Journal of Materials Science and Chemical Engineering
Vol.04 No.10(2016), Article ID:71215,10 pages
10.4236/msce.2016.410004
Electronic Structures and Magnetic Properties of Co-Adsorbed Monolayer WS2
Weiyun Xu
School of Materials Engineering, Shanghai University of Engineering Science, Shanghai, China

Copyright © 2016 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 26, 2016; Accepted: October 11, 2016; Published: October 14, 2016
ABSTRACT
Using the first-principles density functional theory (DFT) calculations, we study the effects of Co adatom on the electronic and magnetic properties of monolayer WS2. The calculations show that, for the high symmetry adsorption sites (Tw, H and Ts) on the surface of monolayer WS2, Co atom prefers Tw site. The p-d hybridization mechanism for the magnetism results in the splitting of the energy levels near the Fermi energy. A total magnetic moment of ~1.0 μB is found in WS2 system with one Co adsorbed and local magnetic moment which mainly focuses on the adsorption site. For Tw adsorption position, we further investigate the formation energy of the ferromagnetic (FM) and the antiferromagnetic (AFM) states under different monolayer coverage (ML) of Co atoms. The FM configurations are relatively stable at 0.50 ML and 1.0 ML. The local density of states (LDOS) and band calculations reveal that both of them present half-metal ferromagnetic materials’ property, which are the important preparation materials for spintronic devices.
Keywords:
First-Principles, Monolayer WS2, Monolayer Coverage (ML), Half-Metal

1. Introduction
Recently, two-dimensional transition metal dichalcogenides (TMDCs), such as MoS2, WS2, MoSe2 and others, have been extensively studied due to their analogous structure with the best known material of this kind, graphene [1] - [3] . They are expected to have unique electronic properties varying from metal to wide-gap semiconductor [4] [5] . Some TMDCs have been proposed and investigated for possible applications, including transistors [6] , photovoltaics [7] , catalysis [8] , hydrogen storage [9] and Li-ion batteries [10] [11] . Recently, great attention has been paid to explore the magnetic properties for the nanoscale spintronic applications by doping foreign atoms [12] - [16] . The electronic structure of the system can be altered, which could lead to appealing character. For instance, a systematic DFT investigation of the absorbed MoS2 monolayer with various atoms, including alkali metals, alkaline earth metals, main group metal, 3d-transition metals, noble metal and nonmetal atoms was performed. Metallic, semimetallic or semiconducting behavior can be found in direct bandgap monolayer MoS2 [17] [18] . In particular, the doping of transition metal atoms (TM) from the IIIB to VIB groups has been confirmed to induce magnetism in nonmagnetic nanomaterials [19] [20] . Mn- doped MoS2 monolayers had been demonstrated to be the potential for engineering a new class of atomically thin dilute magnetic semiconductors [21] .
As one of the typical TMDs, monolayer WS2, is composed of covalently bonded S-W-S with atomic thickness of ~0.7 nm and is weakly bonded together by Van der Waals force forming the crystal [22] [23] . Bulk WS2 is an indirect gap semiconductor with a gap of 1.3 eV, while the gap becomes direct with size of ~2 eV for a single layer. The layers WS2 have a P63/mmc space group symmetry with the W atoms having a trigonal prismatic coordination with the S atoms [2] [24] , which are similar to MoS2 structurally and electronically, as both of them reside in the same column of the periodic table. But WS2 has superior thermal and oxidative stability than that of MoS2. Recent studies show that the monolayers WS2 possess a high in-plane carrier mobility and electrostatic modulation of conductance [25] . The doping of other metallic elements into the monolayer WS2 has been scarcely studied. For example, the Mn-doped WS2 monolayer is found to have a ferromagnetic coupling by a double-exchange mechanism [26] and the properties of V, Nb and Ta substituted WS2 monolayers had been reported under S-rich condition [27] . As a matter of fact, a pristine monolayer WS2 is nonmagnetic; for the low-dimensional systems, atom adsorption is an effective doping way to modify its magnetic and electronic properties [28] [29] . In this paper, we investigated the electronic and magnetic properties of Co-adsorbed monolayer WS2 using the first- principles methods based on density functional theory. Co is typical ferromagnetic metal material, due to the interaction of Co atoms and the WS2 sheets. We need to consider the effect of the WS2 sheet on the Co atom magnetic sequence. We calculated the formation energy of the ferromagnetic (FM) and the antiferromagnetic (AFM) states under the same coverage, to illustrate the structures of Co-adsorbed monolayer WS2 which are ferromagnetic, ensuring the significance and feasibility of the simulation calculation.
