November 10, 2016; Accepted: January 16, 2017; Published: January 19, 2017
ABSTRACT
The parasite Plasmodium falciparum is responsible for the major world scourge malaria, a disease that affects 3.3 billion people worldwide. The development of new drugs is critical because of the diminished effectiveness of current antimalarial agents mainly due to parasitic resistance, side effects and cost. Molecular docking was used to explore structural motifs responsible for the interactions between triose phosphate isomerase (TPI), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and aldolase (ALD) from human and Plasmodium cells with 8 novel sufonylamide derivatives. All the ligands modeled, interact with all three enzymes in the micromolar range. The top ligand (sulfaE) shows a 70-fold increase in selective binding to pfTPI compared to hTPI (dissociation constant-KI of 7.83 μM and 0.177 μM for hTPI and pfTPI respectively), on par with antimalarial drug chloroquine.ALD and GAPDH form complexes with similar binding sites, comprising amino acids of similar chemical properties and polarities. Human TPI and pfTPI bind sulfonamide derivatives using two distinct binding sites and residues. Key residues at the dimer interface of pfTPI (VAL44, SER45, TYR48, GLN64, ASN65, VAL78) form a tight pocket with favorable polar contacts. The affinity with TPI is the most specific, stable, and selective suggesting pfTPI is a candidate for development of antimalarial drugs.
Keywords:
Malaria, Glycolytic Enzymes, AutoDock4.2, Dissociation Constant, Triosephosphate Isomerase, Binding Affinity

1. Introduction
Global mapping data and the World Health Organization indicate that malaria continues to be a major challenge with about 3.3 billion people at risk of exposure (Kehr and Guerra) [1] [2] . According to the 2015 WHO report, about half a million people die annually due to complications from malaria (WHO 2015 report) [3] [4] . The eradication of malaria’s debilitating effects (coma, brain damage, loss of muscle function) through development of insecticides, mosquito vector disruptions, and development of new antimalarial therapeutic agents remains a major priority worldwide [4] [5] . The diminishing number of effective antimalarial drugs, and increasing parasitic resistance to all current antimalarial agents including artemisinins are major reasons that continue to spur the search of cost effective and parasite resistant free analogues [5] [6] [7] [8] .
The main causative agent of malaria, the Plasmodium parasite, places a more than 100 fold demand on glucose requirements for parasitized red bloods relative to uninfected host cells [9] . The absence of a viable Krebs cycle in these parasites and sole dependence on glycolysis for energy needs therefore makes glycolytic pathway enzymes potential targets for antimalarial chemotherapies [10] [11] . We hypothesize that high affinity analogues that can form discriminatory complexes between glycolytic enzymes of parasitic and human species can serve as selective inhibitors of Plasmodium parasite and therefore fight malaria. Structural data available from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB-PDB) show that the quaternary structures of glycolytic enzymes are highly conserved across species, with similar catalytic domains [12] [13] . This similarity in sequences and structural domains is therefore a major concern for the development of ligands that target glycolytic enzymes due to the potential toxicity of the developed ligands because of indiscriminate interactions with glycolytic enzymes from both species. There is evidence however, of subtle yet significant differences between the crystal structures of Plasmodium falciparum and human glycolytic enzymes [13] . According to Read and Dunn the cofactor binding pocket of Plasmodium lactate dehydrogenase is wider when compared to the human isoform [14] [15] . This is due to a five-residue insertion in sequence for the loop that closes the active site during catalysis. Cameron and coworkers have developed a number of azole-based compounds that possess antimalarial activity, and selectively inhibit Plasmodium LDH with submicromolar affinities, based on such molecular level differences [16] . Similar structural differences have also been reported for the enzyme glyceraldehyde-3-phospahe dehydrogenase (GAPDH) [17] . Specifically, a two-residue insertion (Lys-Gly) creates a bulge at the S-loop that lines the cofactor-binding cavity leading to a more constricted opening in pfGAPDH relative to human enzyme. The X-ray crystal structure of pfGAPDH also serendipitously revealed a ligand-binding fold proximal to the pfGAPDH active site [17] [18] . Triose phosphate isomerase (TPI) is another key dimeric enzyme that speeds up the final investment phase of glycolysis and shows structural differences between pfTPI and human TPI. The conserved serine amino acid residue at position 96 close to the active site residues (K12, H95 and E165) is substituted by Phenylalanine (Phe) in pfTPI sequence [19] .
It is therefore essential to determine whether such differences and other unique structural motifs can be used to develop inhibitors that selectively target Plasmodium parasite enzymes, without harming the human host cells. In this study we have used molecular modeling tools to determine the binding affinity and binding modes of three glycolytic enzymes; triosephosphate isomerase (TPI), aldolase (ALD), and glyceraldehyde phosphate dehydrogenase (GAPDH) with 8 novel sulfonamide ligand derivatives. This study will use molecular modeling to identify selective differences in molecular recognition patches observed in the interactions of three glycolytic enzymes from humans and the Plasmodium parasites with sulfonamide derivatives. The specific questions we address include: 1) Which glycolytic enzymes show a strong and selective affinity with the novel ligands? 2) What are structural differences between the binding domains and residues responsible for interactions? 3) How do the binding affinities of new analogues compare with common antimalarial agents? 4) Which sulfonamide derivatives boost selectivity, based on the binding affinities and dissociation constants?
1.1. Molecular Systems
1.1.1. The Glycolytic Pathway Enzymes
The glycolytic pathway is the catabolic process involving a series of enzymes that converts one molecule of glucose into two molecules of pyruvate with the release of ATP for required by cells (Scheme 1).
