Advances in Materials Physics and Chemistry
Vol.05 No.09(2015), Article ID:59454,8 pages
10.4236/ampc.2015.59038
In-Situ Hydroelectrothermal Deposition of Silicate Layers on Stainless Steel Surfaces
Jaybalan Tamahrajah, Axel Brehm
Department of Technical Chemistry, CvO University of Oldenburg, Oldenburg, Germany
Email: j.tamahrajah@uni-oldenburg.de, axel.brehm@uni-oldenburg.de
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 August 2015; accepted 5 September 2015; published 8 September 2015
ABSTRACT
The deposition of zeolites on solid support materials is possible by means of electrochemical methods, impregnation processes, as well as in-situ syntheses. Electrochemical deposition of zeolites has been reported as well, however with readily synthesized zeolite structures. Adhesive deposition of zeolites on stainless steel (S316) has been reported. This report investigates the feasibility of the deposition of silicates by in-situ hydroelectrothermal means. The investigation was done in aqueous solutions of pH = 7 to 13 at different temperatures (25˚C to 70˚C) by linear sweep method. Deposition was done at a saturated H2 atmosphere to ensure prior deposition of thin iron oxide film on the surface and formation of Fe-O-Si-linkages. This was proven by Raman measurement of the samples. Further linear sweep experiments in the presence of silica show monodentate and bidentate Fe-O-Si linkages on the surface, proven by IR-measurements. Presence of dissolved silica was done by UV-Vis with the molybdate yellow method. The best results are achieved at 70˚C at pH 13 and −4 mV (vs Ag/AgCl) or 200 mV (vs SHE). Discontinuous homogeneous layers are found on the stainless steel surface observed by SEM, EDX measurements and electrochemical measurements. Layer discontinuties are caused due to low silica concentration at equilibrium hydrothermal conditions, especially in the absence of silicic acid. All results shown are for the best results achieved except for linear sweep measurements and solubility constants of dissolved silica.
Keywords:
Hydroelectrothermal Deposition, Silica, Stainless Steel, Silicates, Linear Sweep

1. Introduction
Zeolites have been known for their application in heterogeneous catalysis and adsorption purposes [1] . Their adhesion on structured catalytic packings (KATAPAK®) [2] and deposition on electrodes have been proven to have advantages in their respective fields [1] . The deposition with good adhesion must however be guaranteed. The good adhesion achieved in the literature cited above has proven to have a science of its own.
Deposition of dissolved silica on stainless steel surface has been reported in literature for corrosion prevention [3] - [5] . It would be a great advantage for corrosion experts if inexpensive natural amorphous silica can be deposited at higher amount on stainless steel surface and simultaneously extend the hydroelectrothermal deposition to present hydrothermal to zeolite syntheses procedures. As the entropy electrochemically is lower (consider the theoretical efficiency of a fuel cell of 82% compared to a steam cycle of around 40% at best), the temperature for hydrothermal syntheses can be drastically reduced and further increase specific silicic acid species at preferred potentials for rapid crystallization.
IR and Raman measurements of iron oxides with silica have been investigated by Russell [6] , Li et al. [7] . Interaction between silicic acid and and ferrihydrites (siliceous ferrihydrite), ferrous oxides were investigated by Naren et al. [8] , Hazel et al. [9] , Hansen et al. [10] , [11] . Most importantly, Swedlund et al. [12] proved the bidentate Fe-O-Si linkages by means of IR spectroscopy. Swedlund [13] also proved monodendate (at low silicic acid concentrations) and bidentate linkages (at high silicic acid concentrations) form polymerization of silicates on the surface of a substrate. This hence forms a silicate layer via formation of linkages with preformed ferrihydrite layer as observed in Figure 1.
2. Experiments
2.1. UV-Vis-Existence of Dissolved Silica (Silicic Acid) in Sample Solution
In order to ensure the silica deposition, existence of dissolved silica (silicic acid) has to be guaranteed. Hence the concentration of silicic acid as reactant at equilibrium condition has to be measured first. Non-dissociated silicic acid is the reactant in the dissociation of silicic acid and has the formula Si(OH)4. In the solubility reaction, more than 99 % of the silicic acid is in the monomeric form [14] and the polymeric forms are dissociated upon reaction with molybdic acid for photospectrometric measurements [14] .