2. Methods
All calculations were based on the density functional theory using the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof (PBE) method. The electronic wave functions are described by the projector augmented wave (PAW) potential, as carried out in the Vienna ab initio simulation package (VASP) [30] - [32] . The local- density approximation (LDA) were selected as the exchange and correlation potential [33] - [35] . Although the DFT functional cannot describes very accurately all the characteristics of interactions, but the overestimate of the binding energy by LDA is almost compensated by the ignored van der Waals interactions [36] - [38] . A kinetic energy cutoff is set for the plane-wave expansion at 500 eV, the k-point meshes are generated by Gama-center Monkhorst-Pack scheme for integration over the first Brillouin zone [39] . As the Graphene-like WS2 is composed of a single layer of W and S atoms arranged in a two-dimensional (2D) honeycomb lattice, To detailedly explore the properties of Co- adsorbed 2D systems (Co/WS2), a (2 × 2 × 1) WS2 super cell containing 4 W and 8 S atoms are modeled, a more than 20 Å vacuum layer was set between two adjacent WS2 monolayers in the Z-direction to avoid inter-layer interactions. (12 × 12 × 1) k-point meshes are adopted for the structural optimization, density of states, respectively. In each system, all the atomic positions were fully relaxed until the total energy was converged to less than 1 × 10−7 eV/atom and the forces on all atoms were below 0.01 eV/Å.
3. Results and Discussions
3.1. The Adsorption of Co Atom
According to the monolayer hexagonal lattice structure of WS2, we considered three basic types of adsorption sites (Figure 1): TW (top site directly above a W atom), H site (hollow site above the center of hexagons) and TS site (top site directly above a S atom). The height of adsorption is 3.5 Å, it is reasonable to expect the fully relaxation of Co adatom on one of these adsorption sites. In order to discuss the relative stabilities of the systems, the adsorption energy ( ) is defined as:
) is defined as:
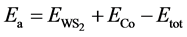 (1)
(1)
 is the total energy of Co/WS2 system,
is the total energy of Co/WS2 system,  is the total energy of the pure WS2 sheet and
is the total energy of the pure WS2 sheet and  is the total energy of an isolated atom. After relaxation, the electron configurations, adsorption energies
is the total energy of an isolated atom. After relaxation, the electron configurations, adsorption energies 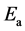 and structural properties for a single Co adsorbed WS2 obtained from our calculations are listed in Table 1. The adsorption energies of Tw-WS2, H-WS2 and Ts-WS2 systems are 3.03 eV, 2.48 eV and 1.44 eV, respectively. They are all positive and the adsorption energies of Tw-WS2 is bigger than the others, which indicates the Co considered can chemically absorb on WS2 monolayer stably and Tw-WS2 is the more stable structure.
and structural properties for a single Co adsorbed WS2 obtained from our calculations are listed in Table 1. The adsorption energies of Tw-WS2, H-WS2 and Ts-WS2 systems are 3.03 eV, 2.48 eV and 1.44 eV, respectively. They are all positive and the adsorption energies of Tw-WS2 is bigger than the others, which indicates the Co considered can chemically absorb on WS2 monolayer stably and Tw-WS2 is the more stable structure.

Figure 1. (a) (b) Top and side views of monolayer WS2, where yellow and orange balls stand for S atoms and W atoms, respectively.
