Open Journal of Biophysics
Vol.2 No.3(2012), Article ID:21420,9 pages DOI:10.4236/ojbiphy.2012.23011
Ab Initio and Density Functional Theory (DFT) Study on Clonazepam
1Department of Physics, DG Vaishnav College, Chennai, India
2Department of Physics, Presidency College, Chennai, India
Email: shylaja_b@ymail.com
Received December 31, 2011; revised February 2, 2012; accepted February 16, 2012
Keywords: FTIR; Ab Initio; DFT; Clonazepam; Vibrational Analysis; Chemical Shift
ABSTRACT
Quantum chemical calculations have been carried out to investigate the vibrational frequencies of clonazepam. The FTIR spectrum of clonazepam was recorded in the region 4000 to 400 cm–1 in the solid phase. The optimized geometry, wave number and intensity of the vibrational bands of clonazepam were obtained by ab initio Restricted Hartree Fock (RHF) and Density Functional Theory (DFT) methods with complete relaxation in the potential energy surface using the 6-31G (d,p) basis set. A complete vibrational assignment aided by the theoretical harmonic frequency analysis is proposed. The observed and the calculated wavenumbers are found to be in good agreement. The experimental spectra also coincide satisfactorily with those of theoretical spectra. Theoretical spectrograms for the IR spectrum were also constructed in RHF and B3LYP levels. In addition to this the RHF and DFT based NMR calculation procedure was used to assign the 1H NMR chemical shift of clonazepam. Theoretical values are compared with the experimental data.
1. Introduction
Vibrational spectroscopy is an important tool for the structural investigation of the organic molecules. Benzodiazepines and their polycyclic derivatives are a very important class of bioactive compounds. Clonazepam is a benzodiazepine derivative drug having anticonvulsant, muscle relaxant and anxiolytic properties. Clonazepam is a benzodiazepine derivative drug with highly potent anticonvulsant, muscle relaxant, and anxiolytic properties. Chemical formula of clonazepam is C15H10ClN3O3. Clonazepam exerts its action by binding to the benzodiazepine site of the GABA receptors, which causes an enhancement of the electric effect of GABA binding on neurons resulting in an increased influx of chloride ions into the neurons. This results in an inhibition of synaptic transmission across the central nervous system [1,2]. In spite of its importance for pharmaceutical purposes, however, the reported spectroscopical studies on clonazepam are scarce. The evolution of density function theory that includes electron correlation in an alternative way has affording opportunities of performing vibrational analysis of moderately large organic molecules. The results from DFT theory with results obtained from experiments has shown that the methods using B3LYP are the most promising in providing correct vibrational wave numbers. A scan of literature survey reveals that to the best of our knowledge, no DFT and RHF calculations of clonazepam have been reported so far.
Hence in this work, a systematic study on the structure and vibrational spectra of clonazepam with the aid of RHF and B3LYP methods, have been carried out.
2. Experimental Details
The Spectroscopic pure samples of clonazepam were procured from reputed pharmaceutical firm in Chennai. Fourier Transform Infrared (FTIR) spectrum was recorded on Bruker IFS 66V spectrophotometer by adopting the KBr pellet technique, in the region 4000 to 400 cm–1. Nuclear magnetic resonance spectrum was recorded using Bruker AVANCE III 500 MHz FT-NMR Spectrometer.
3. Computational Details
All the theoretical computations were performed at RHF and B3LYP levels on a Pentium dual core/1.6 GHz personal computer using the Gaussian 03W program package [3] The geometries were first optimized at the RHF level of theory employing the 6-31G (d,p) basis set. The optimized geometry was used in the vibrational frequency calculations at the RHF and DFT levels to characterize all stationary points as minima. The vibrational frequency assignments were made with a high degree of accuracy with the help Chemcraft software program. The 1H chemical shifts of the title compound were calculated using the keyword NMR in both RHF and DFT calculations with 6-31G (d,p) basis set.