This study will focus on three glycolytic enzymes that catalyse steps 4, 5 and 6 of the glycolytic pathway (Scheme 1). Fructose-1,6-bisphosphate aldolase (ALD) is a homotetrameric enzyme that catalyses the aldol cleavage of fructose-1,6-bisphosphate (F1,
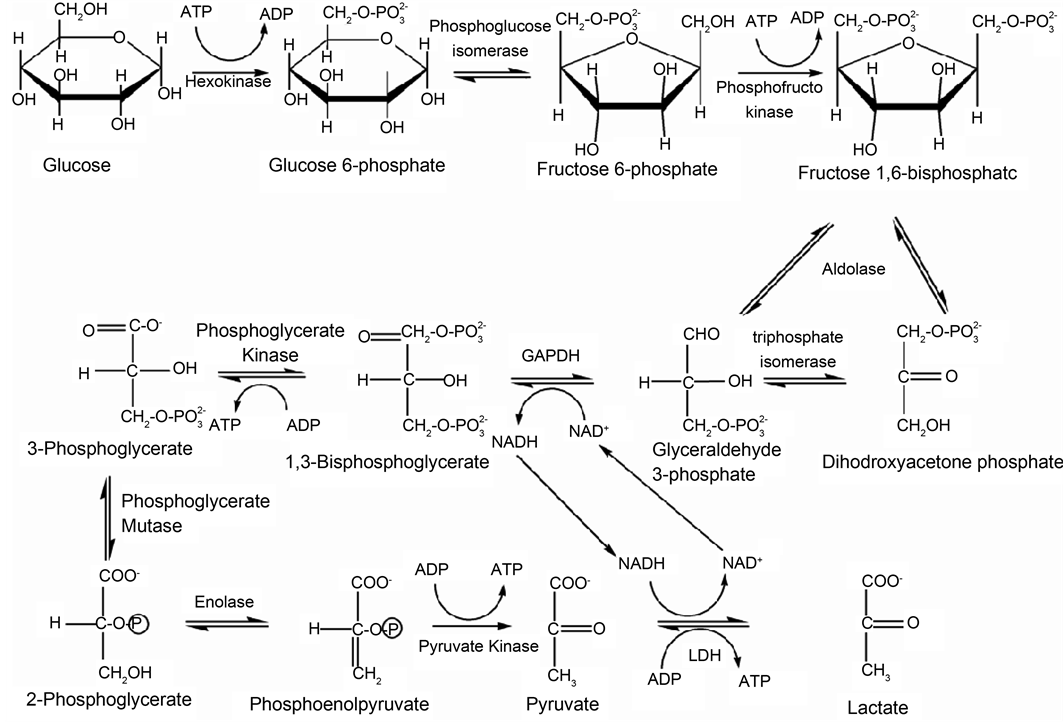
Scheme 1. The glycolytic pathway; metabolic breaking down of glucose to lactate.
6BP) to two triose phosphates, glyceraldehyde-3-phosphate or glyceraldehyde and dihydroxyacetone phosphate (Scheme 1) [20] . The tertiary structures of human aldolase (PDB code 1ALD) and Plasmodium aldolase (PDB code 1A5C) were obtained from the protein data bank. Both structures have a sequence identity of 54% and the RMSD between 317 atom pairs is 0.665 Å [21] . Despite structural similarity (Figure 1(a)), key hydrophobic surface folds and single residue deletion cause a reduction in surface area of Plasmodium aldolase, compared to human aldolase [22] .
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plays an important role in glycolysis and gluconeogenesis by reversibly catalysing the oxidation and phosphorylation of glyceraldehydes-3-phosphate (G3P) to the first energy-rich intermediate 1,3- bisphosphoglycerate (1,3BPG) in glycolysis (Scheme 1) [23] . The tertiary structures of human GAPDH (hGAPDH) and Plasmodium GAPDH (pfGAPDH) were downloaded from the protein data bank with accession codes of 1ZNQ and 1YWG [24] [25] . Both molecules are homotetrameric enzymes of about 150 kDa in molecular mass. In most species, the primary amino acid sequence and physico-chemical properties of GAPDH are conserved, but some active site and cofactor binding site residues differ (Figure 1(b)).
TPI is a key dimeric enzyme that speeds up the final investment phase of glycolysis (Scheme 1). The X-ray crystal structures of hTPI and pfTPI have been determined to atomic resolution and were also downloaded from the RCSB Protein Data Bank, with accession codes 4POC (Roland, 2015) and 2VFI, respectively (Gayathri, 2009). HTPI and PFTPI share a sequence identity of 42%. Despite the 58% difference in sequence

Figure 1. (a) Plasmodium aldolase (red) superimposed on human aldolase (cyan); (b) pfGAPDH (red) superimposed on hGAPDH (cyan); (c) pfTPI (red) and hTPI (cyan) structures aligned. hTPI-Yellow (A46, I48, S96) and pfTPI red (V46, Y48, F96). Active site residues (K12, H95 and E165) in Blue. Different residues in both sequences highlighted.
identity, the three dimensional structure of both molecules have similar structural folds. For example, a root mean square deviation (RMSD) of 0.825 Å is obtained when both enzymes are structurally aligned (Figure 1(c)). HTIM and PFTIM do, however, have some amino acid residues located in key binding motifs with different side chain polarities (Figure 1). Position 96 that is close to TIM active site residues (K12, H95 and E165) in many TIM sequences is usually occupied by serine residue (Ser), but it is replaced by Phenylalanine (Phe) in PFTIM sequence [19] .