3.2. The Influence of the Co-Adsorbed Monolayer WS2
Among the different adsorption sites considered here, the bond lengths dW-S near the adsorption sites have been extended in different levels, the distortion introduced by adsorption can not be ignored, which are used to check the deformation of the WS2 monolayer. From Figure 2 and Table 1, the dW-S near the Tw adsorption position is 2.51 Å, compared with the pure WS2 (2.42 Å), H-WS2 (2.48 Å) and Ts-WS2 (2.42 Å) systems, The change is the most obvious. Suggesting that the interaction between Co adatom and the WS2 sheet is the most powerful, Tw site can be referred to as the favored site. Here, the corresponding vertical equilibrium height “h” represents the distance between the adatom and WS2 hosts, it monotonically increases with increasing atomic size. Since the atom radius rS < rCo < rW, so the “h” of the Tw-WS2, H-WS2 and Ts-WS2 decreases successively. To illustrate the origin of the magnetic properties in Tw-WS2 system, the total density of states (TDOS) and charge density difference are presented in Figure 3 and Figure 4, respectively.
TDOS of pure WS2 and Tw-WS2 monolayers are shown in Figure 3, illustrating that the electronic properties of WS2 sheet is changed markedly upon the Co adatom. For isolated monolayer WS2, there is strong hybridization between the W-d orbitals and the p orbitals of S atoms, leading a strong covalent bonding between W and S, the sharing

Table 1. Calculated geometry of TW-WS2 system, adsorption energy of adatom Ea, total magnetic moment of the system Mtot, local magnetic moment of the Co adatom MCo, W-S bond length dW-S, height of the adsorption h.

Figure 2. (1)-(3) depict the optimized structures of Co atom adsorption on monolayer WS2 sheet ((a): top view, (b): side view. blue balls stand for Co atoms).

Figure 3. The total and partial density of states of clear monolayer WS2 and Tw-WS2 system, the PDOS of Co and the nearby W, S atom.

Figure 4. The electron charge density difference for Tw-WS2 system, where red and green contours stand forgiven or loss electron density region, respectively.
degree of electronic states is high from ~−4.00 to ~−1.00 eV, which forming a strong σ bond; Near the Fermi level of the valence band, the contribution of the electronic states is more from the W atoms with a weaker π bond formed, which is unstable. After a single Co adsorption, the band structure has been modified, especially near the Fermi level, where the conduction band as well as the valence band moves down. The Fermi level of TW-WS2 systems shift up more or less, which shows the impurity states leading from the Co adatom is very distinct in the original state of WS2 host. Since the occupation situation of the spin-up and spin-down states is very uneven, the spin-down states are full occupied while the spin-up states are not. The adsorption of transition metal Co atom induce a magnetic moment about 1.00 μB in WS2, which due to the strong hybridization between the d orbitals (came from the nearby W and Co atoms) and a little from the p orbitals (are from the nearby S atoms). Especially, the impurity states near the Fermi level mainly come from the Co atom.
To further explore the electronic distribution of Tw-WS2 system, the charge density difference ( ) is determined by the following function:
) is determined by the following function:
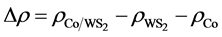 (2)
(2)
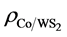 ,
, 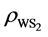 and
and 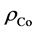 are the charge density of Tw-WS2, WS2 sheet and isolated Co atom, respectively. It clearly shows that the charge density increases in the region between Co adatom and W or S atoms and causes the electron density of surrounding S atoms to loss. This suggests that all of Co, W and S atoms interact with each other. These results are consistent with the analysis of the density of states, illustrating that there is a strong hybridization between Co adatom and the surface of the sheet.
are the charge density of Tw-WS2, WS2 sheet and isolated Co atom, respectively. It clearly shows that the charge density increases in the region between Co adatom and W or S atoms and causes the electron density of surrounding S atoms to loss. This suggests that all of Co, W and S atoms interact with each other. These results are consistent with the analysis of the density of states, illustrating that there is a strong hybridization between Co adatom and the surface of the sheet.