4. Results and Discussion
4.1. Molecular Geometry
The optimized molecular structure for clonazepam in the ground state (in vacuo) was computed by RHF and B3LYP calculations with the 6-31G (d,p) basis set. The calculated geometrical parameters (bond lengths and bond angles) were compared with experimental geometrical parameters of structurally related molecules [4]. The optimized structural parameters obtained from the RHF and B3LYP/6-31G (d,p) calculations and the literature values are listed in Table 1, in accordance with the atom numbering scheme given in Figure 1. The slight deviation in literature data from the computed geometry is probably due to the fact that the intermolecular interactions in the crystalline state are dominant. A statistical treatment of these data shows that, for the bond lengths,
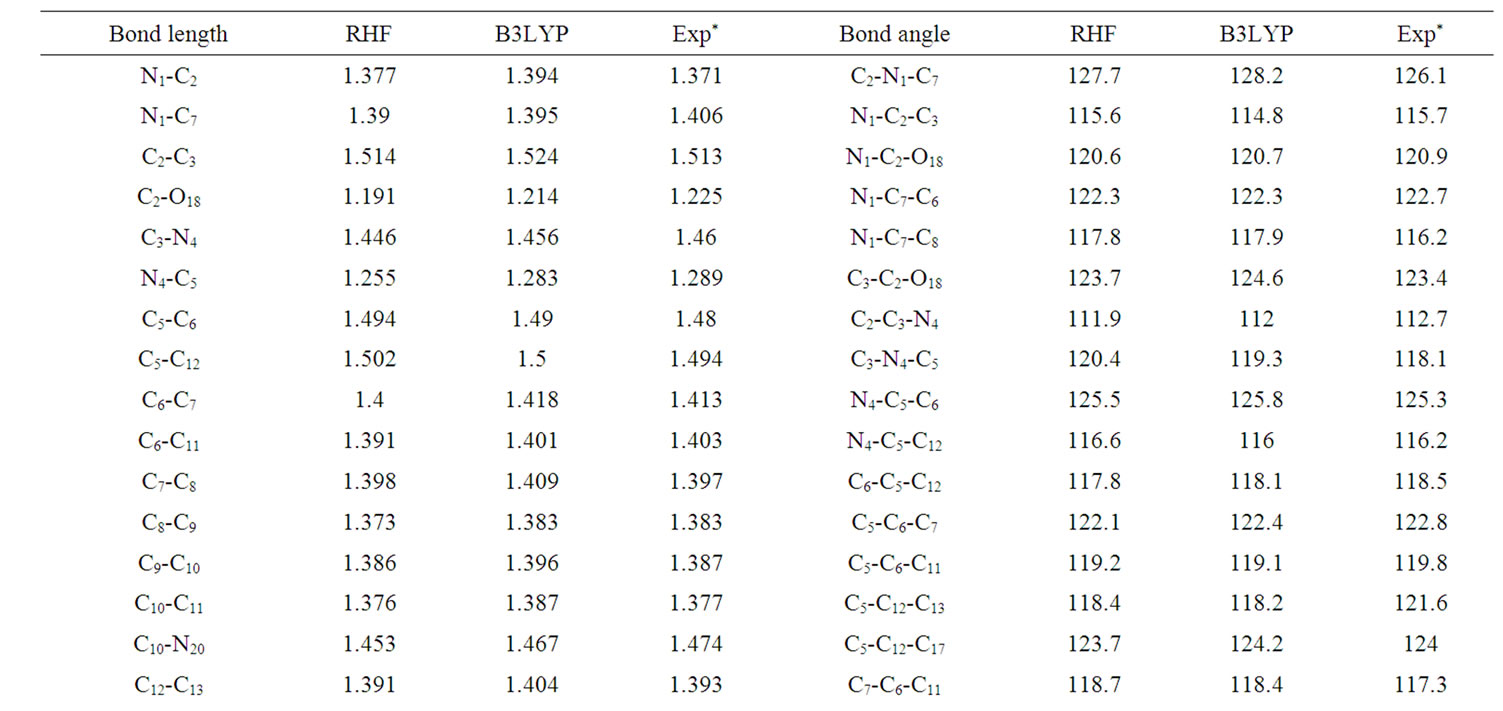

Table 1. Selected bond length (À ) and bond angles (deg) values for clonazepam.
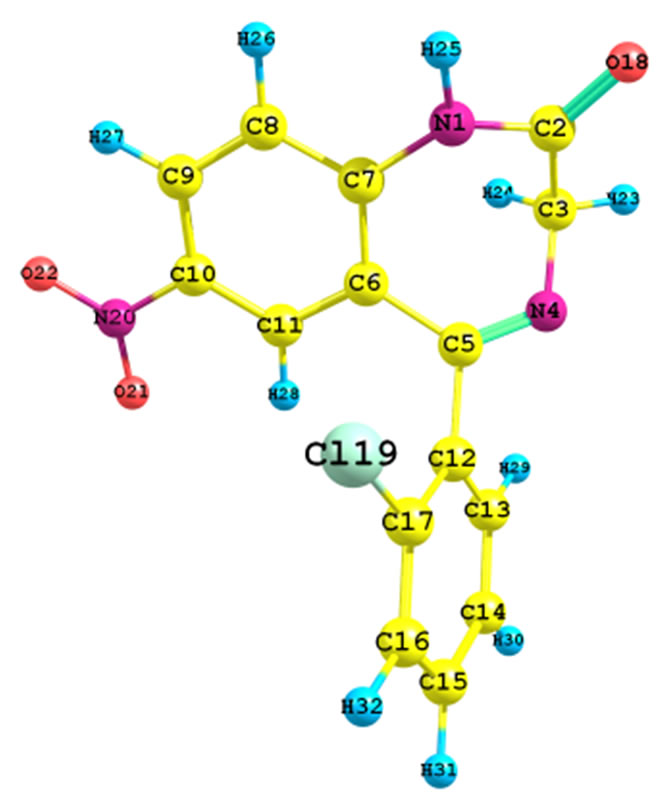
Figure 1. Molecular structure of clonazepam with atom numbering.
B3LYP/6-31G (d,p) is better than the RHF/6-31G (d,p) geometry. It can be noted from the correlation coefficient values that the theoretical predictions are in fairly agreement with the experimental value for bond length and bond angles.