1.1.2. Sulfonamide Derivatives
Aniline and sulfonamide based drugs like sulfanilamide have historically been used to treat bacterial and yeast infections. This is because they target enzymatic reactions in the foliate metabolic pathway producing cellular cofactors important for amino acids and DNA synthesis. For example, sulfadoxine has been shown to interfere with the foliate metabolism, by inhibiting the enzyme dihydropteroate synthase [26] . The combination of sulfadoxine and pyrimethamine has been discontinued due to parasitic resistance. Herein we have developed and tested the selectivity and binding affinity of novel fluorinated sulfonamide derivatives, with branched architectures that show improved affinity compared to sulfanilamide. The derivatives have the same basic structure, but differ from one another based the position of polar fluorine, methoxy and alkyl functional groups. The three-dimensional structures of the ligands tested were built using Guass View, and the geometry optimized with Gaussian 09 using a B3LY0/6-311g basis set [27] . The structures for the ligands tested are shown in Table 1.
2. Computational Methods
The three-dimensional structures of the enzymes used were refined prior to docking using Pymol and Chimera [28] [29] . This involved adding deleted atoms from crystal structures, removing unnecessary ligands and relaxing the enzyme structures. The molecular modeling software AutoDock 4.2 was used to screen the interactions of eight sulfonamide derivatives with the glycolytic enzymes [30] [31] . This blind docking was used to map different binding pockets or sites and determine residues involved in complexes, and binding affinities of the enzymes (Aldolase, GAPDH and TPI) with the ligands. The AutoDockTools (ADT) suite was used to prepare the ligand and receptor structures, add appropriate Gasteiger charges, identify and modify ligand rotatable bonds (Morris, 2009) [32] . A grid-based method was used to enhance the quick evaluation of the binding energy of conformations of the complexes formed. The grid boxes were centered using coordinates of a virtual center of mass atom for the enzymes. Each grid box had dimensions of 120 Å × 120 Å × 120 Å, large enough to allow for sampling of binding domains or pockets on the entire protein surface by the probe ligand atoms. The Lamarckian genetic algorithm was used in configuration space search and binding energy determination of the complexes. The affinity of the docked complexes was described using dissociation constants (Ki) (Equation (1)) and binding energy based on the semiempirical force field expression as described in Equation (2) [32] .
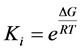 (1)
(1)

Table 1. Structures of the sulfonamide derivatives used in docking and MD simulations together with sulfanilamide.
where Ki (dissociation constant, and i indicates it is also an inhibition constant)
 (2)
(2)
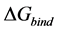 Free Energy of Binding
Free Energy of Binding
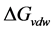 van der Waals potential
van der Waals potential
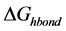 Hydrogen bonding potential
Hydrogen bonding potential
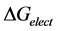 Electrostatic Potential
Electrostatic Potential
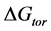 torsional energy
torsional energy
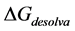 Free Energy of desolvation
Free Energy of desolvation
3. Results
3.1. Binding Energies
In these simulations, 2 × 106 energy evaluations between the sulfonamide derivatives were used to identify binding sites and amino acid residues involved in formation of enzyme ligand complexes. All the sulfonamide ligands screened successfully docked to the three glycolytic enzymes with varying binding affinities. We also wanted to determine whether any of these ligands showed a selective affinity tothe human glycolytic enzymes (ALD, GAPDH and TPI) compared to Plasmodium enzymes. The binding energy difference between the complex and free enzyme and ligands was used to determine strength of interaction (Figure 2).
The relative binding energy data suggest that the sulfonamide derivatives seem to interact more strongly with human aldolase compared to Plasmodium aldolase (Figure 2). For the GAPDH enzyme there is no preference in binding affinity for either species. This is not the case with TPI, where there is strong indication that the ligands show a preference for complex formation with the parasitic enzyme (TPI) as opposed to the human enzyme. The complexes formed with pfTPI are more stable with higher affinity compared to hTPI complexes (Figure 2). This observation is distinctive from the interactions with GAPDH where there is no significant difference in affinity between species enzymes and ALD where the affinity to human enzymes is stronger than Plasmodium enzymes. SulfaC, SulfaH and SulfaE also standout as the ligands with the best binding affinity for all the enzymes modeled.

Figure 2. Relative binding energies between sulfonamide derivatives and glycolytic enzymes (Ald, GAPDH and TPI). Red bars represent Plasmodium enzymes, while the blue bars represent human enzymes.
3.2. Dissociation Constants
The dissociation constants or inhibition constants (Ki) usually indicate the concentration at which binding domains of the enzyme is half filled. In general, a small dissociation constant is an indication of a tightly bound complex. Larger bars signify weak binding, while bars of similar sizes indicate no preference in binding one enzyme over the other. The dissociation constants again show that Plasmodium ALD does not form strong complexes with the sulfonamide derivatives. For example, SulfaE binds human ALD with a Ki of 0.197 uM and 1.64 uM for pfALD (Figure 3). SulfaE binds hGAPDH with Ki of 0.645 uM and 0.331 uM for pfGAPDH. This suggests that the binding between SulfaE and human aldolase is about 8 times stronger than the interaction with pfALD. Similarly, SulfaE shows a slight enhancement in affinity to pfGAPDH compared to hGAPDH by a factor of 2 (Figure 3 and Figure 4). The situation is quite different for TPI, where there is an enhancement factor of 67 for the interaction of sulfaE with pfTPI compared to human TPI (Figure 4). Specifically, SulfaE interacts with pfTPI with a Ki of 0.197 uM and 1.64 uM for pfALD. Sulfa H and SulfaC also have selectivity enhancement factors of 60 and 45, respectively.

Figure 3. Dissociation constant between ligands with ligands and enzymes. The blue bar represents human enzymes and the red bars Plasmodium enzymes.