3.3. The Properties under the Different ML
In order to further study the effect of Co adsorption to the properties of WS2 under the different monolayer coverage (ML). We investigate the electronic and magnetic properties of Tw-WS2 in the 0.5 ML and 1.0 ML (the number ratio of Co:W = 1:2 and 1:1), as shown in Figure 5. Initially, we calculate the energy differences ( ) between the ferromagnetic ground state and the antiferromagnetic state at first, which is defined as following:
) between the ferromagnetic ground state and the antiferromagnetic state at first, which is defined as following:
 (3)
(3)
 and
and  are the total energy of the ferromagnetic ground state and the antiferromagnetic state under the same kind of monolayer coverage. From Table 2, the
are the total energy of the ferromagnetic ground state and the antiferromagnetic state under the same kind of monolayer coverage. From Table 2, the  is positive, the total energy of the ferromagnetic ground state lower than that of the antiferromagnetic state. It is clear that the Co atoms preferentially display ferromagnetic coupling at both the 0.5 ML and 1.00 ML.
is positive, the total energy of the ferromagnetic ground state lower than that of the antiferromagnetic state. It is clear that the Co atoms preferentially display ferromagnetic coupling at both the 0.5 ML and 1.00 ML.
Additional insight into the electronic structure of the Co-adsorbed WS2 monolayer can be obtained from the electronic total density of states (TDOS) displayed in Figure 6. For the 0.5 ML and 1.0 ML, the TDOS shows the impurities state density wave peaks

Figure 5. (a) (b) depict the structures of the ferromagnetic ground state and the antiferromagnetic state under the different Co ML (0.5 ML and 1.0 ML), where green and red contours repre- sent spin-up and spin-down, respectively.
Table 2. Calculated geometry of TW-WS2 system.

Figure 6. The total density of states under 0.5 ML and 1.0 ML (left), the energy band diagram of clear monolayer WS2 and 1.0 ML system.
straddling the Fermi level in the spin down channels, while the spin up channels continues to display an appreciable gap, indicating that the doped monolayer is essentially half-metallic.
In order to illustrate the significant changes caused by Co adsorption in the nature of the monolayer WS2, we calculated the band structure of the pure monolayer WS2 and Tw-WS2 systems (1.0 ML). Form Figure 6, It clearly shows that the band of the spin up and spin down channels are exactly the same, monolayer WS2 is a direct gap semiconductor, the valence band top and the conduction band bottom are located at the K point in the Brillouin zone. The gap is about 1.82 eV, which consistent with the experimental value and theoretical value [26] [27] . For 1.0 ML Tw-WS2, the spin up structure becomes a indirect band gap of 0.99 eV semiconductor, while the spin down channel is characterized by the properties of the metal (Figure 6). Thus, it promises to be a compelling and feasible candidate-halfmetallic material for low-dimensional spintronic devices in the future.
4. Conclusion
We have performed first-principles electronic structure calculations to study the properties of Co-adsorbed monolayer WS2, and found that the Tw site is stable position of the Co adsorbed on the surface of the sheet, and the local magnetic moment is about 0.94 μB focused on the Co atomic adsorption. For the Tw-WS2, we further investigate the stability of the ferromagnetic (FM) and antiferromagnetic (AFM) states under the different monolayer coverage (ML). The calculation results of formation energy show that the ferromagnetic (FM) configurations are more stable at 0.50 ML and 1.0 ML. Both of the local electronic density states (LDOS) and band calculation indicate that two kinds of structure present the half-metal property.