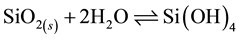 (1)
(1)
 (2)
(2)
where the activity of SiO2 and H2O being unity by definition for a solid and high concentration of H2O in the system investigated. Furthermore, according to Swedlund [12] , monosilicic acid predominantly exists in the presence of ferrihydrites and Fe-ion in both oxidation states and its oxides (>99%) [12] . The method of Iler is recommended and proved to be the most reliable in literature [14] . UV-Vis measurements are done to validate the presence of silicic acid in the sample solution without any presence of iron. Schenk and Weber proved the stability of silica concentration in the presence of iron upon achieving equilibrium [15] . Calibration measurements for neutral solutions are done for 100 ppm, 250 ppm, 500 ppm, 1000 ppm. Calibration measurements for pH = 12 and pH = 13 are done for higher concentrations (100 ppm to 10,000 ppm).

Figure 1. Schematic figure for the formation of silicate layers via interaction of iron oxide with silicic acid.
Calibration solutions are prepared by dissolving 1 g of amorphous silica in 1 M of NaCl solution (solution A) weighing 1 kg. This corresponds to 1000 ppm of silicic acid and the fact that NaCl is added to the solution does not affect the intensity of molybdate complex as only Si(OH)4 fits in the octahedral formed cage of the
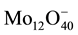 anion complex. Further calibration solutions are prepared by further diluting the solution A two-fold, four-fold and ten-fold with distilled water.
anion complex. Further calibration solutions are prepared by further diluting the solution A two-fold, four-fold and ten-fold with distilled water.
A 96% H2SO4 solution was diluted to a 4% solution (solution B-50 mL) and 5 g of ammonium heptamolybdate tetrahydrate ((NH4)6Mo7O24∙4H2O) is added to 40 ml distilled water and 2.36 g of 25% NH4OH solution and diluted to 50 mL (solution C). Water, solution B, and solution C were mixed with a 5:2:1 ratio (solution D). 8 mL of the solution D were mixed with the 2 mL calibration solution (CS) or the sample solution (SS) containing silica (Solution E). Solution E is diluted 50-fold for neutral solution and 250-fold (Solution F) to reduce the colour intensity to obey to the Beer-Lambert Law [16] .
The solution F is measured from 320 to 700 nm. The value at 410 nm is of great importance as the absorption value has a linear dependence on the concentration of silicic acid at this wavelength. Calibration measurements were done for every sample solution measured from 298.15 K to 443.15 K for pH 7, 12 and 13.
2.2. Linear Sweep Measurement
The electrochemical potential measurement (ECP) of iron was done with and without the presence of silica from 25˚C to 70˚C in a common electrochemical glass cell.
First the existence of the deposition must be proven in a simple setup. Hence, electrochemical linear sweep measurements are done in a simple glass cell. Further investigations will be done in an upcoming publication. The temperature was controlled through an outer jacket with a thermostat. Linear sweep measurements are favored as the reversible corrosion potential was measured without the presence of a passive layer on bare stainless steel. Cyclic voltammetry measurements would measure the potential after the formation of an existing passive layer upon returning from the anodic to the cathodic region, which should be avoided [17] .
A Pt-wire was used as the counter electrode, a 2 cm × 0.2 cm stainless steel probe as the working electrode and the reference electrode used is a Ag/AgCl electrode. The solution (1 × 10−3 mol/dm³ KNO3 solution with 0, 0.052 and 0.52 moles of NaOH to give a pH of 7, 12 and 13) was filled in a glass electrochemical cell and purged with N2 for 30 minutes. Hydrogen is injected to the system for 20 minutes while measuring the open circuit potential (OCP), then the linear sweep measurement is done between the values of −0.8 V to 0.5 V. The presence of the electrolyte does not affect the equilibrium concentration and dissociation constant of dissolved silica [14] [15] .
2.3. SEM and EDX Measurements
EDX-measurements provide the general prove of Fe2O3 for the calcined sample and goethite (FeOOH) for the uncalcined sample as well as silica deposition. EDX measurements together with SEM photos of the iron oxide formation were done in order to coarsely observe evidence of silica deposition under simultaneous iron oxide deposition. After linear sweep measurement, SEM and EDX measurements were made directly. This corresponds to the uncalcined sample. From the EDX Data, intensity of silica on the iron oxide deposition location is identified for both calcined and uncalcined samples. To achieve calcined samples, the uncalcined sample was calcined at 250˚C for 24 hours. Higher temperatures are avoided to prevent the formation of other iron oxide polymorphs such as maghemite [7] . Measurements are identical to the calcined samples and best results are shown.