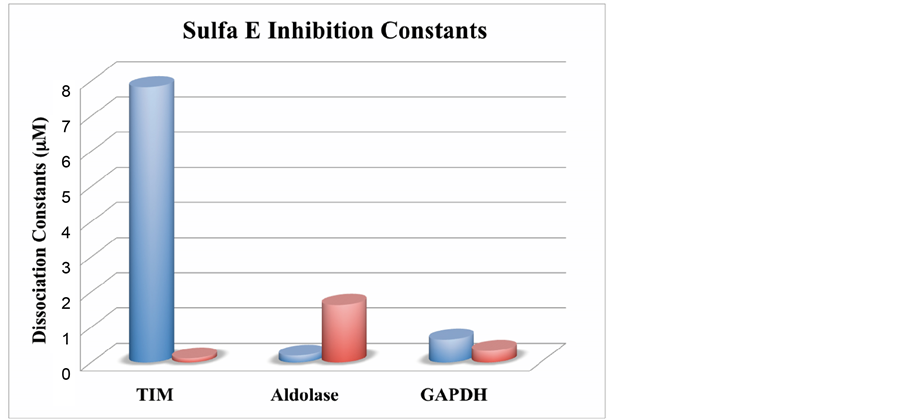
Figure 4. Dissociation constant (Ki) Selectivity Factor for binding of SulfaE with the glycolytic enzymes. The blue cylinders represent human enzymes and the red Plasmodium enzymes.
3.3. Binding Site and Complexes
All the glycolytic enzymes modeled are docked with the sulfonamide derivatives. Aldolase and GAPDH however, do not seem to form complexes that discriminate between human and Plasmodium enzymes. The interaction affinities and dissociations constants do suggest that there is potential of selective binding with TPI enzymes. Due to structural similarity of many glycolytic enzymes, the significance of this observation can be explained by the nature of complexes formed and the residues responsible for interactions. All the sulfonamide ligands screened interact with each enzyme using similar binding sides and residues (Figure 5).
In general, there are two main sites that these ligands use to interact with the glycolytic enzymes. These include an allosteric binding side occurring at the dimer interface or between monomers (Site D), and a site proximal to the active site or cofactor-binding site of the enzyme (site A). Amongst the 100 complexes analyzed for SulfaE docking calculations, site D seems to be the preferred site of binding in hALD, pfALD, hGAPDH, and pfGAPDH (Figure 5 and Figure 6). Specifically, about 90% of all complexes with the GAPDH enzyme form at thedimer/dimer or monomer/monomer interface in both human and parasitic isoforms of GAPDH. The following amino acid residues were most common and responsible for complex formation between sulfaE and GAPDH. These residues include: LEU203, GLN204, ALA238, VAL240, PRO236, THR237, ASN239, SER283, and SER 284 for hGAPDH, and LEU206, PRO239, ILE240, GLY241 and SER207 for pfGAPDH. These amino acids form a binding pocket that is not close to the active site of the molecules. The similarity in location, types, and polarities of the residues also explains why there is no clear distinction in affinity between hGAPDH and pfGAPDH. The second binding domain (site A) occurs less frequently (Figure 6) and comprise of the following residues: PRO124, SER152, CYS153, THR154, THR183, and ASN185. These residues form a groove that is in the vicinity of the NAD cofactor binding domain. The dimer interface binding domain (site D) is also more
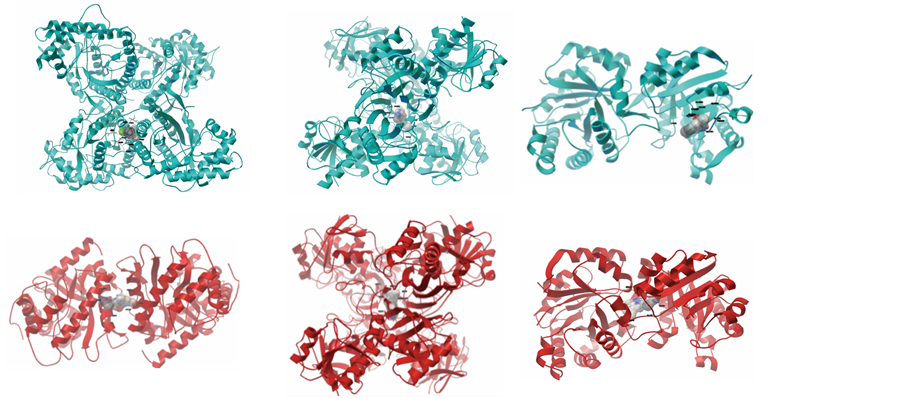
Figure 5. Binding mode of sulfonamide ligands: Top Panel: Human ALD, GAPDH and TPI. Lower Panel: Plasmodium ALD, GAPDH and TPI.
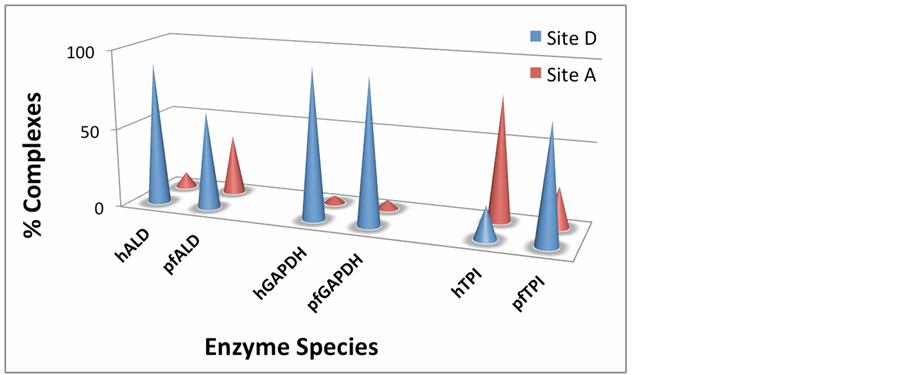
Figure 6. Distribution of Docked Complexes to the identified binding sites for the glycolytic enzymes aldolase (hALD and pfALD), glyceraldehyde phosphate dehydrogenase (hGAPDH and pfGAPDH), and triose phosphate isomerase (hTPI and pfTPI).