Cite this paper
Xu, W.Y. (2016) Electronic Structures and Magnetic Properties of Co-Adsorbed Monolayer WS2. Journal of Materials Science and Chemical Engineering, 4, 32-41. http://dx.doi.org/10.4236/msce.2016.410004
References
- 1. Butler, S.Z., Hollen, S.M., Cao, L., Cui, Y., et al. (2013) Progress, Challenges, and Opportunities in Two-Dimensional Materials beyond Graphene. ACS Nano, 4, 2898-2926.
http://pubs.acs.org/doi/abs/10.1021/nn400280c
http://dx.doi.org/10.1021/nn400280c - 2. Chhowalla, M., Shin, S.H., Eda, G., Li, L.-J., Loh, K.P. and Zhang, H. (2013) The Chemistry of Two-Dimensional Layered Transition Metal Dichalcogenidenanosheets. Nature Nanotechnology, 5, 263-275.
http://www.nature.com/search?journal=nchem&q=10.1038%2FNCHEM.1589 - 3. Liu, H., Han, H. and Zhao, J. (2015) Atomistic Insight into the Oxidation of Monolayer Transition Metal Dichalcogenides: From Structures to Electronic Properties. RSC Advances, 5, 17572-17572.
http://pubs.rsc.org/en/Content/ArticleLanding/RA/2015/C4RA17320A
http://dx.doi.org/10.1039/C4RA17320A - 4. Yazyev, O.V. and Kis, A. (2014) MoS2 and Semiconductors in the Flatland. Materials Today, 18, 1369-7021.
- 5. Ding, Y., Wang, Y., Ni, J., Shi, L., Shi, S. and Tang, W. (2011) First Principles Study of Structural, Vibrational and Electronic Properties of Graphene-Like MX2 (M = Mo, Nb, W, Ta; X = S, Se, Te) Monolayers. Physica B, 406, 2254-2260.
https://www.researchgate.net/publication/229313422
http://dx.doi.org/10.1016/j.physb.2011.03.044 - 6. Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. and Kis, A. (2011) Single-Layer MoS2 Transistors. Nature Nanotechnology, 6, 147-150.
https://www.researchgate.net/publication/49796080
http://dx.doi.org/10.1038/nnano.2010.279 - 7. Wang, Q.H., Kis, A., Kourosh, K.-Z., Coleman, J.N. and Strano, M.S. (2012) Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nature Nanotechnology, 7, 699-712.
http://www.nature.com/doifinder/10.1038/nnano.2012.193
http://dx.doi.org/10.1038/nnano.2012.193 - 8. Drescher, T., Niefind, F., Bensch, W. and Grunert, W. (2012) Sulfide Catalysis without Coordinatively Unsaturated Sites: Hydrogenation, Cis-Trans Isomerization, and H2/D2 Scrambling over MoS2 and WS2. Journal of the American Chemical Society, 134, 18896- 18899.
http://dx.doi.org/10.1021/ja3074903 - 9. Balog, R., et al. (2010) Bandgap Opening in Graphene Induced by Patterned Hydrogen Adsorption. Nature Materials, 9, 315-319. https://www.researchgate.net/publication/41944338
http://dx.doi.org/10.1038/nmat2710 - 10. Shiva, K., Ramakrishna Matte, H.S.S., Rajendra, H.B., Bhattacharyya, A.J. and Rao, C.N.R. (2013) Employing Synergistic Interactions between Few-Layer WS2 and Reduced Graphene Oxide to Improve Lithium Storage, Cyclability and Ratecapability of Li-Ion Batteries. Nano Energy, 2, 787-793.
http://dx.doi.org/10.1016/j.nanoen.2013.02.001 - 11. Zhou, L., Yan, S., Pan, L., et al. (2016) A Scalable Sulfuration of WS2 to Improve Cyclability and Capability of Lithium-Ion Batteries. Nano Research, 9, 857-865.
http://link.springer.com/article/10.1007/s12274-015-0966-9
http://dx.doi.org/10.1007/s12274-015-0966-9 - 12. Yue, Q., Shao, Z., Chang, S. and Li, J. (2013) Adsorption of Gas Molecules on Monolayer MoS2 and Effect of Applied Electric Field. Nanoscale Research Letters, 8, 425.
http://link.springer.com/article/10.1186%2F1556-276X-8-425
http://dx.doi.org/10.1186/1556-276X-8-425 - 13. Rastogi, P., Kumar, S., Bhowmick, S., Agarwal, A. and Chauhan, Y.H. (2014) Doping Strategies for Monolayer MoS2 via Surface Adsorption: A Systematic Study. Journal of Physical Chemistry C, 118, 30309-30314.