4.2. Atomic Charge and 1H NMR Spectral Analysis
The calculation of effective atomic charges plays an important role in the application of quantum mechanical calculations to molecular systems. The charge distribution on different atoms (C, N, O and Cl) for clonazepam from Mulliken population analysis (MPA) procedures using RHF and B3LYP methods is listed in Table 2. From Table 2, it is observed that the calculated Mulliken charge trends are consistent when comparing the RHF and DFT calculations. It is noticed that the carbon atom attached to oxygen and nitrogen atoms (C2, C5, C7 and C10) in both RHF and B3LYP is an electron-deficient atom (i.e. it possesses positive electronic charge due to electronegativity character). The most negative values are those that are present at nitrogen and oxygen, in the order: N1, O18, N4, O22, O21, O12. The remaining carbon atoms possess negative electronic charge. As the carbon atom C2 is attached with both nitrogen and oxygen in either side, it pulls more charge from C2 atom resulting in more positive charge on the site C2 as compared with that of other carbon atoms. The atomic charges on C9 atom become less negative as compared with that other carbon.
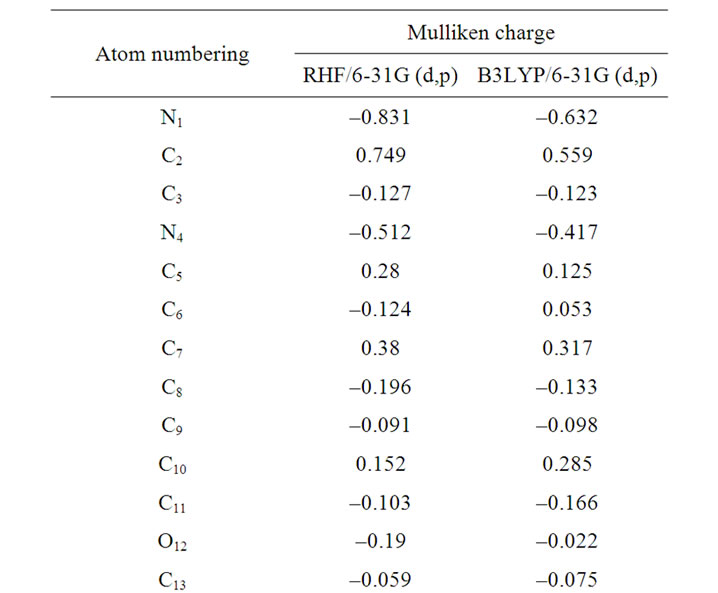
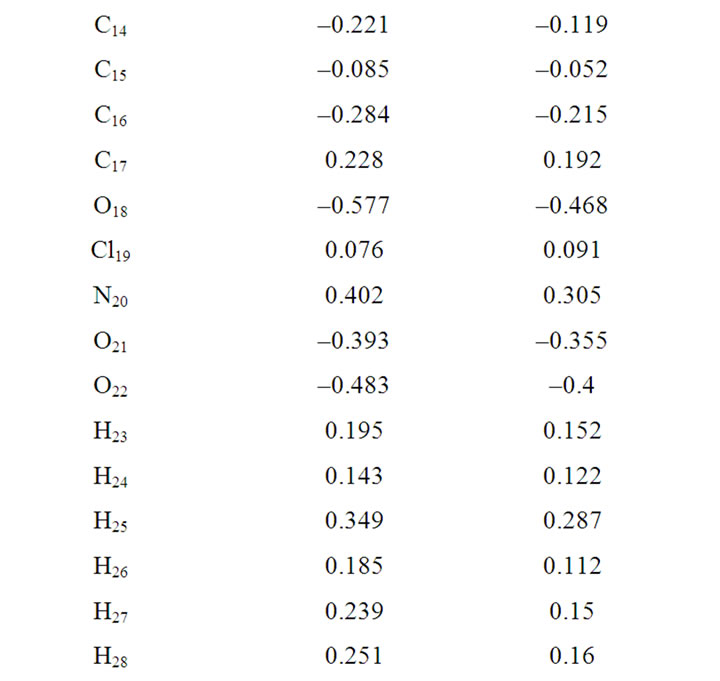
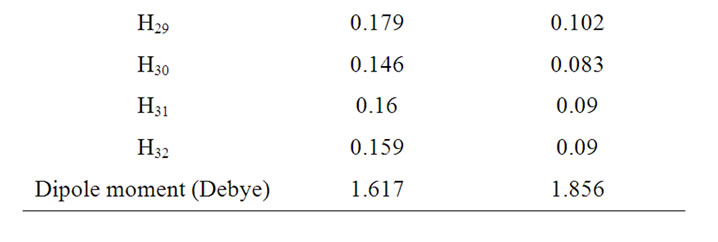
Table 2. Mulliken atomic charges.
From Table 2, it is noticed that calculated atomic charges of clonazepam are not exactly the same, and hence variations in the dipole moment values were observed. The dipole moment value obtained from RHF is small (1.617 Debye), whereas by B3LYP method gives higher dipole moment values (1.856 Debye). This dipole moment value discrepancy is mainly attributed to the variation of atomic charges.
Table 3 gives the predicted chemical shift values obtained by the RHF, DFT and experimental NMR spectrum

Table 3. Experimental and calculated 1H NMR chemical shift for clonazepam.