common in Aldolase, however, the pfALD also shows about 38 % of docked complexes in the site B, with about 5% docked complexes for hALD (Figure 6). Site D residues include: GLU206, TYR213, LYS214, SER217, LEU221, LEU223, ARG258, THR259 for hALD, and HIS171, ALA174, TRP175, CYS218, ALA222 for pfALD. The binding site D residues for hALD are more polar compared to the residues for pfALD. This could explain the stronger affinity of hALD compared with pfALD (Figure 2 and Figure 3).
There is a significant difference in distribution of docked complexes amongst the binding sites D and A in the interactions between all the sulfonamide drug derivatives with hTPI or pfTPI. These differences can explain the selectivity observed in the binding energies and dissociation constants. In docking simulations involving pfTPI, more of the complexes are formed using site D, while site A is preferred in hTPI docking solutions (Figure 6). The favorable interactions observed at the dimer interface (Site D) for pfTPI involve the amino acid residues: VAL44, SER45, TYR48, GLN64, ASN65, VAL78. The collection of polar and smaller hydrophobic residues facing eachother creates a groove at the dimer interface of pfTPI making it more specific and a preferred binding domain. Site A residues responsible for interactions of most complexes in hTPI include: LYS3, LYS237, ALA234, THR213, GLU239, VAL212, GLY209, GLY232 and VAL28.
4. Discussion
The major goal of this study was to computationally determine whether the novel fluorinated sulfonamide ligands selectively interact and hence inhibit glycolytic enzymes of Plasmodium cells as opposed to humans. The broad similarity in structure of glycolytic enzymes amongst different species, has generally limited their use as molecular receptors or targets in the development of antimalarial therapies [33] [34] [35] . The increase in availability of more refined crystal structures of glycolytic enzymes do show subtle changes in amino acid residues, that can lead to significant structural and functional differences in the way these enzymes interact with substrates and their ligands. The Plasmodium parasite’s continuous resistance to many mainstream drugs like chloroquineand artemisinin derivatives is a major bottleneck to the complete eradication of the malaria epidemic using chemotherapeutic agents [6] [7] [36] [37] . The increased application of molecular docking methods in the pharmaceutical industry and academia is a direct result of increase in computer speed, and the reliability of simulation theories and docking software [38] [39] . Molecular docking studies have shown that a number of quinoline-based ligands competitively inhibit the glycolytic enzyme lactate dehydrogenase (LDH), by docking at the nicotinamide adenine dinucleotide (NADH) cofactor binding site of LDH [40] . For example, the crystal structure of Plasmodium LDH complexed with the antimalarial agent chloroquine also suggests an overlap in binding site with the cofactor NADH [41] .
In this study we provide data that shows that sulfonamide ligands are suitable candidates to selectively target glycolytic enzymes. The data involving rigid enzyme targets reveal that all the sulfonamide ligands screened, bind the three glycolytic enzymes (ALD, GAPDH, TPI) with high affinity. The dissociation constants observed for these interactions also fall in the micromolar range, comparable to many current drugs in the market (ref). While these affinities are strong, the interactions with ALD and GAPDH are not selective (Figure 2 and Figure 4). TPI is the only enzyme where there is a significant difference in affinity between the human and Plasmodium enzymes. There is a preference for an allosteric binding site (Site D) in pfTPI for all the sulfonamide ligands tested in this study and a sample of currently used antimalarial agents (ref). The human enzyme on the other hand binds the sulfonamide ligands using site A that is proximal to the active site (Figure 6).
ALD and GAPDH from the both species interact with the ligands using similar binding sites, and the residues all have similar chemical properties (size and polarity) (Figure 6). This explains the lack of enhancement in binding observed between species. The dimer interface of pfTPI with a collection of polar and smaller hydrophobic residues (VAL44, SER45, TYR48, GLN64, ASN65, VAL78) facing eachother form a groove that provides important electrostatics contributions responsible for strong binding (Figure 7). In hTPI, this dimer interface is not the preferred. This may be due to the substitution of key residues (P44, A46, I48) that make this pocket no longer a potential binding site. Amongst the 100 complexes analyzed for hTPI, none of the ligands successfully docked in the vicinity of the dimer interface residues common to those found in site D binding site for pfTPI (Figure 7). The difference in residues involved shows that the residues substitution (V44P, Y48I) successfully changes the polarity of the binding pocket of hTPI thereby affecting the strength of binding with sulfonamide ligands screened. This is an indication that the observed binding site D is unique and specific to pfTPI and is the sole reason for the observed selectivity because of the combination of polar and non-polar groups at the dimer interface responsible for complex formation.

Figure 7. (a) hTPIsulfaE interaction region showing contact residues responsible for polar contacts. Contact residues show in ball and stick and the ligand in surface representation. (b) pfTPIsulfaE interaction region showing contact residues responsible for polar contacts. Contact residues show in ball and stick and the ligand in surface representation.