2.4. Raman Measurements for the Existence of Pristine Iron Oxide
Raman measurement serve well for prove of pristine iron oxide under exposure of silicates as the silicate vibration are not distinct in the Raman vibration but only in the IR vibration mode. Hence, Si-O-Fe-linkages were proven in IR and iron oxide in Raman. The intensity of the radiation is limited to 10 mW with a laser frequency of 785 nm and at rather short integration times of 4 minutes. This parameter is set accordingly to prevent the formation of other oxide polymorphs such as maghemite for the calcined sample [7] . The focus is depicted on the microscope and the CCD camera, which was cooled at −90˚C. The focus was adjusted each time to get the best resolution. Raman measurements were done for the uncalcined sample as well. The intensity of the radiation was 5 mW but longer integration times (6 minutes) with a laser frequency of 488 nm was used for the sample from the electrochemical glass cell. A lower laser frequency was used to prevent oxides formation from goethite and keep the penetration depth lower as the experiments for the electrochemical glass cell was done for a short period (1 hour per experiment).
2.5. IR-Measurements―Determination of Monodendate and Bidendate Fe-O-Si Linkages
Rest deposits on the stainless steel surface were mechanically abraded and washed with distilled water several times and the products collected. The products were dried first at 105˚C for 24 hrs to remove excessive water and further calcined at 250˚C for 24 hrs to remove the rest of the water trapped. Samples were measured before and after calcination. Higher temperatures are not chosen, as this will cause a phase change from hematite to maghemite [7] . Prove of Si-O-H and Fe-O-Si bands are available in literature and its vibration magnitudes are distinguishable by the shifting between the extremes of the Si-O-H band and the Si-O band. This work is able to prove the existence of Fe-O-Si bonds with respect to previously published data by Russell [6] , Li et al. [7] and Swedlund [13] .
3. Results
3.1. UV-Vis Measurements
The results in Figure 2 show the logarithmic equilibrium constant of dissolved silica from Equation (2) with varying reciprocal temperature. The values are corresponds to the low concentration of dissolved silica especially at pH = 7. The solubility of silica increases to values of 10−2 mol/dm3 (pH = 13 and 70˚C), corresponding to 0.6 g/dm3 and provides better distinction of silica deposition on stainless steel. Some inconsistencies of silica deposition occur due to the still insufficient concentration of dissolved silica and heterogeneous surface of the stainless steel. If deposition is proven even at these mild conditions, the deposition should improve at high dissolved silica concentration and homogeneous surface of stainless steel. This will be proven in a later publication.
3.2. Linear Sweep Measurements
The low current density at lower potential is the corrosion potential of Fe2+ from Fe(s) and approaches the standard reduction potential at ideal corrosive conditions, namely pH = 13 and 70˚C [16] . Linear measurements with and without silica showed no signs of passivation at pH = 7 and less passivation at pH = 12. Passivation of iron oxide in Figure 3 occurred slightly below 0.00V (vs Ag/AgCl) at pH = 13 for T = 298.15 K and 333.15 K, whereas it was slightly increased to positive values at T = 343.15 K. However, the linear sweep measurements without dissolved silica in the solution in Figure 3 show less passivation at anodic regions of the Tafel diagram due to instable current densities. Raman measurements prove the existence of mixed pristine iron oxide, which may cause a formation of an instable layer.

Figure 2. Logarithmic equilibrium constants determined from Equation (2) from UV-Vis measurements (data of Greenberg [18] at pH = 13 extrapolated from pH = 10.35 to 10.8. Other data are taken from Elmer et al. [19] and Gunnarson et al. [20] ).
![]()
Figure 3. The linear sweep measurements with varying pH (vertical 7 to 13 from top to bottom) and temperature (horizontal, 25˚C to 70˚C from left to right) for deposition without silica.
The values at −0.04 V (vs Ag/AgCl, ~200 mV vs SHE) correspond to the formation of FeOOH hydroxides on the surface as proven by uncalcined Raman measurements. This is also mentioned by Landolt [21] for corrosion potential for a passive layered stainless steel electrode, hence reducing the current density. Reduction of the current density in the anodic region proves the formation of passive layers, as observed in SEM and EDX images. At dry conditions, Fe2O3 forms as shown by calcined Raman measurements as well. The experiments containing dissolved silica in Figure 4 show potentials at the same region, which concludes the formation of natural goethite FeOOH [21] . Prove of the hydroxide forming Fe-O-Si linkages is given by IR measurements. The passivation with dissolved silica in Figure 4 seems to be more stable than without silica due to the stable current density at −0.04 to 0.0 V (vs Ag/AgCl), giving hints of a stable passivation layer. No potential change occurs as there is no charge transfer occurring during the reaction of FeOOH and Si(OH)4. The OH− is cleaved from the Fe-O layer and forms a monodendate bond with![]() .