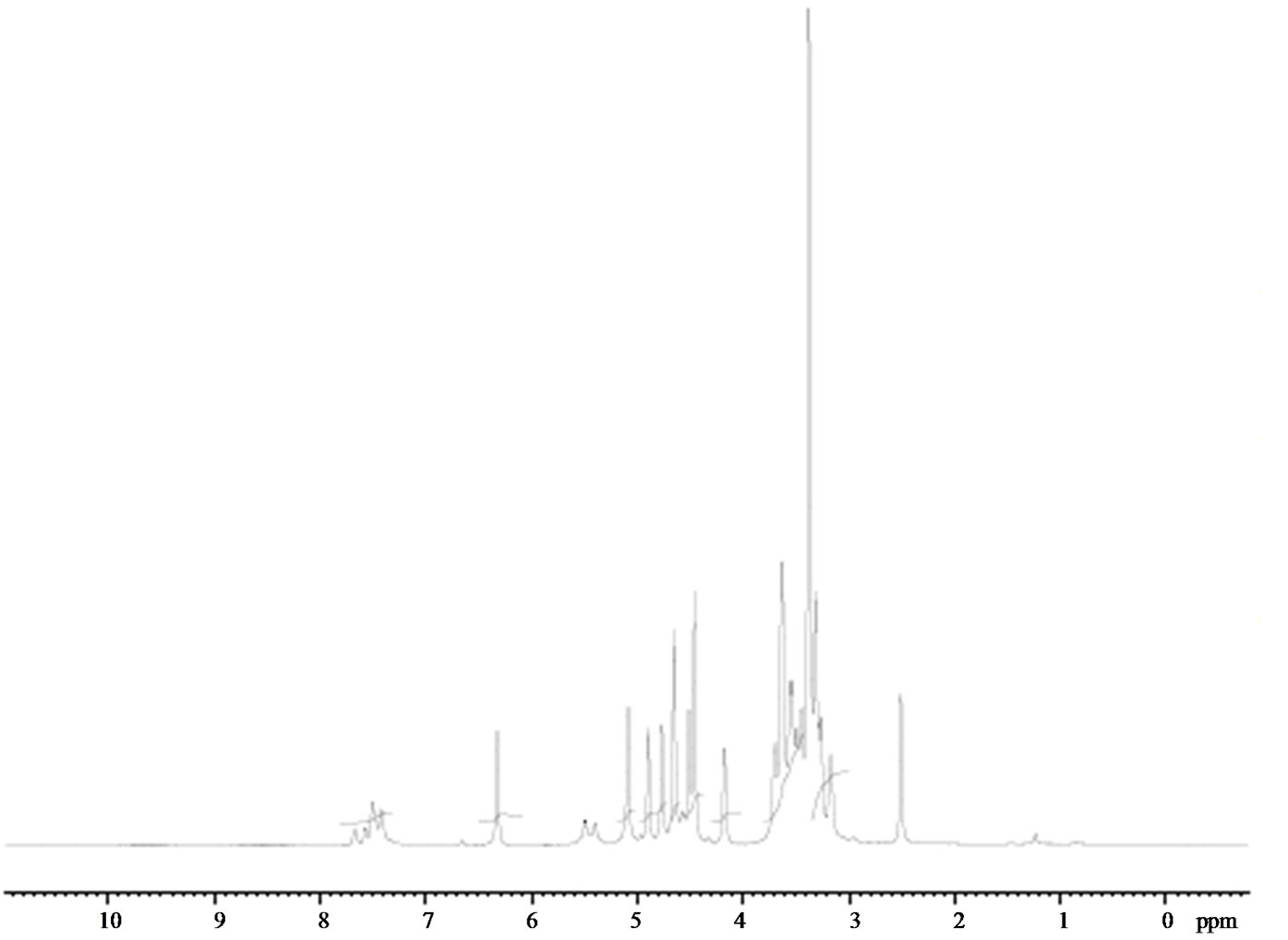
Figure 2. 1H NMR spectrum of clonazepam.
and its assignment along with the shielding values. In general, highly shielded electrons appear downfield and vice versa. The present study reveals that the theoretically predicted chemical shifts slightly deviate from the experimental values. It is observed that the predicted chemical shift values by the B3LYP method are in close agreement with the reported literature values. The spectrum of clonazepam showed a singlet at 3.4 ppm for the proton of the methylene hydrogen (H23 and H24) in the seven membered rings which contradicts the theoretical calculations. Multiplet peaks at 7 - 8 ppm mark the aromatic hydrogen atoms, i.e. in the benzene ring.
4.3. Vibrational Band Assignment
According to theoretical calculations, clonazepam minimum energy structure belongs to the C1 point group symmetry. The molecule has 32 atoms and 90 normal modes of fundamental vibrations. The fundamental modes of these groups are assigned on the basis of group vibrational concept and calculated vibrational frequencies. The harmonic vibrational modes calculated for clonazepam at the RHF and B3LYP levels using the 6-31G (d,p) basis set along with diffuse and polarization function have been summarized in Table 4. The experimentally observed FTIR bands for various modes of vibrations are assigned and presented in Table 4. The fundamental modes of clonazepam were assigned with the aid of animated Chemcraft program. The FTIR spectrum of the title molecule along with theoretically simulated IR spectrum is given in Figure 3. It is noticed that the best representation of low frequency region IR spectrum is obtained in both the methods. From Table 4, both B3LYP and RHF produce the same result which agrees with the experimental values. The scaling factors 0.960 and 0.899 for DFT and RHF values, respectively, have to be necessarily used to find a good agreement with the experimental values. The DFT values were found to be in good agreement with the experimental values after scaling the vibrational frequencies in comparison to RHF values.