The docking binding affinities of the new sulfonamide analogues also compare with affinities of current antimalarial drugs like chloroquine (Figure 8). We compared the dissociation constant for the interactions between some antimalarial drugs (chloroquine, primaquine, mefloquine, and quinine), and the screened sulfonamide ligands in docking simulations with hTPI and pfTPI. The Ki selectivity factor was obtained as a ratio of the dissociation constant for the interaction with hTPI and pfTPI. A large Ki selectivity factor therefore indicates that the ligands bind one receptor with much higher affinity than the other. The ligand with the highest selectivity factor in the docking studies was chloroquine, which is currently used in many regions of the world to treat malaria. The enhancement factor for SulfaE is on par with that of chloroquine, suggesting that this ligand can selectively inhibit glycolytic enzymes TPI. In addition to SulfaE, SulfaC and SulfaH also possess better selectivity factors compared to a number of current antimalarial agents like mefloquine, primaquine, and quinine (Figure 8). The combination of binding energies and dissociation constants suggests that interaction between the novel sulfonamide derivatives with aldolase or GAPDH is not selective. Theenzyme TPI can serve as a suitable target for inhibition with atleast 3 of the sulfonamide derivatives (SulfaC, SulfaH and SulfaE).
5. Conclusion
Eliminating the debilitating effects of the disease malaria remains a major concern of the WHO and many countries in the world. Many antimalarial agents (e.g. Chloroquine) have been used for a number of years to fight malaria without any clear-cut mode of action. The reemergence of parasitic resistance has continued to spur research

Figure 8. Ki selectivity factor for the interaction of some antimalarial drugs and sulfonamide ligands binding with hTPI and pfTPI.
for more effective analogues. In this study, we have shown that eight novel sulfonamide ligands screened can successfully dock to the three glycolytic enzymes ALD, GAPDH and TPI from humans and Plasmodium falciparum. The docking results suggest that glycolytic enzymes generally used two binding sites; site D (the dimer or quaternary structure interface) and site A (site proximal to active site or cofactor binding site). In interactions involving ALD and GAPDH similar binding sites and amino acid residues are responsible for complex formation in the enzymes from both species. The molecular docking results suggest that triose phosphate isomerase (TPI) is a good target for the development of new antimalarial agents. Specifically, we have determined that the current sulfonamide ligands interact with pfTPI and hTPI using different very binding domains. This dimer interface binding site is different from the TPI active site, and may suggest the possibility of noncompetitive or uncompetitive inhibition. SulfaE, SulfaH and SulfaC also stand out as key molecules can be fine-tuned to take advantage of differences in binding domains and residues between pfTPI and HTPI. These sulfonamide derivatives showed an enhancement in dissociation constants on par or greater than some known antimalarial agents. The micromolar range affinity of sulfonamide derivatives is comparable to the affinity of many current antimalarial agents. The sulfonamide derivatives can thus serve as pharmacophores for the development of novel antimalarial drugs. The rigid nature docking is one limitation of this study because dynamic effects of binding during complex formation are not accounted for. Further investigations using molecular dynamics simulations are underway to dynamically test the stability and hydrogen bonding network responsible for complexes. Specifically, it will be important to understand how the flexibility and dynamic motions of both ligand and enzyme will affect the interactions using molecular dynamics simulations. The specificity of binding and inhibitory effects of these ligands also have to be tested using kinetic experiments associated with the pfTPI dimer interface.
Acknowledgements
This work was supported with funds from the GGC STEM Minigrant, and VPASA Seed Fund Program at Georgia Gwinnett College.
Cite this paper
Forlemu, N., Watkins, P. and Sloop, J. (2017) Molecular Docking of Selective Binding Affinity of Sulfonamide Derivatives as Potential Antimalarial Agents Targeting the Glycolytic Enzymes: GAPDH, Aldolase and TPI. Open Journal of Biophysics, 7, 41-57. http://dx.doi.org/10.4236/ojbiphy.2017.71004
References
- 1. Kehr, S., Stum, N., Rahlfs, S., Przyborski, J.M. and Becker, K. (2010) Compartmentation of Redox Metabolism in Malaria Parasites. PLoS Pathogens, 6, 1-12.
https://doi.org/10.1371/journal.ppat.1001242 - 2. Guerra, C.A., Snow, R.W. and Hay, S.I. (2006) Defining the Global Spatial Limits of Malaria Transmission in 2005. Advanced Parasitology, 62, 157-179.
https://doi.org/10.1016/S0065-308X(05)62005-2 - 3. World Health Organization (2016).
http://www.who.int/features/factfiles/malaria/en/index.html - 4. Bloland, B.P. (2014) Drug Resistance in Malaria. 2014 WHO Report, World Health Organization, Chamblee.
- 5. Plowe C.V., Roper, C., Barnwell, J.W., Happi, C.T., Joshi, H.H., Mbacham, W., Meshnick, R.S., Mugittu, K., Naidoo, I., Price, N.R., Schafer, W.R., Sibley, H.C., Sutherland, C.J., Zimmerman, A.P. and Rosenthal, J.P. (2007) World Antimalarial Resistance Network (WARN) III: Molecular Markers for Drug Resistant Malaria. Malaria Journal, 6, 121-131.
https://doi.org/10.1186/1475-2875-6-121 - 6. Bray, P.G., Barrett, P.M., Ward, A.S. and de Koning. P.D. (2003) Pentamidine Uptake and Resistance in Pathogenic Protozoa: Past, Present and Future. Trends Parasitology, 19, 232-239.
https://doi.org/10.1016/S1471-4922(03)00069-2 - 7. Briolant, S., Almeras, L., Belghazi, M., Boucomont-Chapeaublanc, E., Wurtz, N., Fontaine, A., Granjeaud, S., Fusaï, T., Rogier, C. and Pradines, B. (2010) Plasmodium falciparum Proteome Changes in Response to Doxycycline Treatment. Malaria Journal, 9, 141-155.
https://doi.org/10.1186/1475-2875-9-141 - 8. Derbyshire, R.E., Prudencio, M., Mota, M.M. and Clardy, J. (2012) Liver-Stage Malaria Parasites Vulnerable to Diverse Chemical Scaffolds. Proceedings National Academy Science U.S.A, 109, 8511-8516.
https://doi.org/10.1073/pnas.1118370109 - 9. Roth, E.F., Calvin Jr., M., Max-Audit, I., Rosa, J. and Rosa, R. (1998) The Enzymes of the Glycolytic Pathway in Erythrocytes Infected with Plasmodium Falciparum Malaria Parasites. Blood, 72, 1922-1925.