.
However, some discontinuity of the silicate layer can be still observed from SEM and EDX measurements, due to the low concentration of dissolved silica (see UV-Vis measurements). All forthcoming measurements are done for stainless steel surfaces, which have been exposed to dissolved silica only. Measurements on bare iron oxide layers are not feasible due to their low stability as proven by linear sweep methods. Hence the measurements are not representative and do not fulfill the target of strong layer stability for this investigation.
3.3. SEM and EDX Measurements
The brighter silicate layer from the stainless steel sample is shown in Figure 5(a) and Figure 5(b). However, the stainless steel surface is partly uncovered due to the low silicic acid concentration and heterogeneous surface of the stainless steel.
3.4. Raman Measurements
The Raman measurements for calcined and uncalcined samples are shown in Figure 6. Raman spectra are taken from the samples to identify the form of present iron oxide as hematite or goethite in the aqueous solution [7] . The calcined powder shows signs of a mixed pristine Fe2O3.
![]()
Figure 4. The linear sweep measurements with varying pH (vertical 7 to 13 from top to bottom) and temperature (horizontal, 25˚C to 70˚C from left to right) for deposition with silica.
![]() (a) (b)
(a) (b)
Figure 5.The figures (a) and (b) show the SEM and EDX images of the stainless steel sample.
![]() (a) (b)
(a) (b)
Figure 6. Raman measurement of calcined ((a)―green) and uncalcined sample ((b)―green) with references of pristine iron oxide (black and red [22] ) and goethite (blue) [23] .
Raman shifts at 120, 226, 250, ~400, 540, 610, 650, 740, 822, 1175 cm−1, 1250 cm−1 for the hematite type 1 (R060190) and 2 are identified. Additionally a shift at 950 cm−1 for hematite type 2 (R040024) is observed. This proves the existence of the mixed hematite.
The deposition in the electrochemical cell is not calcined and showed prove of goethite at Raman shifts of 247, 300, 400 cm−1 and Fe2O3 in a mixed form with peaks from 120 to 300 cm−1. The goethite and hematite peaks above 650 cm−1 is too low to be observed, however the 400 cm−1 peak gives a good proof for the existence of goethite. This is why the peak is not sharp as the superposition of mixed iron oxides (type 1 and 2) and goethite gives the sharp edge at 120 cm−1 and linear reduction up to 400 cm−1.
Hence it is very safe to take the formation of hematite respectively goethite (existence of Fe2+ ions). Nevertheless, the Raman spectrum shows the development of the oxide form of iron, which is hematite.
3.5. IR Measurements
The IR measurements are shown in Figure 7. The Si-O-H band at 1080 cm−1, gives the existence of the Si-O-H bonds. The band at 940 - 990 cm−1 is the Si-O vibration as reported by Naren et al. [8] . Si(OH)4 binds monodendately or bidendately to FeOOH on the stainless steel surface, causing significant amounts of Si-O-Fe linkages. The higher the silicate amount being ligands to ferrihydrite, the further the band shifts to the lower frequency band. Bands for the presence of water are observed at 3430 cm−1 and 1630 cm−1. The black and red spectra (for low and high calcination temperatures of 105˚C and 250˚C respectively) show a significant band at 990 cm−1. This proves the existence of Si-O-Fe-linkages on the surface. The IR-spectrum for iron oxide (Fe2O3) is shown for comparison of the IR band shift.
4. Discussion
Prove of the existence of dissolved silica is given by the molybdate yellow method of Iler [14] . The linear sweep measurements were done for the sample with and without the presence of silica in order to observe differences in the current densities. The current densities were significantly unstable due to the instability of pristine iron oxide. The defined abrupt decrease in current density at around 0 V vs Ag/AgCl electrode and around 200 mV (vs SHE) gives the defined formation of the passive layer. This shows that silica brings some stability to the passive layer. Hence, the SEM, EDX, Raman and IR measurement were done for the passive layer of the stainless steel for samples which have been exposed to dissolved silica only. From all measurements, the silicate layer can be observed for both calcined and uncalcined samples. However, there are some discontinuities of the silicate layer on the surface of the stainless steel. This could be improved by increasing the concentration of dissolved silica or by providing a homogeneous stainless steel layer and increasing the deposition time.