CC stretching: The ring CC stretching vibrations occur
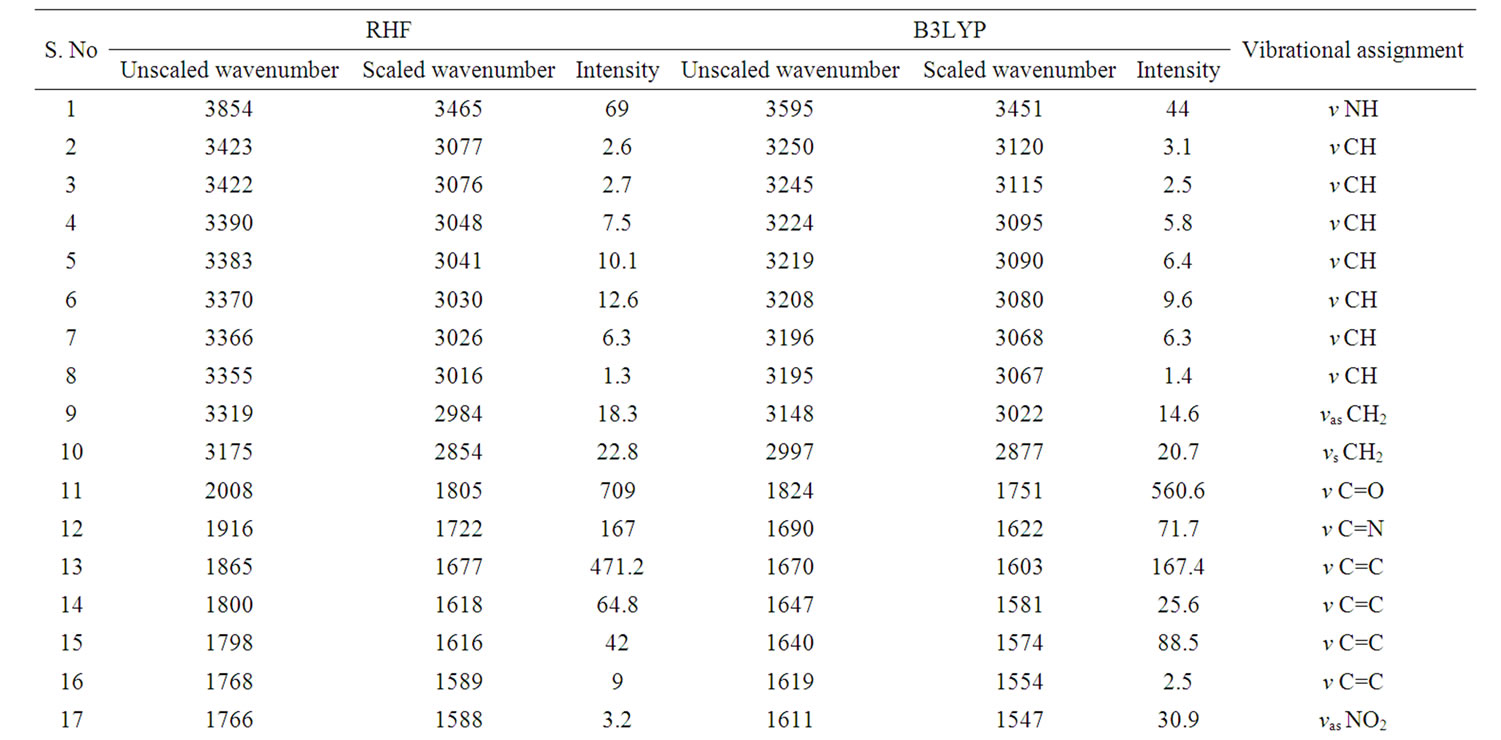

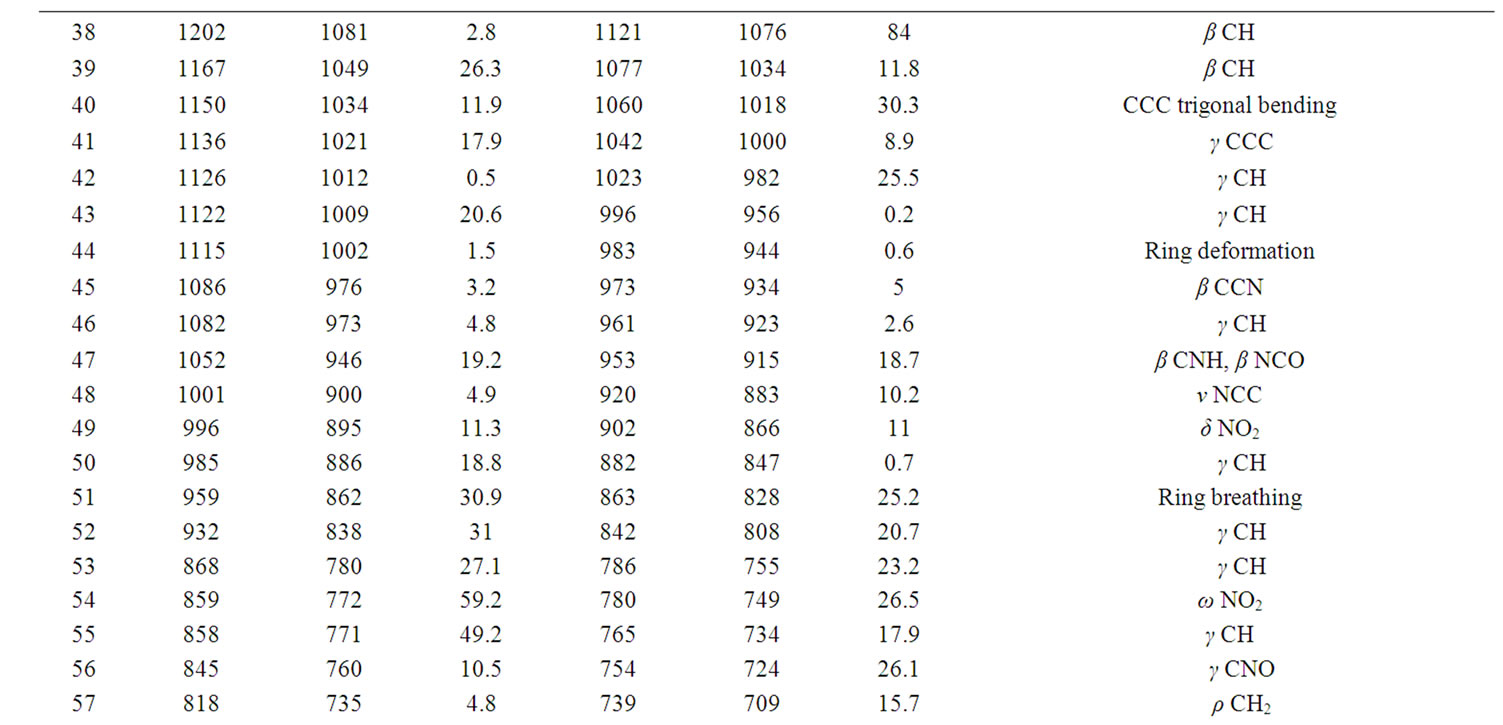