http://dx.doi.org/10.1021/jp510662n - 14. Ataca, C. and Ciraci, S. (2011) Functionalization of Single-Layer MoS2 Honeycomb Structures. Journal of Physical Chemistry C, 115, 13303-13311.
http://dx.doi.org/10.1021/jp2000442 - 15. Dolui, K., Rungger, I., Pemmaraju, C.D. and Sanvito, S. (2013) Possible Doping Strategies for MoS2 Monolayers: An Ab Initio Study. Physical Review B, 88, Article ID: 075420.
http://dx.doi.org/10.1103/PhysRevB.88.075420 - 16. Rastogi, P., Kumar, S., Bhowmick, S., Agarwal, A. and Chauhan, Y.H. (2014) Doping Strategies for Monolayer MoS2 via Surface Adsorption: A Systematic Study. Journal of Physical Chemistry C, 118, 30309-30314.
http://dx.doi.org/10.1021/jp510662n - 17. Chang, J., Larentis, S., Tutuc, E., Register, L.F. and Banerjee, S.K. (2014) Atomistic Simulation of the Electronic States of Adatoms in Monolayer MoS2. Applied Physics Letters, 104, Article ID: 141603.
http://dx.doi.org/10.1063/1.4870767 - 18. Li, X.D., Fang, Y.M., Wu, S.Q. and Zhu, Z.Z. (2015) Adsorption of Alkali, Alkaline-Earth, Simple and 3D Transition Metal, and Nonmetal Atoms on Monolayer MoS2. AIP Advances, 5, Article ID: 057143.
http://dx.doi.org/10.1063/1.4921564 - 19. Zhao, X., Xia, C., Wang, T. and Dai, X. (2016) Electronic and Magnetic Properties of X-Doped (X = Ti, Zr, Hf) Tungsten Disulphide Monolayer. Journal of Alloys and Compounds, 654, 574-579.
http://dx.doi.org/10.1016/j.jallcom.2015.09.160 - 20. Menon, M. and Andriotis, A.N. (2014) Tunable Magnetic Properties of Transition Metal Doped MoS2. Physical Review B, 90, Article ID: 125304.
- 21. Ramasubramaniam, A. (2013) Mn-Doped Monolayer MoS2: An Atomically Thin Dilute Magnetic Semiconductor. Physical Review B, 87, Article ID: 195201.
http://dx.doi.org/10.1103/physrevb.87.195201 - 22. Matte, H.S.S.R., Gomathi, A., Manna, A.K., Late, D.J., Datta, R., Pati, S.K. and Rao, C.N.R. (2010) MoS2 and WS2 Analogues of Graphene. Angewandte Chemie International Edition, 49, 4059-4062.
http://dx.doi.org/10.1002/anie.201000009 - 23. Yang, J., Voiry, D., Ahn, S.J., Kang, D., Kim, A.Y., Chhowalla, M. and Shin, H.S. (2013) Two-Dimensional Hybrid Nanosheets of Tungsten Disulfide and Reduced Graphene Oxide as Catalysts for Enhanced Hydrogen Evolution. Angewandte Chemie International Edition, 52, 13751-13754.
http://onlinelibrary.wiley.com/doi/10.1002/anie.201307475/full
http://dx.doi.org/10.1002/anie.201307475 - 24. Fang, X.P., Hua, C.X., Wu, C.R., Wang, X.F., Shen, L.Y., Kong, Q.Y., Wang, J.Z., Hu, Y.S., Wang, Z.X. and Chen, L.Q. (2013) Synthesis and Electrochemical Performance of Graphene-Like WS2. Chemistry, 19, 5694-5700.
http://dx.doi.org/10.1002/chem.201204254 - 25. Zhao, W.J., Ghorannevis, Z., Chu, L.Q., Toh, M.L., Kloc, C., Tan, P.H. and Eda, G. (2013) Evolution of Electronic Structure in Atomically Thin Sheets of WS2 and WSe2. ACS Nano, 7, 791-797.