5. Conclusion
Hydroelectrothermal deposition of silicate layers has been proven in this investigation. The findings show stable deposition of silicate layers at around −4 mV at 70˚C and pH = 13. IR-measurements proved to be a powerful measurement tool to indicate Fe-O-Si linkages. Raman measurements were important to prove the existence of pristine iron oxide. However, the discontinuities of the deposition are due to the low concentration of dissolved
![]()
Figure 7. IR-Spectra of both low calcination temperature (black) and high calcination temperature samples (red). The IR- Spectrum for Fe2O3 is shown for comparison of the IR band shift.
silica (silicic acid). The improvement of the silicate layer deposition could be improved by increasing the concentration of dissolved silica. An increase in temperature or pH will increase the silicic acid concentration and hence increases the deposition. Longer depositions periods may improve the deposition as well. Further research can be done to hydrothermally deposit silicates and aluminosilicates by integrating electrochemical methods. This is a promising pathway of saving energy for conventional hydrothermal processes concerning silicates.
6. Instruments Used for This Investigation
6.1 UV-Vis Measurements
The UV-Vis measurements were done with a SPEKOL 1000 spectrometer from Analytik Jena.
6.2. Linear Sweep Measurement
The linear sweep method was employed with a potentiostat from AMEL Instruments of the model MOD.7050. The electrochemical glass cell is from the company MetroOHM.
6.3. SEM and EDX Measurements
The SEM sampling was done with a Hitachi S3200-N SEM device with images processed and scanned by the Digital Processing and Scanning System 5 software respectively. EDX measurements were done with the INCAx-act device from the company Oxford with its respective software for image processing. A α-Cu cathode was used for the sampling.
6.4. Raman Measurements
The Raman measurements were done with a Bruker Sentera Raman measurement device.
6.5. IR-Measurements
The IR-measurements were done by Golden gate (ATR) method in a Bruker Sentera 27 Apparatus from 300 cm−1 to 4000 cm−1 with the OPUS V6.5 spectral processing software.
Acknowledgements
The authors thank Prof. Dr. Gunther Wittstock for UV-Vis measurements, Dr. Dereje H. Taffa and Prof. Dr. Michael Wark for providing resources for electrochemical measurements. The authors also thank Florian Loose for assistance in IR measurements and Next Energy Research Institute to access the Raman measurement device. The authors thank the anonymous reviewers for their constructive comments.
Cite this paper
JaybalanTamahrajah,AxelBrehm, (2015) In-Situ Hydroelectrothermal Deposition of Silicate Layers on Stainless Steel Surfaces. Advances in Materials Physics and Chemistry,05,374-382. doi: 10.4236/ampc.2015.59038
References
- 1. Auerbach, S.M., Carrado, K.A. and Dutta P.K. (2003) Handbook of Zeolite Science and Technology. Marcel Dekker Inc., New York and Basel.
http://dx.doi.org/10.1201/9780203911167 - 2. Brehm, A. and Zanter, K.D. (2002) Formation of Zeolite (MFI) Layers on Gauze Wire and Arranged Packing Segment. Chemical Engineering & Technology, 25, 917-920.
http://dx.doi.org/10.1002/1521-4125(20020910)25:9<917::AID-CEAT917>3.0.CO;2-X - 3. Browman, M.G., Robinson, R.B. and Reed, G.D. (1989) Silica Polymerization and Other Factors in Iron Control by Sodium-Silicate and Sodium-Hypochlorite Additions. Environmental Science & Technology, 23, 566-572.
http://dx.doi.org/10.1021/es00063a009 - 4. Vasconcelos, D.C.L., Carvalho, J.A.N., Mantel, M. and Vasconcelos, W.L. (2000) Corrosion Resistance of Stainless Steel Coated with Sol-Gel Silica. Journal of Non-Crystalline Solids, 273, 135-139.
http://dx.doi.org/10.1016/S0022-3093(00)00155-1 - 5. Thim, G.P., Oliveira, M.A.S., Oliveira, E.D.A. and Melo, F.C.L. (2000) Sol-Gel Silica Film Preparation from Aqueous Solutions for Corrosion Protection. Journal of Non-Crystalline Solids, 273, 124-128.