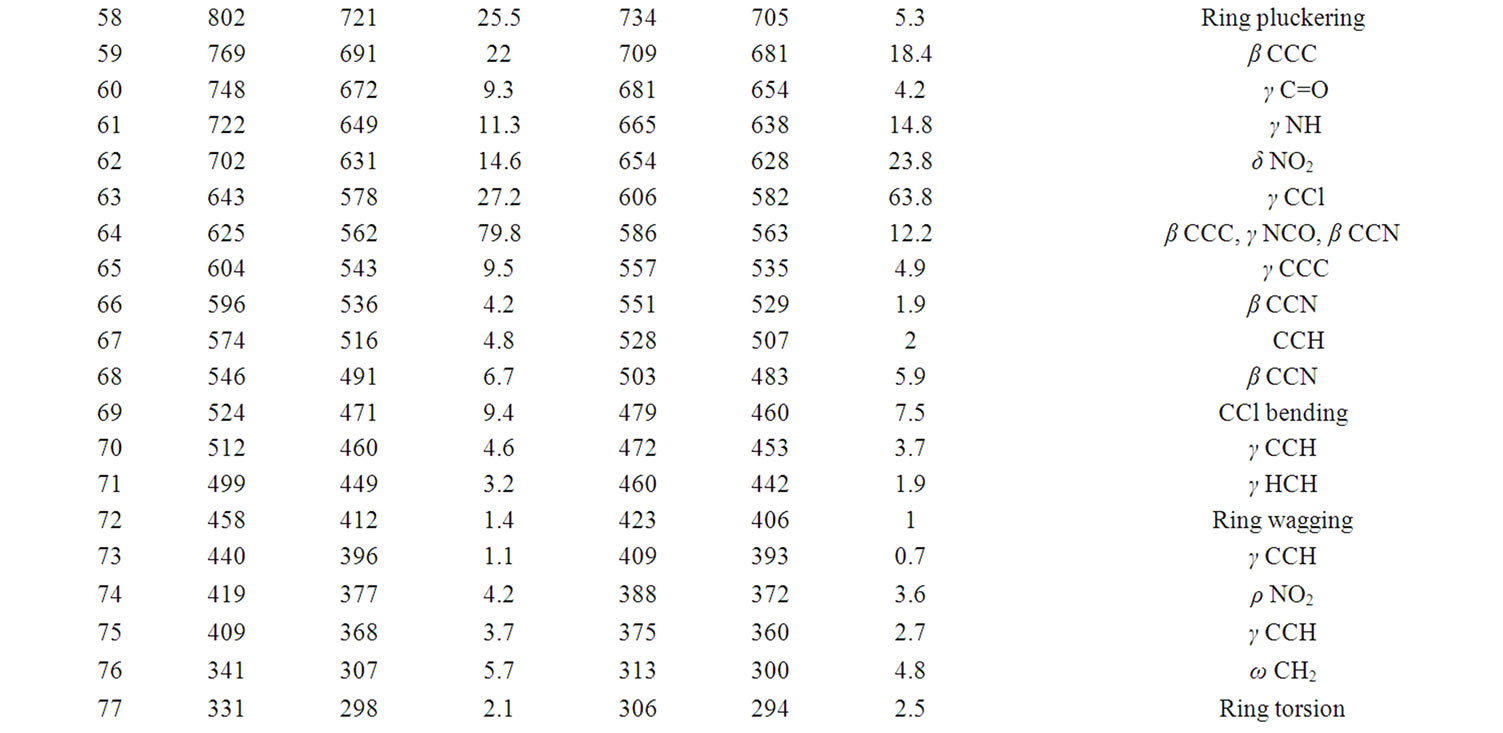

Table 4. Calculated IR wavenumber (cm–1) and intensities (km·mol–1) of clonazepam.