- 10. Verlinde, C.L.M.J., Hannaert, V., Blonski, C., Willson, M., Perie, J.J., Fothergill-Gilmore, L.A., Opperdoes, F.R., Gelb, M.H., Hol, W.G.J. and Michels, P.A.M. (2001) Glycolysis as a Target for the Design of New Anti-Trypanosome Drugs. Drug Resistance, 4, 50-65.
https://doi.org/10.1054/drup.2000.0177 - 11. Kim, J.W. and Dang, C.V. (2005) Multifaceted Roles of Glycolytic Enzymes. Trends in Biochemical Sciences, 30, 142-150.
https://doi.org/10.1016/j.tibs.2005.01.005 - 12. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank. Nucleic Acids Research, 28, 235-242.
https://doi.org/10.1093/nar/28.1.235 - 13. Sameer, S.V., Soumya, S.R., Rajesh, S.G., Suma, S., Hemalatha, B. and Murthy, M.R.N. (1997) Triosephosphate Isomerase from Plasmodium falciparum: The Crystal Structure Provides Insights into Antimalarial Drug Design. Structure, 5, 751-761.
https://doi.org/10.1016/S0969-2126(97)00230-X - 14. Read, J.A., Winter, V.J., Eszes, C.M., Sessions, R.B. and Brady, R.L. (2001) Structural Basis for Altered Activity of M-and H-Isozyme Forms of Human Lactate Dehydrogenase. Proteins, 43, 175-185.
- 15. Dunn, C.R., Banfield, M.J., Barker, J.J., Higham, C.W., Moreton, K.M., Turgut-Balik, D., Brady, R.L. and Holbrook, J.J. (1996) The Structure of Lactate Dehydrogenase from Plasmodium falciparum Reveals a New Target for Anti-Malarial Design. Nature Structural Biology, 3, 912-915.
https://doi.org/10.1038/nsb1196-912 - 16. Cameron, A., Read, J., Tranter, R., Winter, J.V., Sessions, B.R., Brady, L., Vivas, L., Easton, A., Kendrick, H., Croft, S.L., Barros, D., Lavandera, J.L., Risco, F., Garci-Ochoa, S., Gamo, F.J., Sanz, L., Leon, L., Ruiz, J.R., Gabarro, R., Mallo, A. and Gomez de las Heras, F. (2004) Identification and Activity of a Series of Azole-Based Compounds with Lactate Dehydrogenase-Directed Anti-Malarial Activity. Journal Biological Chemistry, 279, 31429-31439.
https://doi.org/10.1074/jbc.M402433200 - 17. Robien, M.A., Bosch, J., Buckner, F.S., Van Voorhis, W.C., Worthey, E.A., Myler, P., Mehlin, C., Boni, E.E., Kalyuzhniy, O., Anderson, L., Lauricella, A., Gulde, S., Luft, J.R., DeTitta, G., Caruthers, M.J., Hodgson, K.O., Soltis, M., Zucker, F., Verlinde, M.J., Merritt, A.E. and Schoenfeld, W.L. (2006) Crystal Structure of Glyceraldehyde-3-Phosphate Dehydrogenase from Plasmodium falciparum at 2.25 Å Resolution Reveals Intriguing Extra Electron Density in the Active Site. PROTEINS: Structure Function Bioinformatics, 62, 570-577.
https://doi.org/10.1002/prot.20801 - 18. Cowan-Jacob, S.W., Kaufmann, M., Anselmo, A.N., Stark, W. and Gruetter, M.G. (2003) Structure of Rabbit-Muscle Glyceraldehyde-3-Phosphate Dehydrogenase. Acta Crystallographica Section D Structural Biology, D59, 2218-2227.
https://doi.org/10.1107/s0907444903020493 - 19. Parthasarathy, S., Ravindra, G., Balaram, H., Balaram, P. and Murthy, M.R.N. (2002) Structure of the Plasmodium falciparum Triosephosphate Isomerase-Phosphoglycolate Complex in Two Crystal Forms: Characterization of Catalytic Loop Open and Closed Conformations in the Ligand-Bound State. Biochemistry, 41, 13178-13188.
https://doi.org/10.1021/bi025783a - 20. Gamblin, S.J., Davies, G.J., Grimes, J.M., Jackson, R.M., Littlechild, J.A. and Watson, H.C. (1991) Activity and Specificity of Human Aldolases. Journal of Molecular Biology, 219, 573-576.
https://doi.org/10.1016/0022-2836(91)90650-U - 21. Kim, H., Certa, U., Dobeli, H., Jakob, P. and Hol, W.G. (1998) Crystal Structure of Fructose-1,6-Bisphosphate Aldolase from the Human Malaria Parasite Plasmodium falciparum. Biochemistry, 37, 4388-4396.
https://doi.org/10.1021/bi972233h - 22. Njabon, N.E. (2005) Brownian Dynamics Simulations of the Interactions between Lactate Dehydrogenase (LDH) and G-or F-Actin. University of North Dakota, Grand Forks.