http://dx.doi.org/10.1021/nn305275h - 26. Zhao, X., Dai, X.Q., Xia, C.G., Wang, T.X. and Peng, Y.T. (2015) Electronic and Magnetic Properties of Mn-Doped Monolayer WS2. Solid State Communications, 215-216, 1-4.
http://dx.doi.org/10.1016/j.ssc.2015.05.003 - 27. Li, H., Liu, S., Huang, S., Yin, D., Li, C. and Wang, Z. (2016) Impurity-Induced Ferromagnetism and Metallicity of WS2 Monolayer. Ceramics International, 42, 2364-2369.
http://dx.doi.org/10.1016/j.ceramint.2015.10.033 - 28. Ma, D. and Yang, Z. (2011) First Principles Studies of Pb Doping in Graphene: Stability, Energy Gap, and Spin-Orbit Splitting. New Journal of Physics, 13, Article ID: 123018.
http://dx.doi.org/10.1088/1367-2630/13/12/123018 - 29. Dai, X., Li, Y., Xie, M., Hu, G., Zhao, J. and Zhao, B. (2011) Structural Stability and Electronic, Magnetic Properties of Ge Adsorption on Defected Graphene: A First-Principles Study. Physica E, 43, 1461-1464.
http://dx.doi.org/10.1016/j.physe.2011.04.006 - 30. Kresse, G. and Hafner, J. (1993) Ab Initio Molecular Dynamics for Open-Shell Transition Metals. Physical Review B, 48, 13115-13118.
http://dx.doi.org/10.1103/PhysRevB.48.13115 - 31. Kresse, G. and Furthmüller, J. (1996) Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Computational Materials Science, 6, 15-50.
http://dx.doi.org/10.1016/0927-0256(96)00008-0 - 32. Kresse, G. and Furthmüller, J. (1996) Efficient Iterative Schemes for ab initio Total-Energy Calculations Using a Plane-Wave Basis Set. Physical Review B, 54, 11169-11186.
http://dx.doi.org/10.1103/PhysRevB.54.11169 - 33. Perdew, J.P. and Zunger, A. (1981) Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems. Physical Review B, 23, 5048-5079.
http://dx.doi.org/10.1103/PhysRevB.23.5048 - 34. Okamoto, Y. and Miyamoto, Y. (2001) Ab Initio Investigation of Physisorption of Molecular Hydrogen on Planar and Curved Graphenes. Journal of Physical Chemistry B, 105, 3470-3474.
http://dx.doi.org/10.1021/jp003435h - 35. Ao, Z., Jiang, Q., Zhang, R., Tan, T. and Li, S. (2009) Al Doped Graphene: A Promising Material for Hydrogen Storage at Room Temperature. Journal of Applied Physics, 105, Article ID: 074307.
http://dx.doi.org/10.1063/1.3103327 - 36. Zhao, S.J., Xue, J.M. and Kang, W. (2014) Gas Adsorption on MoS2 Monolayer from First-Principles Calculations. Chemical Physics Letters, 595-596, 35-42.
http://dx.doi.org/10.1016/j.cplett.2014.01.043 - 37. Hu, A.M., Wang, L.L., Meng, B. and Xiao, W. (2015) Ab Initio Study of Magnetism in Nonmagnetic Metal Substituted Monolayer MoS2. Solid State Communications, 220, 67-71.
http://dx.doi.org/10.1016/j.ssc.2015.07.011 - 38. Perdew, J.P. and Zunger, A. (1981) Self-Interaction Correction to Density-Functional Approximations for Many-Electron Systems. Physical Review B, 23, 5048-5079.
http://dx.doi.org/10.1103/PhysRevB.23.5048 - 39. Monkhorst, H.J. and Pack, J.D. (1976) Special Points for Brillouin-Zone Integrations. Physical Review B, 13, 5188-5192.
http://dx.doi.org/10.1103/PhysRevB.13.5188