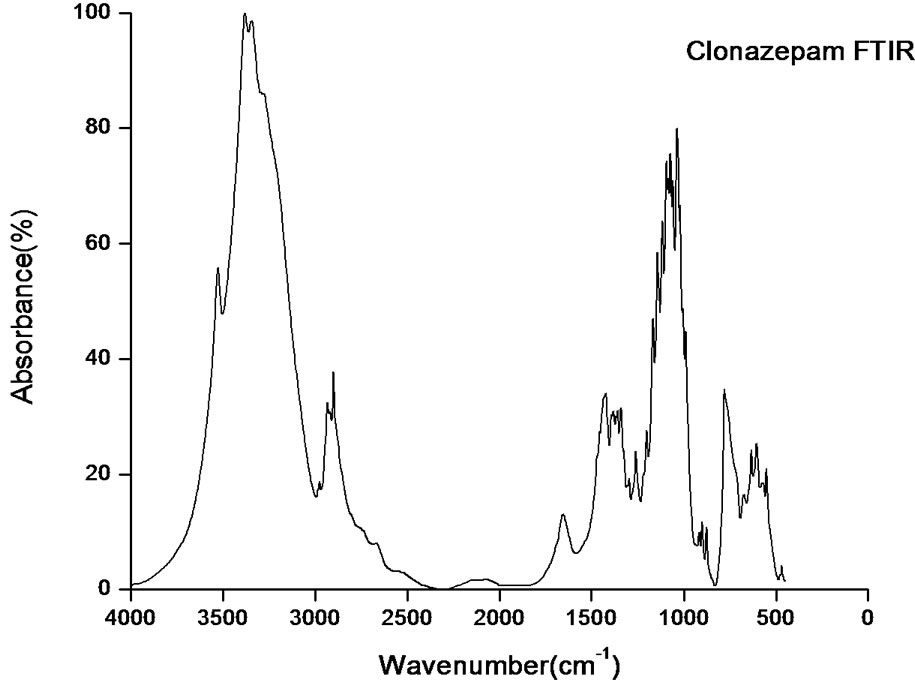
Figure 3. RHF, DFT simulated and experimental FTIR spectrum of clonazepam.

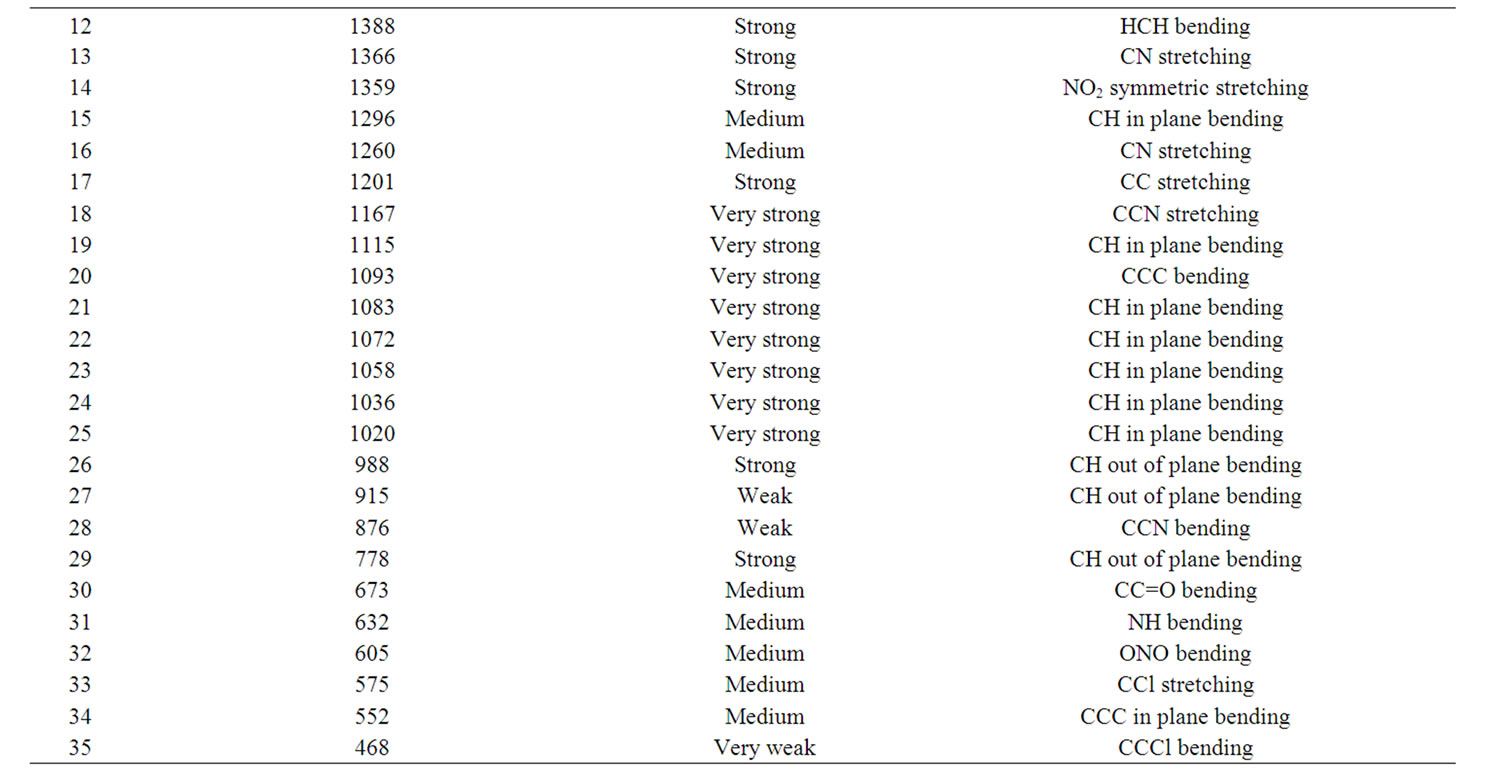
Table 5. FTIR band assignment of clonazepam.