- 23. Ismail, S.A. and Park, H.W. (2005) Crystal Structure of Human Liver GAPDH. Acta Crystallographica, D61, 1508-1513.
- 24. Satchell, J.F., Malby, R.L., Luo, C.S., Adisa, A., Alpyurek, A.E., Klonis, N., Smith, B.J., Tilley, L. and Colman, P.M. (2005) Structure of Glyceraldehyde-3-Phosphate Dehydrogenase from Plasmodium falciparum. Acta Crystallographica, D61, 1213-1221.
- 25. Kent, M. (2000) Advanced Biology. Oxford University Press, Oxford.
- 26. Matondo, S.I., Temba, G.S., Kavishe, A.A., Kauki, S.J., Zwetselaar, M., Reyburn, H. and Kavishe, R. (2014) High Levels of Sulphadoxine-Pyrimethamine Resistance Pfdhfr-Pfdhps Quintuple Mutations: A Cross Sectional Survey of Six Regions in Tanzania. Malaria Journal, 13, 152.
https://doi.org/10.1186/1475-2875-13-152 - 27. Dennington, R., Keith, T. and Millam, J. (2009) Gauss View, Version 5. Semichem Inc., Shawnee Mission.
- 28. Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C. and Ferrin, T.E. (2004) UCSF Chimera—A Visualization System for Exploratory Research and Analysis. Journal of Computational Chemistry, 25, 1605-1612.
https://doi.org/10.1002/jcc.20084 - 29. DeLano, W.L. (2002) The PyMOL Molecular Graphics System. Delano Scientific, San Carlos.
- 30. Morris, G.M., Goodsell, D.S., Halliday, R.S., Huey, R., Hart, W.E., Belew, R.K. and Olson, A.J. (1998) Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. Journal of Computational Chemistry, 19, 1639-1662.
https://doi.org/10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B - 31. Morris, G.M., Huey, R., Lindstrom, W., Sanner, M.F., Belew, R.K., Goodsell, D.S. and Olson, A.J. (2009) AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. Journal of Computational Chemistry, 30, 2785-2791.
https://doi.org/10.1002/jcc.21256 - 32. Huey, R., Morris, G.M., Olson, A.J. and Goodsell, D.S. (2006) A Semi-Empirical Free Energy Force Field with Charge-Based Desolvation. Journal of Computational Chemistry, 28, 1145-1152.
https://doi.org/10.1002/jcc.20634 - 33. Deck, L.M., Royer, R.E., Chamblee, B.B., Hernandez, V.M., Malone, R.R., Torres, J.E., Hunsaker, L.A., Piper, R.C., Makler, M.T. and Vander Jagt, D.L. (1998) Selective Inhibitors of Human Lactate Dehydrogenases and Lactate Dehydrogenase from the Malarial Parasite Plasmodium falciparum. Journal of Medicinal Chemistry, 41, 3879-3887.
https://doi.org/10.1021/jm980334n - 34. Alam, A., Neyaz, M.K. and Hasan, I.S. (2014) Exploiting Unique Structural and Functional Properties of Malarial Glycolytic Enzymes for Antimalarial Drug Development. Malaria Research and Treatment, 2014, Article ID: 451065.
https://doi.org/10.1155/2014/451065 - 35. Gayathri, P., Banerjee, M., Vijayalakshmi, A., Balaram, H., Balaram, P. and Murthy, M.R.N. (2009) Biochemical and Structural Characterization of Residue 96 Mutants of Plasmodium falciparum Triosephosphate Isomerase: Active-Site Loop Conformation, Hydration and Identification of a Dimer-Interface Ligand-Binding Site. Acta Crystallographica, 65, 847-857.
- 36. Harinasuta T., Lasserre R., Bunnag, D., Leimer, R. and Vinijanont, S. (1985) Trials of Mefloquine in Vivax and of Mefloquine plus “Fansidar” in Falciparum Malaria. The Lancet, 325, 885-888.
https://doi.org/10.1016/S0140-6736(85)91670-8 - 37. Huy, T.N., Kamei, K., Kondo, Y., Serada, S., Kanaori, K., Takano, R., Tajima, K. and Hara, S. (2002) Effect of Antifungal Azoles on the Heme Detoxification System of Malarial Parasite. Journal of Biochemistry, 131, 437-444.
https://doi.org/10.1093/oxfordjournals.jbchem.a003119 - 38. Seeliger, D. and De Groot, L.B. (2010) Ligand Docking and Binding Site Analysis with PyMOL and Autodock/Vina. Journal of Computer Aided Molecular Design, 24, 417-422.
https://doi.org/10.1007/s10822-010-9352-6 - 39. Schneider, G. and Bohm, H. (2002) Virtual Screening and Fast Automated Docking Methods. Drug Discovery Today, 7, 64-70.
https://doi.org/10.1016/S1359-6446(02)00004-1 - 40. Waingeh, F.W., Groves, T.D. and Eberle, J.A. (2013) Binding of Quinoline-Based Inhibitors to Plasmodium falciparum Lactate Dehydrogenase: A Molecular Docking Study. Open Journal of Biophysics, 3, 285-290.
https://doi.org/10.4236/ojbiphy.2013.34034 - 41. Read, J.A., Wilkinson, K.W., Tranter, R., Sessions R.B. and Brady, R.L. (1999) Chloroquine Binds Binding Site of Plasmodium falciparum in the Cofactor m Lactate Dehydrogenase. Journal of Biological Chemistry, 274, 10213-10218.
https://doi.org/10.1074/jbc.274.15.10213