http://dx.doi.org/10.1016/S0022-3093(00)00125-3 - 6. Russell, J.D. (1979) Infrared Spectroscopy of Ferrihydrite: Evidence for the Presence of Structural Hydroxyl-Groups. Clay Minerals, 14, 109-114.
http://dx.doi.org/10.1180/claymin.1979.014.2.03 - 7. Li, Y.S., Church, J.S. and Woodhead, A.L. (2012) Infrared and Raman Spectroscopic Studies on Iron Oxide Magnetic Nano-Particles and Their Surface Modifications. Journal of Magnetism and Magnetic Materials, 324, 1543-1550.
http://dx.doi.org/10.1016/j.jmmm.2011.11.065 - 8. Naren, G., Ohashi, H., Okaue, Y. and Yokoyama, T. (2013) Adsorption Kinetics of Silicic Acid on Akaganeite. Journal of Colloid and Interface Science, 399, 87-91.
http://dx.doi.org/10.1016/j.jcis.2013.02.032 - 9. Hazel, F., Schock, R.U. and Gordon, M. (1949) Interaction of Ferric Ions with Silicic Acid. Journal of the American Chemical Society, 71, 2256-2257.
http://dx.doi.org/10.1021/ja01174a516 - 10. Hansen, H.C.B., Wetche, T.P., Raulundrasmussen, K. and Borggaard, O.K. (1994) Stability-Constants for Silicate Adsorbed to Ferrihydrite. Clay Minerals, 29, 341-350.
http://dx.doi.org/10.1180/claymin.1994.029.3.05 - 11. Hansen, H.C.B., Rabenlange, B., Raulundrasmussen, K. and Borggaard, O.K. (1994) Monosilicate Adsorption by Ferrihydrite and Goethite at pH 3-6. Soil Science, 158, 40-46.
http://dx.doi.org/10.1097/00010694-199407000-00005 - 12. Swedlund, P.J., Miskelly, G.M. and McQuillan, A.J. (2009) An Attenuated Total Reflectance IR Study of Silicic Acid Adsorbed onto a Ferric Oxyhydroxide Surface. Geo-chimica et Cosmochimica Acta, 73, 4199-4214.
http://dx.doi.org/10.1016/j.gca.2009.04.007 - 13. Swedlund, P.J. and Webster, J.G. (1999) Adsorption and Polymerisation of Silicic Acid on Ferrihydrite, and Its Effect on Arsenic Adsorption. Water Research, 33, 3413-3422.
http://dx.doi.org/10.1016/S0043-1354(99)00055-X - 14. Iler, R.K. (1979) The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties, and Biochemistry. John Wiley and Sons Ltd., New York.
- 15. Schenk, J.E. and Weber Jr., J.T. (1968) Chemical Interactions of Dissolved Silica with Iron(II) and Iron(III). Journal (American Water Works Association), 60, 199-212.
- 16. Atkins, P.W. and De Paula, J. (2005) The Elements of Physical Chemistry. Oxford University Press, New York.
- 17. Kelly, R.G., Scully, J.R., Shoesmith, D.W. and Buchheit, R.G. (2003) Electrochemical Techniques in Corrosion Science and Engineering. Marcel Dekker Inc, New York, Basel.
- 18. Greenberg, S.A. and Price, E.W. (1957) The Solubility of Silica in Solutions of Electrolytes. Journal of Physical Chemistry, 61, 1539-1541.
http://dx.doi.org/10.1021/j150557a019 - 19. Elmer, T.H. and Nordberg, M.E. (1958) Solubility of Silica in Nitric Acid Solutions. Journal of the American Ceramic Society, 41, 517-520.
http://dx.doi.org/10.1111/j.1151-2916.1958.tb12907.x - 20. Gunnarsson, I. and Arnorsson, S. (2000) Amorphous Silica Solubility and the Thermodynamic Properties of H4SiO°4 Degrees in the Range of 0° to 350°C at Psat. Geochimica et Cosmochimica Acta, 64, 2295-2307.
http://dx.doi.org/10.1016/S0016-7037(99)00426-3 - 21. Landolt, D. (2007) Corrosion and Surface Chemistry of Metals. EPFL Press, Taylor and Francis Group LLC, Lausanne.
http://dx.doi.org/10.1201/9781439807880 - 22. RRUFF (2015) Hematite Raman Spectral Data.
http://rruff.info/Hematite - 23. RRUFF (2015) Goethite Raman Spectral Data.
http://rruff.info/Goethite/R120086