in the region 1625 to 1430 cm–1. Mohan and Settu [5] have identified the IR bands at 1470, 1484, 1561 and 1575 cm–1 in closely related compound of benzodiazepines due to aromatic CC stretching vibrations. Neville and Beckstead [6] have identified the IR bands at 1488 and 1452 cm–1 in clonazepam. Based on these factors, the FTIR bands at 1490, 1455 cm–1 are assigned to CC stretching vibrations. The calculated bands at B3LYP and RHF levels in the same region are in excellent agreement with experimental observation in FTIR spectrum of clonazepam. As is seen from Table 5, the predicted vibrational bands by both RHF and B3LYP agree well with the observed ones.
CH stretching: The aromatic structure shows the presence of CH stretching vibrations in the region 3100 to 3000 cm–1. Krishnakumar [7] identified the strong CH stretching absorption of methylene group centered on 2925 cm–1. According to Neville and Beckstead [6] the CH stretching is observed at 3104 and 3060 cm–1 in clonazepam. Hence, in the present study, the FTIR band at 3070 and 2976 cm–1 is assigned to CH stretching vibration. The CH stretching mode of vibrations due to methylene is identified and presented in the Table 4. The calculated wavenumbers through RHF and DFT techniques are in good agreement with the experimental data.
C=O stretching: The carbonyl stretching frequency is very sensitive to the factors that disturb the nature of carbonyl group and its precise frequency is characteristic of the type of the carbonyl compound being studied. This region is considered as a very important region by organic chemists. The band due to C=O stretching vibration is observed in the region 1850 - 1550 cm–1 due to tautomerism, pyrmidines substituted with hydroxyl groups are generally in the keto form and therefore have a strong band due to carbonyl group [8]. According to Neville and Beckstead [6] the C=O stretching is observed at 1693 cm–1. Therefore in this present study the C=O stretching vibration was observed at 1654 cm–1 in FTIR is in excellent agreement with theoretically predicted frequency obtained in B3LYP/6-31G (d,p).
5. Conclusion
Attempts have been made in the present work for the proper vibrational band assignments for the compound clonazepam from FTIR spectrum. The difference between observed and calculated wavenumbers values of most of the fundamental modes is very small. Any discrepancy noted between the observed and the calculated vibrational band contrary to the experimental values recorded in the presence of intermolecular interactions. Therefore the assignments made at higher levels of theory with only reasonable deviations from the experimental values seem to be correct.
REFERENCES
- J. H. Skerritt and G. A. Johnston, “Enhancement of GABA Binding by Benzodiazepines and Related Anxiolytics,” European Journal of Pharmacology, Vol. 89, No. 3-4, 1983, pp. 193-198. doi:10.1016/0014-2999(83)90494-6
- P. F. Lehoullier and M. K. Ticku, “Benzodiazepine and β-Carboline Modulation of GABA-Stimulated 36Cl-Influx in Cultured Spinal Cord Neurons,” European Journal of Pharmacology, Vol. 135, No. 2, 1987, pp. 235-238. doi:10.1016/0014-2999(87)90617-0
- M. J. Frisch, et al., “Gaussian 03, B05,” Gaussian Inc., Wallingford, 2003.
- G. Gilli, V. Bertolasi and M. Sacerdoti, “7-Nitro-1,3-Dihydro-5-Phenyl-2H-1,4-Benzodiazepin-2-One (Nitrazepam),” Acta Crystallographica Section B, Vol. 33, 1977, pp. 2664-2667. doi:10.1107/S0567740877009157
- S. Mohan and K. Settu, “Vibrational Spectra and Analysis of 1,2,3-Benzotriazole,” Indian Journal of Pure and Applied Physics, Vol. 31, No. 11, 1993, pp. 850-854.
- G. A. Neville and H. D. Beckstead, “Fourier Transform Raman and Infrared Study of Nitrazepam, Nimetazepam, Clonazepam and Flunitrazepam,” Vibrational Spectroscopy, Vol. 1, No. 3, 1991, pp. 287-297. doi:10.1016/0924-2031(91)85006-9
- V. Krishnakumar and S. Seshadri, “Scaled Quantum Chemical Calculations and FT-IR, FT-Raman Spectral Analysis of 2-Methylpiperazine,” Spectrochimica Acta Part A, Vol. 68, No. 3, 2007, pp. 833-838. doi:10.1016/j.saa.2006.12.067
- R. Meenakshi, L. Jaganathan, S. Gunasekaran and S. Srinivasan, “Density Functional Theory, Restricted HartreeFock Simulations and Vibrational Spectroscopic studies of Nicorandil,” Molecular Simulation, Vol. 36, No. 6, 2010, pp. 425-433. doi:10.1080/08927020903583822